

Abstract
The purpose of this study was to investigate the pre- and postsynaptic mechanisms that contribute to synaptic facilitation in the myenteric plexus of the trinitrobenzene sulphonic acid-inflamed guinea-pig distal colon. Intracellular recordings of evoked fast excitatory postsynaptic potentials (fEPSPs) in myenteric S neurons were evaluated, and the density of synaptic terminals was morphometrically analysed by transmission electron microscopy. In inflamed tissue, fEPSPs were reduced to control levels by the protein kinase A (PKA) inhibitor, H89, but H89 did not affect the fEPSPs in control tissue. This PKA activation in inflamed tissue did not appear to involve 5-HT4 receptors because the antagonist/inverse agonist, GR 125487, caused comparable decreases of fEPSPs in both tissues. Inhibition of BK channels with iberiotoxin did not alter the fEPSPs in inflamed tissue, but increased the fEPSPs in control tissue to the amplitude detected in inflamed tissue. During trains of stimuli, run-down of EPSPs was less extensive in inflamed tissue and there was a significant increase in the paired pulse ratio. Depolarizations in response to exogenous neurotransmitters were not altered in inflamed tissue. These inflammation-induced changes were not accompanied by alterations in the pharmacological profile of EPSPs, and no changes in synaptic density were detected by electron microscopy. Collectively, these data indicate that synaptic facilitation in the inflamed myenteric plexus involves a presynaptic increase in PKA activity, possibly involving an inhibition of BK channels, and an increase in the readily releasable pool of synaptic vesicles.
A basic tenet of neuroscience is that synaptic strength governs the effectiveness of interneuronal signalling. In the hippocampus, synaptic facilitation through long-term potentiation is thought to underlie increased effectiveness of signalling in the context of learning and memory (Kandel, 2001). In autonomic pathways, ganglionic long-term potentiation has also been described and is thought to have important regulatory or homeostatic functions (Alkadhi et al. 2005). In the enteric nervous system (ENS), the third division of the autonomic nervous system, located in the wall of the gastrointestinal tract, fast synaptic transmission in the form of excitatory postsynaptic potentials (EPSPs) is critical for interneuronal signalling and, in turn, appropriate patterns of motility and secretion.
Alteration of synaptic transmission can affect gut function. For example, blockade of nicotinic acetylcholine receptors inhibits reflex-activated motility (Tonini et al. 2001) and secretion (Kellum et al. 1999; Sun et al. 2000). Furthermore, an augmentation or inhibition in the amplitude of fast excitatory postsynaptic potentials (fEPSPs) can affect gut function. 5-Hydroxytryptamine-4 (5-HT4) receptor agonists, which have presynaptic facilitory effects (Kilbinger & Wolf, 1992; Pan & Galligan, 1994; Galligan et al. 2003), promote motility and enhance secretion (Grider et al. 1998; Stoner et al. 1999; Ito et al. 2006; Weber et al. 2006), whereas opioid receptor agonists, which have presynaptic inhibitory actions (Cherubini et al. 1985), suppress motility and secretion (Culpepper-Morgan et al. 1988; Schulzke et al. 1990; Shahbazian et al. 2002). Hence, proper fidelity of synaptic signals is necessary for appropriate co-ordination of the intrinsic circuitry within the ENS, and modification of these signals can alter gut function. Recently, strikingly altered synaptic properties have been described in enteric neurons under inflamed conditions.
In the intestines of the guinea-pig, identification of the function of a given neuron can be based on its electrical, morphological and neurochemical characteristics (Furness, 2006). Two types of neurons can be identified: AH neurons, which are thought to act as intrinsic sensory neurons and interneurons (Bertrand et al. 1997; Furness et al. 1998; Kunze & Furness, 1999; Wood, 2006), typically receive slow, but not fast synaptic input; and S neurons, which can function as mechanosensory neurons, interneurons and motor neurons, receive fast and slow synaptic input (Bornstein et al. 1994; Wood, 1994a; Brookes, 2001; Spencer & Smith, 2004; Wood, 2006). Synaptic activities of these neurons are altered by inflammation. In inflamed intestines, fEPSPs can be elicited in AH neurons, and the fEPSPs in S neurons are substantially augmented compared with control preparations (Palmer et al. 1998; Linden et al. 2003b; Lomax et al. 2005). Understanding the mechanisms of synaptic facilitation within the myenteric plexus will help elucidate how the intrinsic circuitry of the ENS, and motility, are affected by inflammation, as well as providing a unique model of synaptic plasticity.
Plasticity leading to fEPSP facilitation in the myenteric plexus can involve a variety of changes at pre- or postsynaptic sites. The goal of this study was to investigate potential mechanisms that could contribute to synaptic plasticity in the myenteric plexus. Data reported here indicate that fEPSP facilitation in the myenteric plexus involve presynaptic mechanisms of protein kinase A activation and an increase in the readily releasable pool of synaptic vesicles.
Methods
Animals
Experiments were performed on Hartley guinea-pigs (Charles River, Montreal, Canada) of either sex, weighing 250–350 g, housed in cages with soft bedding. The animals had access to food and water ad libitum and were maintained at 23–24°C on a 12 h–12 h light–dark cycle. Inflammation was generated in the colon of guinea-pigs anaesthetized with isoflurane (induced at 4%, maintained at 1.5% in oxygen) by 0.3 ml of trinitrobenzene sulphonic acid (TNBS; 25 mg ml−1) in 30% ethanol delivered into the lumen of the colon through a polyethylene catheter inserted rectally 7 cm proximal to the anus. Control animals remained naïve until tissue collection, which is appropriate since there are no differences in the neuronal properties between saline-injected and naïve animals (Linden et al. 2003b). Animals were maintained in a controlled environment for 6 days after TNBS administration. Animals are sometimes less active during the first 24–48 h after TNBS administration, but they do continue to eat and drink. No analgesics were administered in these studies because these compounds directly affect intestinal neuromuscular function and would therefore confound the results. At the time of tissue collection, animals were anaesthetized with isoflurane and exsanguinated. The severity of colitis was assessed in two ways: changes in the weight of the animals and scoring of macroscopic damage (Linden et al. 2003a). Macroscopic scoring was based on intestinal wall thickness and the presence and extent of adhesions, ulceration, hyperaemia and diarrhoea. All animals treated with TNBS lost weight and exhibited macroscopic damage scores that were consistent with previous reports (Linden et al. 2003a,b). The University of Vermont Animal Care and Use Committee approved all methods used in this study.
Electrophysiological recordings
Experiments were performed on the distal colon, identified as the part of the colon between the hypogastric flexure and the pelvic brim, removed and place in iced Krebs solution (mm: NaCl, 121; KCl, 5.9; CaCl2, 2.5; MgCl2, 1.2; NaHCO3, 25; NaH2PO4, 1.2; and glucose, 8; aerated with 95% O2–5% CO2; all from Sigma, St Louis, MO, USA). The tissue was then placed in a Sylgard-coated dissecting dish with ice-cold Krebs solution containing nifedipine (5 μm) and atropine (200 nm) to eliminate smooth muscle contraction. The tissue was cut open along the mesenteric border, and the mucosa, submucosa and circular muscle of the colon were subsequently removed with forceps to expose the myenteric plexus on the longitudinal smooth muscle. The preparation was then moved to a 2.5 ml recording chamber.
Preparations were continuously perfused at 10 ml min−1 with Krebs solution containing nifedipine and atropine, maintained at 37°C. Glass microelectrodes used for recording were filled to the shoulder with 1.0 m KCl, and the remainder filled with 2.0 m KCl, and had resistances in the range of 50–150 MΩ. Myenteric ganglia were visualized at ×200 with Hoffman modulation contrast optics through an inverted microscope (Nikon Diaphot, Melville, NY, USA), and individual myenteric neurons were randomly impaled. Transmembrane potential was measured with an Axoclamp-2A amplifier (Axon Instruments, Union City, CA, USA), and electrical signals were acquired and analysed using PowerLab Chart (version 5.01, ADInstruments, Castle Hill, NSW, Australia). Synaptic activation of neurons was elicited by direct stimuli applied to fibre tracts in interganglionic connectives with monopolar extracellular electrodes made from Teflon-insulated platinum wire (a single pulse of 0.5 ms). Cells were deemed unhealthy if they had an input resistance below 50 MΩ or had an action potential that peaked at a level less than 0 mV, and were excluded from the study.
Using criteria previously described for classifying neurons in the guinea-pig small intestine (Bornstein et al. 1994; Wood, 1994b), S neurons were identified by the existence of fEPSPs and the lack of a shoulder on the repolarizing phase of the action potential. The amplitude of the maximal fEPSP was acquired while injecting hyperpolarizing current to hold the membrane potential of the neuron to approximately −90 mV to avoid action potentials. Analysis of fEPSP duration was performed on fEPSPs with a smooth contour (not compound) at maximal amplitude. The duration was measured as the time from the point of half-depolarization to the point of half-repolarization.
Assessment of postsynaptic response by exogenous neurotransmitter application
The neurotransmitters adenosine triphosphate (ATP) and acetylcholine (ACh) were exogenously applied by pressure microejection onto S neurons of control and inflamed tissue that exhibited an electrically evoked fEPSP to assess postsynaptic responsiveness. A micropipette containing 100 μm of either ATP or ACh was placed ∼150 μm from the impaled neuron, and a 20–500 ms application of either neurotransmitter was applied in order to obtain a maximal response from an individual neuron that was consistent. The maximal amplitude of the response was compared between control and inflamed intestines.
Pharmacological analysis of neurotransmission
Drugs used in the present studies were purchased from Sigma (St Louis, MO, USA) and were applied to the circulating Krebs solution for at least 5 min prior to evoking fEPSPs. For the pharmacological study, the nicotinic cholinergic antagonist hexamathonium (100 μm) was first applied with subsequent application of the P2X antagonist, pyridoxalphosphate-6-azophenyl-2′,4′-disulphonic acid (PPADS; 10 μm). Averages of at least five fEPSP amplitudes before and after antagonist application were used to evaluate the effects of cholinergic and purinergic antagonists. Fast EPSPs were deemed sensitive to the antagonists if they blocked at least 80% of the fEPSP amplitude. Other drugs used were the 5-HT4 antagonist/inverse agonist, GR 125487 (100 nm), the protein kinase A (PKA) inhibitor, H89 (10 μm), the cyclic adenosine monophosphate (cAMP) activator, forskolin (10 μm), the inactive forskolin analogue, 1,9-dideoxyforskolin (10 μm), and the large-conductance Ca2+-activated K+ (BK) channel blocker, iberiotoxin (100 nm). In these experiments, maximal fEPSPs before and after drug application were evaluated.
To determine whether GR 125487 has a postsynaptic effect on fEPSP amplitudes, the neurotransmitters ACh and ATP were exogenously applied by microejection onto S neurons from control preparations. A micropipette containing 1 mm of each transmitter was placed ∼150 μm from the impaled neuron. Responses to either transmitter were obtained using a 20–100 ms duration pulse, which generated a consistent response in Krebs solution that was comparable in amplitude to the average control fEPSP (∼20 mV). Responses were then obtained with the same microejection parameters in the presence of GR 125487 (100 nm).
Analysis of neurotransmitter release properties
Pairs of stimuli were applied at 50 ms latency in control and inflamed tissue. The fEPSP amplitudes were measured, and the ratio of the second fEPSP amplitude to the first fEPSP amplitude was reported. Paired pulse depression (PPD) was determined to occur if the ratio was less than 1, and paired pulse facilitation (PPF) was determined to occur if the ratio was greater than 1. A train of 20 stimuli at 0.5, 5, 10 and 20 Hz was applied to control and inflamed tissues. At least 1 min was allowed to elapse between trains of different frequencies to avoid responses to previous trains affecting responses to subsequent trains. The amplitudes of the second to the twentieth EPSPs were normalized to the first fEPSP.
Electron microscopy
Distal colons from eight animals in each group were removed and dissected into layers as described above. Tissue was placed in 0.1 m phosphate buffer containing 3% paraformaldehyde and 2.5% glutaraldehyde for 1 h at 4°C and then washed three times for 5 min each in 0.1 m phosphate-buffered saline. It was then placed in Millonig's buffer (Electron Microscopy Sciences, Hatfield, PA, USA) containing 1% OsO4 for 45 min at 4°C and subsequently washed three times for 5 min each in Millonig's buffer. Tissues were dehydrated with increasing concentrations of ethanol, and finally embedded in Spurr's resin between two slides coated with liquid release agent (Electron Microscopy Sciences, Hatfield, PA, USA). These tissues were then examined with a light micrscope to identify areas of interest before being sectioned for electron microscopy. Ganglia were cut from the whole mount with a razor, and mounted on blocks. Thin (60–80 nm) sections were taken using a Reichert-Jung Ultracut E ultramicrotome, collected on Ni grids and contrasted with uranyl acetate and lead citrate. Random sections from both animal groups were examined on a JEOL 1210 transmission electron microscope. Montages of the surfaces of one to five cells per preparation were obtained. Neurons were identified by a euchromatic nuclear appearance with a smooth nuclear envelope and an abundance of ribosomes in the cytoplasm. Synaptic contacts were recognized by membrane-bound structures containing vesicles directly apposing the membranes of these neurons (Gabella, 1972). Only sections of neurons containing a visible nucleus and a minimum of 20 μm of the membrane that could be imaged were included in the study. All micrographs were analysed using MetaMorph software (Molecular Devices Corp., Sunnyvale, CA, USA) for determining the percentage of the total membrane of each neuron covered by presynaptic nerve terminals, the number of synaptic contacts per micrometre, and the average area of each discernable nerve terminal. The investigator remained blinded until all measurements were complete.
Data analyses
Statistical analyses were performed using GraphPad Prism software (version 4.0a for Macintosh, GraphPad Software, San Diego, CA, USA). For all experiments, except for trains and antagonist experiments, differences between inflamed and control tissues were determined by Student's unpaired t test for unpaired data and Student's paired t test for paired data. For train experiments, differences between the two tissues and differences between pulses were determined using a two-way ANOVA with repeated measures. For BK channel and PKA experiments, differences between control and inflamed neurons and pre- and postdrug application were determined using a two-way ANOVA with repeated measures. For the ligand-gated ion channel antagonist experiments, differences in the proportions of the types of fEPSPs and differences in transmitter contributions were determined using χ2 analysis. Values of n represent the number of cells recorded from or imaged under each condition. A P value < 0.05 was considered to be statistically significant. All data are presented as means ±s.e.m. for n cells.
Results
Intracellular microelectrode recordings were obtained from a total of 192 S neurons (101 from 62 control colons and 95 from 57 inflamed colons). The S neurons were identified by lack of a detectable shoulder on the repolarizing portion of action potentials, and the electrophysiological properties of neurons from control animals were comparable to previous studies (Wade & Wood, 1988a,b; Lomax et al. 1999; Wada-Takahashi & Tamura, 2000; Tamura et al. 2001; Linden et al. 2003b). Neurons in both control and inflamed tissue samples had comparable electrical properties (resting membrane potential, control −50 ± 1 mV, inflamed −52 ± 1 mV, P = 0.14; input resistance, control 95 ± 1 MΩ, inflamed 88 ± 6 MΩ, P = 0.47). Furthermore, fibre tract stimulation evoked fast EPSPs that had significantly greater amplitude in inflamed compared with control tissue (control 23 ± 1 mV, inflamed 31 ± 1 mV; P < 0.0001). These data are consistent with our previous report (Linden et al. 2003b). No differences in the duration of the maximal fEPSPs were detected between control and inflamed tissues (control 8.5 ± 0.5 ms, inflamed 9.7 ± 0.5 ms, P = 0.13; n = 10 for each group).
Inflammation-induced synaptic facilitation does not involve recruitment of additional neurotransmitters
Release of additional neurotransmitters is one property that can contribute to the facilitation of fEPSPs in the inflamed colon. There are four types of fEPSPs within the guinea-pig myenteric plexus, described on the basis of their contributing neurotransmitters: purely nicotinic; a mixed nicotinic/purinergic; a mixed nicotinic/purinergic/unidentified; and mixed nicotinic/unidentified (LePard et al. 1997; Galligan, 2002; Nurgali et al. 2003). Previously, we reported that the facilitated fEPSPs of the submucosal plexus in inflamed tissue involve a recruitment of additional neurotransmitters in inflamed tissue, converting fEPSPs from primarily cholinergic to having an additional purinergic or serotonergic component (Lomax et al. 2005). Therefore, the pharmacology of myenteric synapses was investigated with the nicotinic receptor antagonist, hexamethonium (100 μm), and the purinergic P2X receptor antagonist, PPADS (10 μm), to determine whether the neurochemistry of fEPSPs is altered in TNBS-induced colitis. In these studies, the same four distinct types of fibre tract stimulated/evoked fEPSPs described above were detected in control and inflamed tissue samples (Fig. 1). No change in the proportions of these types of synapses was detected in control versus TNBS-induced colitis tissue (Fig. 1). Furthermore, no changes in the contributions of the nicotinic or purinergic components of the individual EPSP amplitudes were detected, even though the fEPSPs in the inflamed colon were facilitated. These findings suggest that, unlike the submucosal plexus, the inflammation-induced synaptic plasticity leading to facilitation of fEPSPs in the myenteric plexus does not involve neurotransmitter recruitment.
Figure 1. The pharmacological profiles of myenteric fEPSPs are not altered in the inflamed tissue.
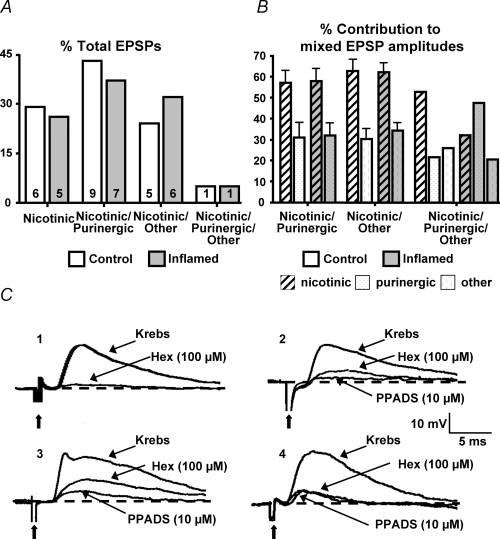
A, bar graph illustrating the percentage of total fEPSPs that are purely nicotinic, nicotinic/purinergic, nicotinic/purinergic/other, or nicotinic/other in control (open bars) and inflamed colons (grey bars) with n values of each type within each bar. B, mean +s.e.m. representing the percentage contribution of individual neurotransmitters to the total amplitude of mixed EPSPs in control (open bars) and inflamed colons (grey bars; n = 21 for control, n = 19 for inflamed; P = 0.63, χ2 analysis). C, representative traces from control tissue of the four types of fEPSPs from the myenteric plexus: (1) nicotinic; (2) nicotinic/purinergic; (3) nicotinic/purinergic/other; and (4) nicotinic/other. Dashed lines represents baseline of EPSP amplitude. All traces are on the same voltage and time scale. Bold arrows are pointing at the stimulus artifact.
Inhibition of protein kinase A decreases fEPSPs in inflamed tissue
Synaptic plasticity can involve activation of PKA, which has been shown to facilitate vesicular release in hippocampal neurons (Trudeau et al. 1996) and in myenteric neurons (Galligan et al. 2003). Inflammation may cause increased activity of PKA, resulting in increased sensitivity of the amplitudes of the fEPSPs in the inflamed tissue to a PKA inhibitor. Application of the PKA inhibitor, H89 (10 μm), had no effect on fEPSP amplitude in the control tissue (Fig. 2), whereas in the inflamed tissue the fEPSP amplitude was significantly decreased to control levels after H89 application (Fig. 2). Conversely, if PKA activity is increased in the myenteric plexus of the inflamed tissue, than an activator of adenylate cyclase, which activates PKA, should not have as great an effect on fEPSP amplitudes as it should in control tissue. Application of forskolin (10 μm), an adenylate cyclase activator, increased fEPSP amplitudes in control tissue to a significantly greater extent than in the inflamed tissue (Fig. 3). In contrast, the inactive analogue 1,9-dideoxyforskolin (10 μm) had no effect in either the control (n = 4; Krebs 25 ± 2 MV, 1,9-dideoxyforskolin 23 ± 2 MV; P = 0.11, Student's paired t test) or inflamed tissue (n = 3; Krebs 36 ± 4 MV, 1,9-dideoxyforskolin 34 ± 3 MV; P = 0.10, Student's paired t test). These data suggest that the synaptic plasticity in the inflamed guinea-pig colon involves an increase in PKA activity, which leads to the facilitated fEPSPs.
Figure 2. The PKA inhibitor, H89, attenuates EPSPs in inflamed, but not control, tissue.
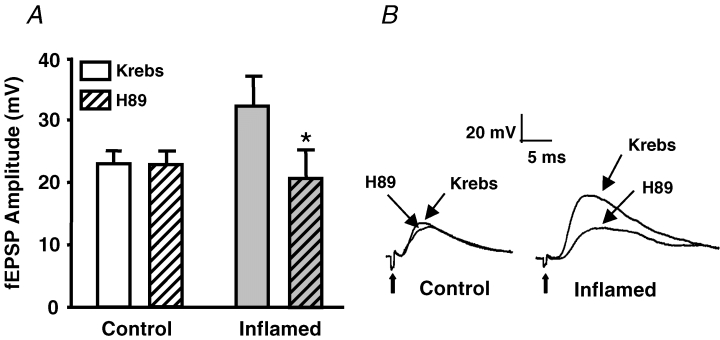
A, mean +s.e.m. of the fEPSP amplitudes from control (open bars) and inflamed colons (grey bars) in Krebs solution and in Krebs solution with protein kinase inhibitor H89 (10 μm; n = 5 for control, n = 6 for inflamed; *P < 0.05 compared with before H89, Student's paired t test). B, representative traces illustrating the differences in amplitudes and sensitivity of fEPSPs to H89 from control and inflamed colons. Traces are on the same voltage and time scales. Bold arrows are pointing to the stimulus artifact.
Figure 3. Inflamed tissue is less sensitive to forskolin than control tissue.
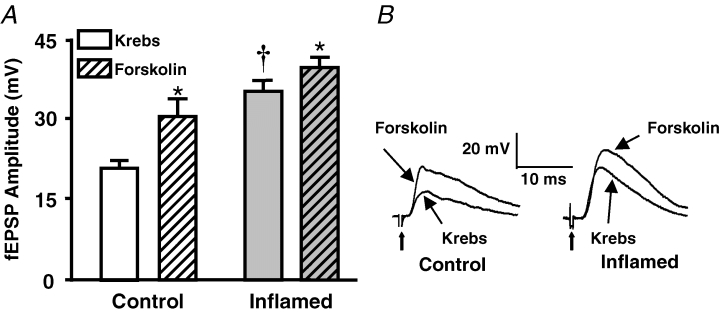
A, mean +s.e.m. of the fEPSP amplitudes from control (open bars) and inflamed colons (grey bars) in Krebs solution and in Krebs solution with the adenylate cyclase activator, forskolin (10 μm; n = 5 for both control and inflamed; *P < 0.05 compared with before forskolin, Student's paired t test; †P < 0.05 different from control, Student's unpaired t test). B, representative traces illustrating the difference in amplitudes and sensitivities of fEPSPs from control and inflamed colons. Traces are on the same voltage and time scales. Bold arrows are pointing to the stimulus artifact.
Inflammation-induced synaptic plasticity does not involve the presynaptic 5-HT4 receptor
Facilitation of fEPSPs within the ENS can involve activation of presynaptic 5-HT4 receptors, which are coupled to cAMP, and lead to PKA activation (Kilbinger & Wolf, 1992; Pan & Galligan, 1994; Galligan et al. 2003). The increased sensitivity of the fEPSP amplitudes in inflamed tissue to a PKA inhibitor may be because inflammation induces increased expression or activity of the 5-HT4 receptors. Therefore, the augmented fEPSPs in inflamed tissue would display a greater degree of sensitivity to an antagonist/inverse agonist than the fEPSPs in control tissue. Application of the 5-HT4 antagonist/inverse agonist, GR 125487 (100 nm; Claeysen et al. 1999, 2000), led to a comparable decrease in the amplitude of the fEPSP in control and inflamed tissue (12 ± 4% in control, 11 ± 4% in inflamed; n.s.; Fig. 4). To determine whether GR 125487 acted postsynaptically, exogenous ACh and ATP were applied to individual neurons from control tissue in both Krebs solution and Krebs solution with GR 125487. No differences were detected in the responses to either transmitter (ACh + Krebs 19 ± 3 mV, ACh + GR 125487 20 ± 3 mV, P = 0.67, Student's paired t test; ATP + Krebs 9.0 ± 2 mV, ATP + GR 125487 10 ± 2 mV; n = 4 for both, P = 0.25, Student's paired t test). These data suggest that the 5-HT4 receptor does not contribute to the synaptic facilitation that occurs in the inflamed colon.
Figure 4. The 5-HT4 receptor antagonist/inverse agonist, GR 125487, causes a slight inhibition of the fast EPSP in both control and inflamed tissue.
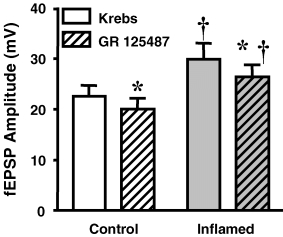
Mean +s.e.m. of the fEPSP amplitudes from control (open bars) and inflamed colons (grey bars) in Krebs solution and in Krebs solution with the 5-HT4 receptor antagonist/inverse agonist, GR 125487 (100 nm; n = 7 for control, n = 10 for inflamed; †P < 0.05 different from control animals, Student's unpaired t test for Krebs data, and two-way ANOVA with repeated measures for GR 125487 data; *P < 0.05 compared with before GR 125487, Student's paired t test).
Synaptic plasticity may involve alterations in the large-conductance Ca2+-activated K+ (BK) channel
Within nerve terminals, BK channels can play a significant role in the termination of the action potential (Gho & Ganetzky, 1992; Bielefeldt & Jackson, 1994) and, in turn, transmitter release. Inflammation-induced synaptic plasticity may involve a decreased function of this channel, which can lead to facilitation of fEPSPs. We therefore investigated the sensitivity of fEPSPs to the selective BK channel blocker, iberiotoxin (100 nm; Wanner et al. 1999). In control tissue the fEPSP amplitude increased significantly, whereas in the inflamed tissue iberiotoxin had no effect on the fEPSP amplitude (Fig. 5). These findings suggest that inflammation-induced synaptic plasticity within the colon may involve an attenuation of the BK channel current, which can contribute to synaptic facilitation.
Figure 5. The BK channel blocker, iberiotoxin, facilitates the EPSP in control, but not inflamed, tissue.
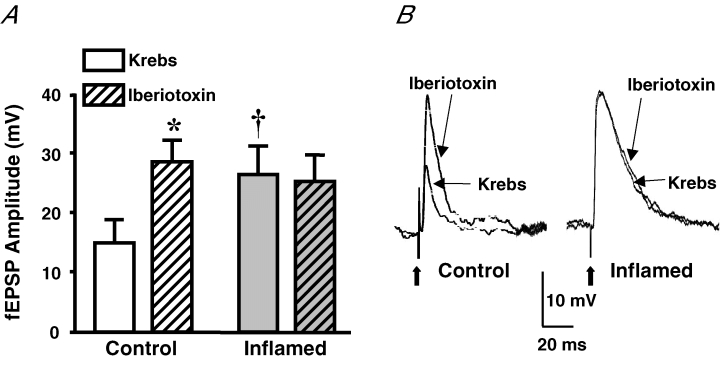
A, mean +s.e.m. of the fEPSP amplitudes from control (open bars) and inflamed colons (grey bars) in Krebs solution and in Krebs solution with the BK channel blocker, iberiotoxin (100 nm; n = 5 for both control and inflamed; *P < 0.05 compared with before iberiotoxin, Student's paired t test; †P < 0.05 different from control, Student's unpaired t test). B, representative traces illustrating the difference in amplitudes and sensitivities of fEPSPS from control and inflamed colons. Traces are on the same voltage and time scales. Bold arrows are pointing to the stimulus artifact.
Synaptic plasticity within the inflamed colon involves changes in the inherent release properties of the nerve terminal
Synaptic plasticity can involve changes in both the size of the readily releasable pool (RRP) of vesicles and the rate of recycling. The responsiveness of synaptic events to repetitive stimuli at high frequency can provide insight into these features of synaptic transmission. Therefore, fEPSPs were evaluated in response to 20 consecutive stimuli delivered at frequencies of 0.5, 5, 10 and 20 Hz in control and inflamed tissues. In the small intestine, fEPSP amplitudes undergo complete run-down at frequencies ≥ 5 Hz (Ren & Galligan, 2005). In the distal colon complete run-down never occurred at any of the frequencies tested. The maximal run-down of EPSPs was observed at a stimulus frequency of 20 Hz in both control and inflamed tissue, and run-down was less extensive in colitis (Fig. 6). These findings suggest that the synaptic plasticity that occurs in the inflamed distal colon is associated with increasing the RRP.
Figure 6. Synaptic run-down is less extensive in inflamed tissue than in control tissue.
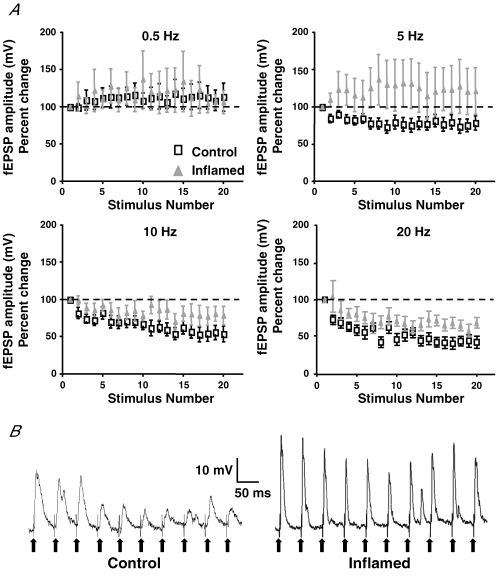
A, mean ±s.e.m. of the percentage change in amplitudes of fEPSPs compared with the first amplitude in control (open squares) and inflamed colons (grey triangles) at 0.5, 5, 10 and 20 Hz. Note how the run-down properties appear to change at 5 Hz, but a significant difference in run-down between control and inflamed tissues was only detected at 20 Hz (0.5 Hz, n = 15 for control, n = 16 for inflamed; 5 Hz, n = 13 for both control and inflamed; 10 Hz, n = 11 for control, n = 13 for inflamed; and 20 Hz, n = 11 for control, n = 13 for inflamed; 20 Hz, P < 0.05, two-way ANOVA with repeated measures). B, representative traces of the first 10 evoked fEPSPs during a train stimulation, illustrating the difference in run-down between control and inflamed colons at 20 Hz. Both traces are on the same voltage and time scales. Each bold arrow is pointing to stimulus artifact.
A paired pulse protocol can also be used to evaluate potential changes in the release properties of the RRP of synaptic vesicles (Thomson, 2000). Paired pulse depression (PPD), designated by a paired pulse ratio (PPR) lower than 1, is indicative of a relatively high probability of release and can be reflective of a low RRP. Alternatively, an increase in the PPR, closer to one, can indicate a decrease in the probability of release and may be reflective of a relatively large RRP. It has been previously reported that PPD occurs in the small intestine owing to relatively low RRP (Ren & Galligan, 2005). In the normal distal colon, PPD was detected, whereas in tissue from TNBS-treated animals, the PPR was significantly greater than that detected in control tissue, with a value of approximately one (Fig. 7). These data suggest that synaptic plasticity of the inflamed colon involves an increase in the RRP of synaptic vesicles.
Figure 7. Inflammation causes an increase in the paired pulse ratio.
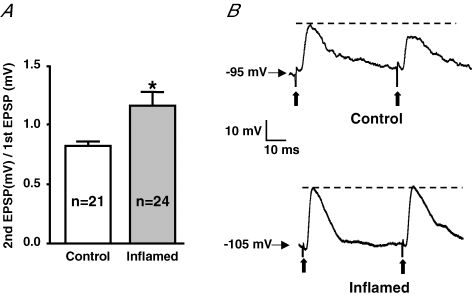
A, mean +s.e.m. of the ratio of the second fEPSP to the first fEPSP at the 50 ms latency from control (open bars) and inflamed colons (grey bars; (*P < 0.05 different from control, Student's unpaired t test). B, representative traces illustrating PPD in control and no change in the EPSP amplitudes in the inflamed colons in response to pairs of stimuli. Traces are on the same voltage and time scales. Bold arrows indicate the stimulus artifact.
Synaptic plasticity of the inflamed colon is not associated with changes of the postsynaptic neuron
Synaptic facilitation could also involve changes in the postsynaptic cell, leading to increased responsiveness to neurotransmitters released from presynaptic terminals. To test whether this plasticity involves postsynaptic sensitization, the principal mediators of fast EPSPs in the colon, ACh and ATP, were exogenously applied to impaled neurons by brief pressure microejection, and responses were evaluated. The responsiveness of colonic myenteric neurons to both neurotransmitters was comparable in normal versus inflamed tissue (Fig. 8). These findings suggest that synaptic facilitation that results from the inflammation-induced plasticity in the myenteric plexus does not involve enhanced sensitivity of the postsynaptic neuron to the neurotransmitters ACh or ATP and is consistent with the notion that the fEPSP facilitation in the inflamed tissue results from presynaptic changes.
Figure 8. Exogenous neurotransmitters evoke comparable depolarizations in neurons from control and inflamed preparations.
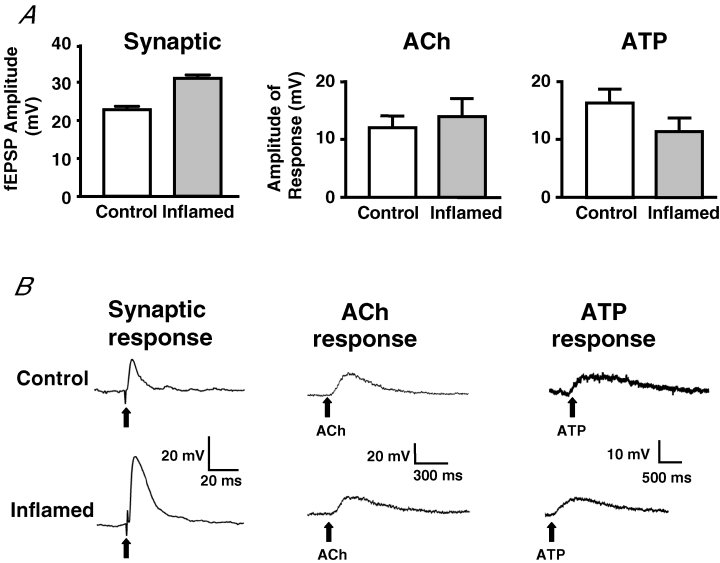
A, bar graphs of mean +s.e.m. illustrating the fEPSP amplitudes for all neurons in the study and the maximum amplitude of response in S neurons in response to exogenously applied neurotransmitters ACh or ATP in control (open bar) and in inflamed colons (grey bar). (fEPSPs amplitudes, n = 93 for control, n = 95 for inflamed, P < 0.0001; ACh, n = 12 for control, n = 9 for inflamed; ATP, n = 10 for both control and inflamed, P > 0.1, Student's unpaired t test.) B, representative traces from control (top) and inflamed colons (bottom), with the synaptic response in the first column and the response to exogenously applied neurotransmitters ACh (second column) and ATP (third column). The time and voltage scales are displayed for each trace. Bold arrows point to stimulus artifact in synaptic response trace and point of neurotransmitter application in response to transmitter traces.
Inflammation-induced synaptic plasticity does not involve an increase in the synaptic contact density
Synaptic plasticity can involve an increase in the number and/or density of synaptic contacts per neuron (Zhang et al. 2003; Morishima & Kawaguchi, 2006). Inflammation of the distal colon is associated with a loss of 20% of myenteric neurons (Linden et al. 2005), which could lead to a rearrangement of the neuropil, resulting in an alteration in the concentration of synaptic contacts on the remaining neurons. Therefore, we used transmission electron microscopy to evaluate whether there were any changes in synaptic density. Montages of neuronal surfaces were constructed from a total of 31 neurons (n = 15 control; n = 16 inflamed) from eight control and eight TNBS-treated guinea-pig colons. No difference was observed between control and inflamed tissue with regard to the percentage of the total membrane of each neuron directly apposed by presynaptic nerve terminals (Fig. 9). Furthermore, no differences were detected in the number of synaptic contacts per micrometre or in the average area of each discernable nerve terminal (Fig. 9). These data suggest that inflammation-induced synaptic facilitation does not involve an increase in the number of synaptic contacts per neuron.
Figure 9. No change in synaptic contact density is observed in the inflamed tissue.
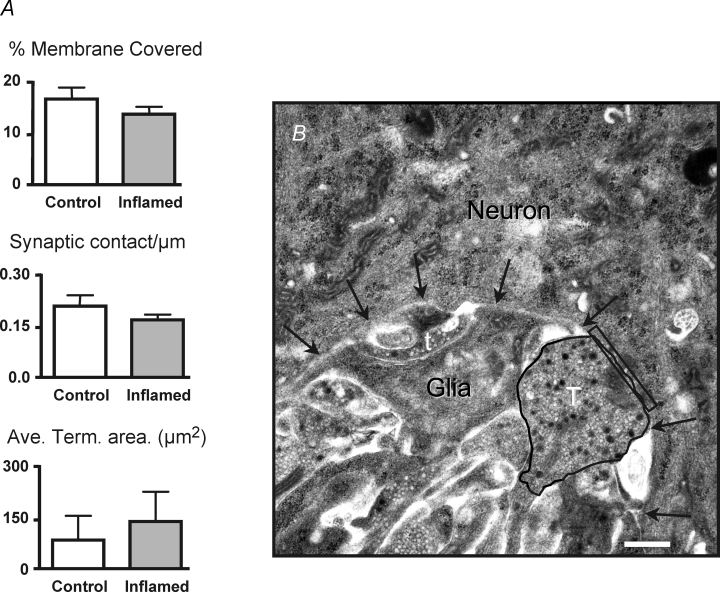
A, bar graphs of mean +s.e.m. of the percentage of postsynaptic membrane directly apposing presynaptic terminals, number of synaptic contacts per micrometre and average area of all discernable nerve terminals in control (open bar) and inflamed colons (grey bar; number of contacts, n = 15 for control, n = 16 for inflamed; percentage membrane covered, n = 15 for control, n = 16 for inflamed; and nerve terminal area, n = 8 for control, n = 14 for inflamed; P > 0.2, Student's unpaired t test). B, electron micrograph of a representative nerve terminal (T) that is contacting a neuron in the field, and a terminal (t) that is not considered to be contacting the postsynaptic neuron. The nerve terminal is circled to illustrate how terminal area was measured. The rectangle denotes the region of the postsynaptic neuronal membrane that is directly apposing the nerve terminal, and the arrows are pointing to the neuronal cell membrane. Scale bar represents 500 nm.
Discussion
The goal of this study was to investigate the potential mechanisms contributing to the synaptic plasticity that leads to facilitation of fEPSPs in the colonic myenteric plexus in response to inflammation (Linden et al. 2003b, 2004). We recently reported that recruitment of additional neurotransmitters leads to inflammation-induced facilitation of EPSPs in the submucosal plexus (Lomax et al. 2005). Here, we report that inflammation-induced synaptic facilitation in the myenteric plexus does not involve an alteration in the pharmacology of the synapse or an alteration in the number of synapses, but rather involves other presynaptic changes to pre-existing terminals that augment transmitter release. Two distinct presynaptic changes have been identified in this study: (1) an increased sensitivity to a PKA inhibitor, reflecting an increase in PKA activity in terminals of inflamed tissue, leading to a possible inhibition of BK channels; and (2) a less extensive synaptic run-down in inflamed preparations, which is indicative of an increase in the releasable pool of synaptic vesicles.
Previous studies of both central and peripheral synapses have demonstrated that synaptic transmission can be facilitated by PKA activation in the presynaptic nerve terminal. For example, PKA activation contributes to long-term potentiation in hippocampal mossy fibre–CA3 synapses (Weisskopf et al. 1994), in cerebellar granule cell–Purkinje cell synapses (Salin et al. 1996; Chen & Regehr, 1997; Linden & Ahn, 1999) and in corticostriatal synapses (Spencer & Murphy, 2002). Furthermore, Galligan and colleagues have demonstrated that PKA activation mediates 5-HT4 receptor-activated synaptic facilitation in the myenteric plexus. In the present study, the PKA inhibitor, H89, decreased the amplitude of fEPSPs in inflamed tissue to a level comparable to that observed in control tissue. The PKA inhibitor did not affect the fEPSP in control preparations, as previously described in the ileum (Galligan et al. 2003), suggesting that basal PKA activity is relatively low in myenteric nerve terminals. These data indicate that inflammation is associated with an increase in PKA activity.
Increased PKA activity is often associated with the activation of G-protein-coupled receptors. In the myenteric plexus, activation of 5-HT4 receptors leads to increased transmitter release (Kilbinger & Wolf, 1992; Pan & Galligan, 1994) via an activation of PKA (Galligan et al. 2003). Therefore, we used the 5-HT4 receptor antagonist/inverse agonist GR 125487 to test whether inflammation-induced synaptic facilitation involves 5-HT4 receptor activation. In the presence of GR 125487, the amplitudes of fEPSPs were suppressed to the same degree in both control and inflamed tissues, indicating that an increase in 5-HT4 receptor activation probably does not contribute to inflammation-induced synaptic facilitation. It is worth noting that certain splice variants of the 5-HT4 receptor (5-HT4e,f) exhibit constitutive activity (Bockaert et al. 2004). Since there was a slight, but significant, decrease in the fEPSP amplitude in the presence of the inverse agonist, it is possible that constitutive activity of the 5-HT4 receptor contributes to the amplitude of myenteric fEPSPs in both control and inflamed tissues.
Investigation into how PKA is activated in the nerve terminals of inflamed tissue remains to be conducted. One possibility is that the proinflammatory cytokine, tumour necrosis factor α (TNF-α), is involved. Tumour necrosis factor α is upregulated during TNBS-induced colitis (Khan et al. 2002), and it has been shown to activate quiescent rat sensory neurons through a PKA-dependent pathway (Zhang et al. 2002). Another possibility is that activation of cyclo-oxygenase 2 (COX-2), and related eicosinoids, is involved in colitis-induced synaptic facilitation, since COX-2 inhibition reverses or restores the excitability of AH neurons to a normal level (Linden et al. 2004). However, fEPSPs are still augmented in animals that have been treated with TNBS plus a COX-2 inhibitor (Linden et al. 2004).
Once PKA is activated within nerve terminals, it can alter neurotransmitter release via actions on ion channels. For example, PKA can phophorylate BK channels (Tian et al. 2003), resulting in activation or inhibition of channel activity (Widmer et al. 2003; Tian et al. 2004); however, this depends on the type of BK channel present, since ‘low’-activity BK channels are activated by PKA and ‘high’-activity BK channels are inhibited by PKA (Widmer et al. 2003). Since the BK channel contributes to the repolarization of the nerve terminal, BK channel activity influences the duration of the action potential, which can influence the amount of transmitter release (Gho & Ganetzky, 1992; Bielefeldt & Jackson, 1994). In the present study, application of the selective BK channel blocker, iberiotoxin, facilitated fEPSPs in control tissue, but had no effect in the inflamed tissue. An interpretation of these results is that these channels are already compromised in the inflamed preparation. This would seem to indicate that PKA activation leads to the inhibition of the BK channel, which could contribute to the facilitation of fEPSPs. In contrast, if the BK channel is inhibited in nerve terminals of the inflamed colon, one might expect to detect a prolonged EPSP owing to the prolonged action potential in the presynaptic nerve terminal; however, there was no significant difference in the duration of the maximal fEPSPs between groups. Given that evoked EPSPs are likely to be comprised of neurotransmitter release from multiple nerve terminals synapsing on the impaled neuron, it is possible that changes in duration of transmitter release from individual nerve terminals is not reflected by a change in the duration of an evoked synaptic event. Elucidation of the type of BK channel present in the myenteric plexus could support or refute this possible action of PKA activation. An alternative interpretation of these results is that the maximum amount of neurotransmitter release is occurring during inflammation, and therefore the fEPSP amplitude cannot be increased further by iberiotoxin. This is probably not the case, however, since application of forskolin resulted in an increase in the amplitude of the fEPSP in inflamed tissue and indicates that the fEPSP is not at its maximum during inflammation.
Alterations in the inherent properties of neurotransmitter release may also contribute to the inflammation-induced plasticity that leads to facilitation of fEPSPs. Postsynaptic potentials activated by trains of stimuli can reflect the relative size of the RRP of synaptic vesicles within the presynaptic nerve terminal. Unlike the myenteric plexus of the small intestine (Ren & Galligan, 2005), complete run-down was not observed at any frequency of stimulation, which is consistent with data from the distal colon of the rat (Browning & Lees, 1996) and previous studies in the distal colon of the guinea-pig (Wade & Wood, 1988b). Additionally, less extensive run-down was observed in the inflamed tissue, and is indicative of a larger RRP. If there is a larger RRP, then the run-down should be less extensive because less recycling of vesicles must occur to replenish those that have been released during previous stimuli (Fernandez-Alfonso & Ryan, 2004). Further support for a larger RRP comes from evaluation of the PPRs. Consistent with the behaviour of fEPSPs in the myenteric plexus of the small intestine (Ren & Galligan, 2005), PPD was detected in the normal colon, but not in inflamed tissue where the PPR was significantly increased compared with that of control tissue. These findings are consistent with the concept that the RRP is increased, and therefore an equal amount of vesicles are released in the presence of elevated Ca2+ during the second stimulus. Regardless, the less extensive run-down in the colon compared with the small intestine suggests that the rate of vesicle recycling may be faster in the distal colon.
Another potential mechanism contributing to inflammation-induced facilitation of fEPSPs could be recruitment of additional neurotransmitters. Indeed, this form of plasticity appears to account for synaptic facilitation in the submucosal plexus of the TNBS-inflamed distal colon. In the submucosal plexus fEPSPs are almost entirely nicotinic (Frieling et al. 1991; Lomax et al. 2001), whereas a significant hexamethonium-insensitive component of the fESPs is detected in inflammation, and this component is blocked by P2X and/or 5-HT3 receptor antagonists (Lomax et al. 2005). Within the myenteric plexus, the pharmacology of the fEPSP in normal tissue is more complex. In the ileum, ACh, ATP and serotonin clearly contribute to fEPSPs (LePard et al. 1997; Galligan, 2002; Nurgali et al. 2003). In the distal colon, ACh and ATP are known to play a role (LePard et al. 1997; Galligan, 2002; Nurgali et al. 2003), and it is possible that serotonin is also involved (Nurgali et al. 2003) and may represent the ‘unidentified’ neurotransmitter described by Galligan and colleagues (LePard et al. 1997). Changes in the contributions of individual neurotransmitters owing to inflammation would therefore be identified by changes in the sensitivity to purinergic and/or nicotinic receptor antagonists. Data presented here show that there are no alterations in the sensitivities to the purinergic and/or nicotinic receptor antagonists, indicating that the synaptic plasticity associated with the facilitated fEPSPs in the myenteric plexus during inflammation differs from that in the submucosal plexus.
An increase in the number of synapses or in the amount of postsynaptic membrane in contact with nerve terminals can affect fEPSP amplitude (Zhang et al. 2003; Morishima & Kawaguchi, 2006); therefore, synaptic rearrangement is a mechanism by which fEPSP amplitudes could be increased in TNBS colitis. Since the myenteric plexus is a multisynaptic pathway (Li & Furness, 2000) and there is a 20% loss of myenteric neurons in guinea-pigs during colitis (Linden et al. 2005), axonal sprouting and synaptic rearrangement of the neuropil is likely to occur. However, analysis of synaptic contacts by electron microscopy indicates that the number of synapses per neuron and the amount of membrane immediately adjacent to presynaptic nerve terminals remain unchanged in TNBS-induced colitis. It should be noted that myenteric ganglia consist of a heterogeneous population of neurons. In this study, we did not evaluate synaptic densities in subpopulations of neurons, so it is possible that changes in synaptic density in particular subpopulations were missed. That being said, the significant overlap of values between the two populations indicates that this is not the case. Collectively, these data suggest that the inflammation-induced synaptic facilitation of the fEPSPs primarily involves changes within pre-existing nerve terminals. It is possible that with the neuronal loss there may be a replacement of lost terminals, resulting in no net change in the amount of synaptic contacts. However, the finding that the mean area of nerve terminals contacting myenteric neurons does not change in the inflamed colon suggests that this does not occur.
Data reported here do not support a postsynaptic site of plasticity contributing to synaptic facilitation. We investigated potential changes to the postsynaptic neuron by evaluating the responsiveness of the impaled neuron to exogenous neurotransmitters. No change was detected in the amplitude of the depolarization response to exogenously applied ACh or ATP. It should be noted that no change in the response to pressure microejection of these transmitters might affect perisynaptic, along with synaptic receptors. This would indicate no global change in the expression of postsynaptic ionotropic receptors without an indication of whether a change in the density of receptors within the synapse is present. That being said, multiple lines of evidence are presented that support our hypothesis that synaptic plasticity occurring during TNBS-induced colitis does involve presynaptic mechanisms.
In conclusion, TNBS-induced colitis results in synaptic facilitation via presynaptic mechanisms that involve increased activity of PKA and an increase in the RRP of synaptic vesicles within pre-existing nerve terminals, but not a change in the pharmacology of synaptic transmission or in synaptic contact density. Since fEPSPs are a primary means of interneuronal communication in the enteric neural circuitry, such a change in the strength of synaptic signals could contribute to the dysmotility that is observed in the inflamed colon. Collectively, these data demonstrate that an inflammatory environment can affect multiple mechanisms within presynaptic nerve terminals, leading to facilitation of fEPSPs.
Acknowledgments
The authors are grateful for the insights and contributions of Dr Rodney Parsons, and we are also grateful to Mrs Nicole Bishop of the Microscopy Imaging Center at the University of Vermont College of Medicine for her assistance with tissue processing for electron microscopy. This work was supported by NIH grants DK62267, NS26995 (G.M.M.), and Ruth L. Kirschstein National Research Service Award F31 NS055512-01 from NINDS (E.M.K.), and a grant from the Crohn's and Colitis Foundation of Canada (K.A.S., G.M.M.). K.A.S. is an Alberta Heritage Foundation for Medical Research Medical Scientist.
References
- Alkadhi KA, Alzoubi KH, Aleisa AM. Plasticity of synaptic transmission in autonomic ganglia. Prog Neurobiol. 2005;75:83–108. doi: 10.1016/j.pneurobio.2005.02.002. [DOI] [PubMed] [Google Scholar]
- Bertrand PP, Kunze WA, Bornstein JC, Furness JB, Smith ML. Analysis of the responses of myenteric neurons in the small intestine to chemical stimulation of the mucosa. Am J PhysiolGastrointest Liver Physiol. 1997;273:G422–G435. doi: 10.1152/ajpgi.1997.273.2.G422. [DOI] [PubMed] [Google Scholar]
- Bielefeldt K, Jackson MB. Phosphorylation and dephosphorylation modulate a Ca2+-activated K+ channel in rat peptidergic nerve terminals. J Physiol. 1994;475:241–254. doi: 10.1113/jphysiol.1994.sp020065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bockaert J, Claeysen S, Compan V, Dumuis A. 5-HT4 receptors. Curr Drug Targets CNS Neurol Disord. 2004;3:39–51. doi: 10.2174/1568007043482615. [DOI] [PubMed] [Google Scholar]
- Bornstein JC, Furness JB, Kunze WA. Electrophysiological characterization of myenteric neurons: how do classification schemes relate? J Auton Nerv Syst. 1994;48:1–15. doi: 10.1016/0165-1838(94)90155-4. [DOI] [PubMed] [Google Scholar]
- Brookes SJ. Classes of enteric nerve cells in the guinea-pig small intestine. Anat Rec. 2001;262:58–70. doi: 10.1002/1097-0185(20010101)262:1<58::AID-AR1011>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- Browning KN, Lees GM. Myenteric neurons of the rat descending colon: electrophysiological and correlated morphological properties. Neuroscience. 1996;73:1029–1047. doi: 10.1016/0306-4522(96)00118-2. [DOI] [PubMed] [Google Scholar]
- Chen C, Regehr WG. The mechanism of cAMP-mediated enhancement at a cerebellar synapse. J Neurosci. 1997;17:8687–8694. doi: 10.1523/JNEUROSCI.17-22-08687.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherubini E, Morita K, North RA. Opioid inhibition of synaptic transmission in the guinea-pig myenteric plexus. Br J Pharmacol. 1985;85:805–817. doi: 10.1111/j.1476-5381.1985.tb11079.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claeysen S, Sebben M, Becamel C, Bockaert J, Dumuis A. Novel brain-specific 5-HT4 receptor splice variants show marked constitutive activity: role of the C-terminal intracellular domain. Mol Pharmacol. 1999;55:910–920. [PubMed] [Google Scholar]
- Claeysen S, Sebben M, Becamel C, Eglen RM, Clark RD, Bockaert J, Dumuis A. Pharmacological properties of 5-hydroxytryptamine4 receptor antagonists on constitutively active wild-type and mutated receptors. Mol Pharmacol. 2000;58:136–144. doi: 10.1124/mol.58.1.136. [DOI] [PubMed] [Google Scholar]
- Culpepper-Morgan J, Kreek MJ, Holt PR, LaRoche D, Zhang J, O'Bryan L. Orally administered κ as well as μ opiate agonists delay gastrointestinal transit time in the guinea pig. Life Sci. 1988;42:2073–2077. doi: 10.1016/0024-3205(88)90120-8. [DOI] [PubMed] [Google Scholar]
- Fernandez-Alfonso T, Ryan TA. The kinetics of synaptic vesicle pool depletion at CNS synaptic terminals. Neuron. 2004;41:943–953. doi: 10.1016/s0896-6273(04)00113-8. [DOI] [PubMed] [Google Scholar]
- Frieling T, Cooke HJ, Wood JD. Synaptic transmission in submucosal ganglia of guinea pig distal colon. Am J Physiol Gastrointest Liver Physiol. 1991;260:G842–G849. doi: 10.1152/ajpgi.1991.260.6.G842. [DOI] [PubMed] [Google Scholar]
- Furness JB. The Enteric Nervous System. Malden: Blackwell Publishing; 2006. [Google Scholar]
- Furness JB, Kunze WAA, Bertrand PP, Bornstein JC. Intracellular recording from myenteric neurons of the guinea-pig ileum that respond to stretch. J Physiol. 1998;506:827–842. doi: 10.1111/j.1469-7793.1998.827bv.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabella G. Fine structure of the myenteric plexus in the guinea-pig ileum. J Anat. 1972;111:69–97. [PMC free article] [PubMed] [Google Scholar]
- Galligan JJ. Ligand-gated ion channels in the enteric nervous system. Neurogastroenterol Motil. 2002;14:611–623. doi: 10.1046/j.1365-2982.2002.00363.x. [DOI] [PubMed] [Google Scholar]
- Galligan JJ, Pan H, Messori E. Signalling mechanism coupled to 5-hydroxytryptamine4 receptor-mediated facilitation of fast synaptic transmission in the guinea-pig ileum myenteric plexus. Neurogastroenterol Motil. 2003;15:523–529. doi: 10.1046/j.1365-2982.2003.00428.x. [DOI] [PubMed] [Google Scholar]
- Gho M, Ganetzky B. Analysis of repolarization of presynaptic motor terminals in Drosophila larvae using potassium-channel-blocking drugs and mutations. J Exp Biol. 1992;170:93–111. doi: 10.1242/jeb.170.1.93. [DOI] [PubMed] [Google Scholar]
- Grider JR, Foxx-Orenstein AE, Jin JG. 5-Hydroxytryptamine4 receptor agonists initiate the peristaltic reflex in human, rat, and guinea pig intestine. Gastroenterology. 1998;115:370–380. doi: 10.1016/s0016-5085(98)70203-3. [DOI] [PubMed] [Google Scholar]
- Ito M, Weber E, Hamel C, Bernhard M, Marcaletti S, Bassilana F, Pfannkuche H. Different expression of 5-HT4 receptor (5-HT4R) transcripts correlates with the functional responses of tegaserod on chloride/water secretion in the human ileum and colon in vitro. Gastroenterology. 2006;130:A544. [Google Scholar]
- Kandel ER. The molecular biology of memory storage: a dialogue between genes and synapses. Science. 2001;294:1030–1038. doi: 10.1126/science.1067020. [DOI] [PubMed] [Google Scholar]
- Kellum JM, Albuquerque FC, Stoner MC, Harris RP. Stroking human jejunal mucosa induces 5-HT release and Cl– secretion via afferent neurons and 5-HT4 receptors. Am J Physiol Gastrointest Liver Physiol. 1999;277:G515–G520. doi: 10.1152/ajpgi.1999.277.3.G515. [DOI] [PubMed] [Google Scholar]
- Khan I, Al-Awadi FM, Thomas N, Haridas S, Anim JT. Cyclooxygenase-2 inhibition and experimental colitis: beneficial effects of phosphorothioated antisense oligonucleotide and meloxicam. Scand J Gastroenterol. 2002;37:1428–1436. doi: 10.1080/003655202762671314. [DOI] [PubMed] [Google Scholar]
- Kilbinger H, Wolf D. Effects of 5-HT4 receptor stimulation on basal and electrically evoked release of acetylcholine from guinea-pig myenteric plexus. Arch Pharmacol. 1992;345:270–275. doi: 10.1007/BF00168686. [DOI] [PubMed] [Google Scholar]
- Kunze WA, Furness JB. The enteric nervous system and regulation of intestinal motility. Annu Rev Physiol. 1999;61:117–142. doi: 10.1146/annurev.physiol.61.1.117. [DOI] [PubMed] [Google Scholar]
- LePard KJ, Messori E, Galligan JJ. Purinergic fast excitatory postsynaptic potentials in myenteric neurons of guinea pig: distribution and pharmacology. Gastroenterology. 1997;113:1522–1534. doi: 10.1053/gast.1997.v113.pm9352854. [DOI] [PubMed] [Google Scholar]
- Li ZS, Furness JB. Inputs from intrinsic primary afferent neurons to nitric oxide synthase-immunoreactive neurons in the myenteric plexus of guinea pig ileum. Cell Tissue Res. 2000;299:1–8. doi: 10.1007/s004419900125. [DOI] [PubMed] [Google Scholar]
- Linden DJ, Ahn S. Activation of presynaptic cAMP-dependent protein kinase is required for induction of cerebellar long-term potentiation. J Neurosci. 1999;19:10221–10227. doi: 10.1523/JNEUROSCI.19-23-10221.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linden DR, Chen JX, Gershon MD, Sharkey KA, Mawe GM. Serotonin availability is increased in mucosa of guinea pigs with TNBS-induced colitis. Am J Physiol Gastrointest Liver Physiol. 2003a;285:G207–G216. doi: 10.1152/ajpgi.00488.2002. [DOI] [PubMed] [Google Scholar]
- Linden DR, Couvrette JM, Ciolino A, McQuoid C, Blaszyk H, Sharkey KA, Mawe GM. Indiscriminate loss of myenteric neurones in the TNBS-inflamed guinea-pig distal colon. Neurogastroenterol Motil. 2005;17:751–760. doi: 10.1111/j.1365-2982.2005.00703.x. [DOI] [PubMed] [Google Scholar]
- Linden DR, Sharkey KA, Ho W, Mawe GM. Cyclooxygenase-2 contributes to dysmotility and enhanced excitability of myenteric AH neurones in the inflamed guinea pig distal colon. J Physiol. 2004;557:191–205. doi: 10.1113/jphysiol.2004.062174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linden DR, Sharkey KA, Mawe GM. Enhanced excitability of myenteric AH neurones in the inflamed guinea-pig distal colon. J Physiol. 2003b;547:589–601. doi: 10.1113/jphysiol.2002.035147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lomax AE, Bertrand PP, Furness JB. Electrophysiological characteristics distinguish three classes of neuron in submucosal ganglia of the guinea-pig distal colon. Neuroscience. 2001;103:245–255. doi: 10.1016/s0306-4522(00)00545-5. [DOI] [PubMed] [Google Scholar]
- Lomax AE, Mawe GM, Sharkey KA. Synaptic facilitation and enhanced neuronal excitability in the submucosal plexus during experimental colitis in guinea-pig. J Physiol. 2005;564:863–875. doi: 10.1113/jphysiol.2005.084285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lomax AE, Sharkey KA, Bertrand PP, Low AM, Bornstein JC, Furness JB. Correlation of morphology, electrophysiology and chemistry of neurons in the myenteric plexus of the guinea-pig distal colon. J Auton Nerv Syst. 1999;76:45–61. doi: 10.1016/s0165-1838(99)00008-9. [DOI] [PubMed] [Google Scholar]
- Morishima M, Kawaguchi Y. Recurrent connection patterns of corticostriatal pyramidal cells in frontal cortex. J Neurosci. 2006;26:4394–4405. doi: 10.1523/JNEUROSCI.0252-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nurgali K, Furness JB, Stebbing MJ. Analysis of purinergic and cholinergic fast synaptic transmission to identified myenteric neurons. Neuroscience. 2003;116:335–347. doi: 10.1016/s0306-4522(02)00749-2. [DOI] [PubMed] [Google Scholar]
- Palmer JM, Wong-Riley M, Sharkey KA. Functional alterations in jejunal myenteric neurons during inflammation in nematode-infected guinea pigs. Am J Physiol Gastrointest Liver Physiol. 1998;275:G922–G935. doi: 10.1152/ajpgi.1998.275.5.G922. [DOI] [PubMed] [Google Scholar]
- Pan H, Galligan JJ. 5-HT and 5-HT receptors mediate inhibition and facilitation of fast synaptic transmission in enteric neurons. Am J Physiol Gastrointest Liver Physiol. 1994;266:G230–G238. doi: 10.1152/ajpgi.1994.266.2.G230. [DOI] [PubMed] [Google Scholar]
- Ren J, Galligan JJ. Dynamics of fast synaptic excitation during trains of stimulation in myenteric neurons of guinea-pig ileum. Auton Neurosci. 2005;117:67–78. doi: 10.1016/j.autneu.2004.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salin PA, Malenka RC, Nicoll RA. Cyclic AMP mediates a presynaptic form of LTP at cerebellar parallel fiber synapses. Neuron. 1996;16:797–803. doi: 10.1016/s0896-6273(00)80099-9. [DOI] [PubMed] [Google Scholar]
- Schulzke JD, Fromm M, Riecken EO, Reutter W. Enkephalin affects ion transport via the enteric nervous system in guinea-pig ileum. Eur J Clin Invest. 1990;20:182–191. doi: 10.1111/j.1365-2362.1990.tb02267.x. [DOI] [PubMed] [Google Scholar]
- Shahbazian A, Heinemann A, Schmidhammer H, Beubler E, Holzer-Petsche U, Holzer P. Involvement of μ- and κ-, but not δ-, opioid receptors in the peristaltic motor depression caused by endogenous and exogenous opioids in the guinea-pig intestine. Br J Pharmacol. 2002;135:741–750. doi: 10.1038/sj.bjp.0704527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spencer JP, Murphy KP. Activation of cyclic AMP-dependent protein kinase is required for long-term enhancement at corticostriatal synapses in rats. Neurosci Lett. 2002;329:217–221. doi: 10.1016/s0304-3940(02)00659-6. [DOI] [PubMed] [Google Scholar]
- Spencer NJ, Smith TK. Mechanosensory S-neurons rather than AH-neurons appear to generate a rhythmic motor pattern in guinea-pig distal colon. J Physiol. 2004;558:577–596. doi: 10.1113/jphysiol.2004.063586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoner MC, Arcuni JC, Lee JA, Kellum JM. A selective 5-HT4 receptor agonist induced cAMP-mediated Cl– efflux from rat colonocytes. Gastroenterology. 1999;118:A2827. [Google Scholar]
- Sun Y, Fihn BM, Jodal M, Sjovall H. Effects of neural blocking agents on motor activity and secretion in the proximal and distal rat colon: evidence of marked segmental differences in nicotinic receptor activity. Scand J Gastroenterol. 2000;35:380–388. doi: 10.1080/003655200750023949. [DOI] [PubMed] [Google Scholar]
- Tamura K, Ito H, Wade PR. Morphology, electrophysiology, and calbindin immunoreactivity of myenteric neurons in the guinea pig distal colon. J Comp Neurol. 2001;437:423–437. doi: 10.1002/cne.1293. [DOI] [PubMed] [Google Scholar]
- Thomson AM. Facilitation, augmentation and potentiation at central synapses. Trends Neurosci. 2000;23:305–312. doi: 10.1016/s0166-2236(00)01580-0. [DOI] [PubMed] [Google Scholar]
- Tian L, Coghill LS, McClafferty H, MacDonald SH, Antoni FA, Ruth P, Knaus HG, Shipston MJ. Distinct stoichiometry of BKCa channel tetramer phosphorylation specifies channel activation and inhibition by cAMP-dependent protein kinase. Proc Natl Acad Sci U S A. 2004;101:11897–11902. doi: 10.1073/pnas.0402590101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian L, Coghill LS, MacDonald SH, Armstrong DL, Shipston MJ. Leucine zipper domain targets cAMP-dependent protein kinase to mammalian BK channels. J Biol Chem. 2003;278:8669–8677. doi: 10.1074/jbc.M211661200. [DOI] [PubMed] [Google Scholar]
- Tonini M, Spelta V, De Ponti F, De Giorgio R, D'Agostino G, Stanghellini V, Corinaldesi R, Sternini C, Crema F. Tachykinin-dependent and -independent components of peristalsis in the guinea pig isolated distal colon. Gastroenterology. 2001;120:938–945. doi: 10.1053/gast.2001.22526. [DOI] [PubMed] [Google Scholar]
- Trudeau LE, Emery DG, Haydon PG. Direct modulation of the secretory machinery underlies PKA-dependent synaptic facilitation in hippocampal neurons. Neuron. 1996;17:789–797. doi: 10.1016/s0896-6273(00)80210-x. [DOI] [PubMed] [Google Scholar]
- Wada-Takahashi S, Tamura K. Actions of reactive oxygen species on AH/type 2 myenteric neurons in guinea pig distal colon. Am J Physiol Gastrointest Liver Physiol. 2000;279:G893–G902. doi: 10.1152/ajpgi.2000.279.5.G893. [DOI] [PubMed] [Google Scholar]
- Wade PR, Wood JD. Electrical behavior of myenteric neurons in guinea pig distal colon. Am J Physiol Gastrointest Liver Physiol. 1988a;254:G522–G530. doi: 10.1152/ajpgi.1988.254.4.G522. [DOI] [PubMed] [Google Scholar]
- Wade PR, Wood JD. Synaptic behavior of myenteric neurons in guinea pig distal colon. Am J Physiol Gastrointest Liver Physiol. 1988b;255:G184–G190. doi: 10.1152/ajpgi.1988.255.2.G184. [DOI] [PubMed] [Google Scholar]
- Wanner SG, Koch RO, Koschak A, Trieb M, Garcia ML, Kaczorowski GJ, Knaus HG. High-conductance calcium-activated potassium channels in rat brain: pharmacology, distribution, and subunit composition. Biochemistry. 1999;38:5392–5400. doi: 10.1021/bi983040c. [DOI] [PubMed] [Google Scholar]
- Weber E, Bernhard M, Pfannkuche H. Tegaserod stimulates 5-HT4 (5-HT4R)-mediated epithelial chloride/water secretion through cAMP/PKA activation but not through cAMP/EPAC or Ca2+-dependent signaling in the rat colon in vitro. Gastroenterology. 2006;130:A545. [Google Scholar]
- Weisskopf MG, Castillo PE, Zalutsky RA, Nicoll RA. Mediation of hippocampal mossy fiber long-term potentiation by cyclic AMP. Science. 1994;265:1878–1882. doi: 10.1126/science.7916482. [DOI] [PubMed] [Google Scholar]
- Widmer HA, Rowe IC, Shipston MJ. Conditional protein phosphorylation regulates BK channel activity in rat cerebellar Purkinje neurons. J Physiol. 2003;552:379–391. doi: 10.1113/jphysiol.2003.046441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood JD. Application of classification schemes to the enteric nervous system. J Auton Nerv Syst. 1994a;48:17–29. doi: 10.1016/0165-1838(94)90156-2. [DOI] [PubMed] [Google Scholar]
- Wood JD. Physiology of the enteric nervous system. In: Johnson LR, editor. Physiology of the Gastrointestinal Tract. New York: Raven Press; 1994b. pp. 423–482. [Google Scholar]
- Wood JD. Cellular neurophysiology of enteric neurons. In: Johnson LR, editor. Physiology of the Gastrointestinal Tract. Burlington, MA, USA: Academic Press; 2006. pp. 629–683. [Google Scholar]
- Zhang JM, Li H, Liu B, Brull SJ. Acute topical application of tumor necrosis factor α evokes protein kinase A-dependent responses in rat sensory neurons. J Neurophysiol. 2002;88:1387–1392. doi: 10.1152/jn.2002.88.3.1387. [DOI] [PubMed] [Google Scholar]
- Zhang H, Wainwright M, Byrne JH, Cleary LJ. Quantitation of contacts among sensory, motor, and serotonergic neurons in the pedal ganglion of Aplysia. Learn Mem. 2003;10:387–393. doi: 10.1101/lm.63903. [DOI] [PMC free article] [PubMed] [Google Scholar]