

Abstract
The extracellular space of the brain contains γ-aminobutyric acid (GABA) that activates extrasynaptic GABAA receptors mediating tonic inhibition. The source of this GABA is uncertain: it could be overspill of vesicular release, non-vesicular leakage, reverse transport, dying cells or glia. Using a novel approach, we simultaneously measured phasic and tonic inhibitory currents and assessed their correlation. Enhancing or diminishing vesicular GABA release in hippocampal neurons caused highly correlated changes in the two inhibitions. During high-frequency phasic inhibitory bursts, tonic current was also enhanced as shown by simulating the summation of IPSCs and by recordings in knockout mice devoid of tonic inhibitory current. When vesicular release was reduced by blocking action potentials or the vesicular GABA transporter, phasic and tonic currents decreased in a correlated fashion. Our results are consistent with most of hippocampal tonic inhibitory current being mediated by GABA released from the very vesicles responsible for activating phasic inhibition.
GABA (γ-aminobutyric acid) is found in the extracellular space of the CNS in concentrations ranging from 0.2 to 2.5 μm (Tossman et al. 1986; Lerma et al. 1986; Ding et al. 1998; Kuntz et al. 2004). Extra- and peri-synaptic GABAA receptors (GABAARs) are in a preferred position to be activated by the low levels of ambient GABA, due to their high GABA affinity in contrast to the lower affinity of synaptic GABAARs (Saxena & MacDonald, 1994; Mody, 2001; Brown et al. 2002; Farrant & Nusser, 2005). The high-affinity extrasynaptic GABAARs consist of specific subunit combinations differentially expressed in various brain regions. These include the δ subunit-containing GABAARs of dentate gyrus and cerebellar granule cells, cortical and thalamic neurons (Nusser et al. 1998; Sur et al. 1999a; Pirker et al. 2000; Nusser & Mody, 2002; Stell et al. 2003; Sun et al. 2004; Jia et al. 2005; Cope et al. 2005; Drasbek & Jensen, 2006), and the α5 subunit-containing GABAARs in CA1 and CA3 pyramidal cells (PCs) (Sperk et al. 1997; Caraiscos et al. 2004; Glykys & Mody, 2006). The current mediated by these extrasynaptic receptors has been termed tonic inhibition (Brickley et al. 1996; Farrant & Nusser, 2005), which is highly sensitive to the extracellular GABA concentration ([GABA]). It is enhanced when ambient [GABA] is increased by blocking GABA transporters, by adding GABA to the aCSF to mimic that normally present in the extracellular space, or by preventing GABA degradation (Nusser & Mody, 2002; Stell & Mody, 2002; Wu et al. 2003; Glykys & Mody, 2006).
Various sources have been proposed for the normal amounts of GABA found in the extracellular space. These include: astrocytic release (Liu et al. 2000; Kozlov et al. 2006), reversal of GABA transporter (Gaspary et al. 1998; Richerson & Wu, 2003), non-vesicular release as well as action potential-mediated release (Attwell et al. 1993; Brickley et al. 1996; Zoli et al. 1999; Rossi et al. 2003; Bright et al. 2007). The cerebellar granule cells (CGCs) show a tonic current early in development that depends on action potential firing (Kaneda et al. 1995; Brickley et al. 1996). However, this source of extracellular GABA decreases during development and in adult CGCs tonic currents are mediated by action potential-independent mechanisms (Wall & Usowicz, 1997; Rossi et al. 2003). Yet, the inhibitory inputs onto CGCs are part of a unique synaptic structure, the glial ensheathed glomerulus. Thus, transmitter release, diffusion and overspill may be unique to this highly specialized structure (Brickley et al. 1996; Wall & Usowicz, 1997; Rossi & Hamann, 1998; Mitchell & Silver, 2000).
In the rest of the brain, where less specialized GABA synapses are the norm, it is unclear whether GABA released by action potential-dependent mechanisms can actually increase the level of tonic inhibitory current and be correlated with phasic inhibitory current. It has been recently shown in glomerular synapses of the dorsal lateral geniculate nucleus (dLGN) thalamic relay neurons that tonic inhibition does depend on the global level of inhibititory activity and vesicular release (Bright et al. 2007). In most cases a proper correlation between tonic and phasic currents cannot be established due to the low frequency of spontaneous inhibitory postsynaptic (phasic) currents (sIPSCs) recorded at room temperature and because of the methods used for the analysis of phasic and tonic inhibitory currents. We have developed a method to simultaneously measure both mean currents (Imean) over an arbitrarily chosen epoch of time. Our experiments show highly correlated tonic and phasic inhibitory currents in the hippocampus under both increased and reduced vesicular GABA release. Thus, a substantial amount of tonic inhibition in this brain region is mediated by action potential-dependent vesicular GABA release.
Methods
Slice preparation
Adult (ages: 34–98 days old) male C57/Bl6, and Gabra5/Gabrd−/− double knockout mice on C57/Bl6 background were obtained from our breeding colonies maintained by the UCLA Division of Laboratory Animal Medicine. Mice were anaesthetized with halothane and decapitated according to a protocol approved by the UCLA Chancellor's Animal Research Committee. The brain was removed and placed in ice-cold artificial cerebrospinal fluid (aCSF) containing (mm): NaCl (126), KCl (2.5), CaCl2 (2), MgCl2 (2), NaH2PO4 (1.25), NaHCO3 (26) and d-glucose (10) with pH 7.3–7.4 when bubbled with 95% O2 and 5% CO2. Coronal brain slices, 350 μm thick, were cut with a Leica VT1000S Vibratome (Leica Microsystems, Wetzlar, Germany) in aCSF containing 3 mm kynurenic acid (Sigma). Slices were placed into an interface holding chamber at room temperature that was gradually heated up to 30 ± 1°C. Slices were stored in the holding chamber for at least 1 h before being transferred to the recording chamber. For the concanamycin A experiments, slices were pre-incubated for the indicated times in submerged chambers.
Whole-cell recordings
Dentate gyrus molecular layer interneurons and CA1 PCs were visually identified by IR-DIC videomicroscopy (Olympus, Melville, NY, USA; BX51WI, 40× water immersion objective) and recorded with an Axopatch 200A amplifier (Molecular Devices Corporation, Sunnyvale, CA, USA). Slices were continuously perfused (∼4–5 ml min−1) at 32–34°C with 95% O2 and 5% CO2 saturated aCSF containing kynurenic acid (3 mm) and other drugs as indicated. Microelectrodes (3–5 MΩ) contained the following internal solution (mm): CsCl (140), MgCl2 (1), Hepes (10), EGTA (0.1), NaCl (4), MgATP (2), NaGTP (0.3) pH ∼7.27, ∼275 mosmol l−1. For CA1 PCs, QX-314 (5 mm) was included in the internal solution. Whole-cell voltage-clamp recordings were performed at −70 mV. Series resistance and whole-cell capacitance were estimated from fast transients evoked by a 5 mV voltage command step and were compensated to 70–80% using lag values of 7 μs. Recordings were discontinued if series resistance increased by more than 25% through an experiment, or the compensated resistance surpassed 25 MΩ at any time during the experiment. Drugs were perfused after obtaining a stable control recording period of at least 1 min. Sucrose was applied by low-pressure puffs controlled by a Picospritzer II (General Valve Corporation, Fairfield, NJ, USA) through a micropipette positioned close to the soma of the recorded cell.
Data acquisition and analysis
All recordings were low-pass filtered at 3 kHz and digitized on-line at 10 kHz using a PCI-MIO 16E-4 data acquisition board (National Instruments, Austin, TX, USA).
Tonic current measurement
A custom-written macro running under IGOR Pro v5.02 (Lake Oswego, OR, USA) was used to perform the following analyses. The entire digitized recording was loaded and seal tests were removed from the traces. An all-point histogram was plotted for every 10 000 points and smoothed by Savitzky–Golay algorithm to obtain the peak value. A Gaussian was fitted to the part of the distribution not skewed by synaptic events from a point 1 or 3 pA to the left of the peak value (automatically determined by the macro) to the rightmost (largest) value of the histogram distribution (Fig. 2B). The mean of the fitted Gaussian was considered to be the mean holding current. This process was repeated for the entire recording. All baseline mean values thus obtained were plotted and detrended by a linear fit obtained from a segment of the baseline condition and extended to the whole recording to correct for potential steady shifts during the recordings. Usually, 20–30 s periods were used for the control and drug conditions and a 10–15 s period was analysed in the presence of bicuculline methiodide (BMI). In a given neuron, we obtained the magnitude of the tonic current by subtracting the baseline current recorded in the presence of BMI.
Figure 2. Simultaneous measurement of phasic and tonic currents using the Imean method.

A, whole-cell voltage-clamp recording from a CA1 PC (Vh=−70 mV) in the presence of 3 mm kynurenic acid and 5 μm GABA with three numbered periods of 1 s each consisting of 10 000 points (1 and 2 are control periods, 3 in the presence of 100 μm BMI). Lower panels are showing, on an expanded time scale, the periods used to calculate tonic and phasic currents in B. B, Gaussian fits to the all-points histograms of the indicated periods. The peak of the Gaussian denotes the mean tonic current while all the points outside of the Gaussian distribution (skewed to the left) constitute the phasic current (see Methods for details). Insets: higher magnifications of the corresponding graphs. C, phasic (○) and tonic (•) current measurements (performed every second) showing their variability over time. Holding current is indicated on the left axis, the right axis shows the measured Imean. Note the larger tonic Imean compared with the phasic Imean throughout the recording. Numbers indicate the corresponding time periods in A and B.
Phasic current determination
The baseline mean current was subtracted bringing the mean of the Gaussian to 0 pA. The cumulative sum of the 10 000 data points (an epoch of 1 s) was calculated and divided by the number of points in that segment of time. Thus, the data points belonging to the Gaussian distribution (baseline noise) summed to 0, while everything below (i.e. outside) the baseline noise (generated by small and large IPSCs) summed to yield the phasic Imean (Fig. 2C).
Detection and measurement of sIPSCs
All sIPSCs were detected in 30–60 s recording segments. Event frequency, 10–90% rise time (RT10-90) and weighted decay time constant (τw) values were measured. Detection and analysis were performed using a custom-written LabView-based software (EVAN).
Burst analysis
A region of interested (ROI; range, 0.8–2.2 s) was selected from the recorded trace corresponding to the duration of a given burst induced by the sucrose puff. For each ROI, all spontaneous events were detected and the mean phasic current was calculated. The baseline current amplitude was determined as the value of the point before the event was detected. All data are shown as mean ± standard error of the mean (s.e.m.). Statistical significance was assessed by Student's paired t test, Student's unpaired t test assuming unequal variances, Wilcoxon matched-pairs signed-ranks test and one-way ANOVA. The level of significance was set at P < 0.05. Cross-correlations were normalized by the following equation: 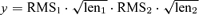 , where RMS represents the root mean square and len1 and len2 represent the total number of points in the segments of the phasic and tonic currents, respectively.
, where RMS represents the root mean square and len1 and len2 represent the total number of points in the segments of the phasic and tonic currents, respectively.
Simulation of IPSCs
A custom-written macro running under IGOR Pro v5.02 was used. The variables entered were: peak amplitude, RT10-90, τw and their corresponding standard deviation, frequency, trace length (was kept constant at 15 s) and baseline current standard deviation. The actual rise time (RT) was obtained from the RT10-90. Next, depending on the frequency of events to be simulated, the peak amplitudes were generated using a log-normal distribution of random peaks constrained by the user-supplied values of mean peak amplitude and its standard deviation. Rise and decay times were generated by a Gaussian distribution around their user-supplied values for mean and standard deviation. Next, the peak amplitude and RT were sorted to obtain a hyperbolic relation. The events were generated at a resolution of Δt = 10−4 s by the sum of two equations: (1) the rising phase of the event was generated by: y1=[0.8 (−Amp)/RT]t and (2) the decay of the event was generated by: y2= Amp (exp−(t−tmax)/τ) where Amp is the peak amplitude of the event to be generated, RT is the 10–90% rise time, τ is τw, tmax is the time of the peak of the event, and t is time. A random noise based on the calculated single channel noise was added to decay phase of the event. The single channel noise was calculated as σ2=i− (i2/N) where σ is the variance, i is the single channel current and N is the number of channels obtained by dividing the peak amplitude of the IPSC by i (2.2 pA) and multiplying the result by 1.25 to account for an average open probability (Popen) of 0.8. A baseline current with a standard deviation specified by the user corresponding to the variance observed in recorded cells during periods with no IPSCs present, and a mean value of 0 pA was generated at a resolution of Δt = 10−4 s. The simulated events were then summed with the baseline current trace by adding each point of the simulated event to the baseline starting at time points chosen from an exponential distribution of interevent intervals generated by the following equation: y = (1/frq) ln(RND), where frq is the event frequency supplied by the user and RND is a random number between 0 and 1.
Single channel simulations
Currents mediated by α1β3δ-containing GABAARs were simulated by using Channel Laboratory V2.030536 (Synaptosoft, Inc., Decatur, GA, USA) based on a published kinetic model (Haas & MacDonald, 1999)
Drugs
GABA (Sigma) at a concentration of 5 μm was added to the aCSF where indicated, and slices were pre-incubated in this solution for ∼5 min before starting the experiment. 1-[2-[[(diphenylmethylene)imino]oxy]ethyl]-1,2,5,6-tetrahydro-3-pyridinecarboxylic acid hydrochloride (NO-711), concanamycin A (Folimycin), gabazine (SR95531) and bicuculline methiodide (BMI) were purchased from Sigma, while tetrodotoxin (TTX) was obtained from EMD Biosciences, Inc. (San Diego, CA, USA).
Results
Decrease in phasic and tonic inhibitory currents in CA1 PCs in the absence of action potentials and presynaptic Ca2+ entry
Action potential-mediated GABA release onto CGCs early in development influences the level of tonic inhibitory current in these neurons (Kaneda et al. 1995; Brickley et al. 1996). We wanted to determine if tonic inhibitory currents of CA1 PCs, which do not posses a specialized glomerulus, are also regulated by action potential-mediated GABA release. Whole-cell voltage-clamp recordings (Vh=−70 mV) were performed in CA1 PCs in the presence of 3 mm kynurenic acid to block ionotropic glutamate receptors and 7.5 μm NO-711 to block the GABA transporter
GAT-1, which acts as a sink for GABA, and enhance the extracellular level of GABA in the slice (Nusser & Mody, 2002; Jensen et al. 2003). After recording a control period, action potential-mediated GABA release and Ca2+ entry into the terminals were blocked by perfusing 0.5 μm TTX and 50 μm CdCl2. Following perfusion of TTX + CdCl2 the frequency and peak amplitudes of sIPSCs showed a gradual decrease (frequency: control, 26.6 ± 3.03 Hz versus TTX, 21.3 ± 2.88 Hz, P = 0.003; peak amplitude: control, 45.1 ± 4.59 pA versus TTX, 37.4 ± 3.45 pA, P = 0.025; n = 6, paired t test; Fig. 1A and B) indicating that vesicular GABA release was reduced. The tonic inhibitory current was also reduced (by 65% from 39.1 ± 11.8 to 13.5 ± 2.8 pA; n = 6, P = 0.031, Wilcoxon matched-pairs signed-ranks test; Fig. 1A and C). According to our data, both phasic and tonic inhibitory currents are decreased in CA1 PCs when vesicular GABA release is reduced by blocking action potentials and Ca2+ entry into the terminals. However, a true correlation between the changes in the two types of inhibition can only be accomplished if they can be measured during short time intervals (e.g. 1 s) instead of determining their properties during long (e.g. 60 s) recording segments as done in the past (Nusser & Mody, 2002). We thus devised a method allowing for the simultaneous measurement of phasic and tonic currents during short (1 s) time epochs which we called Imean.
Figure 1. Concomitant reduction in phasic and tonic inhibitions after blocking action potential-dependent vesicular release.

A, whole-cell voltage-clamp recording from a CA1 PC (Vh=−70 mV) in the presence of 3 mm kynurenic acid and 7.5 μm NO-711. Horizontal lines over the recording denote the time of drug perfusions. Right panel: Gaussian fits to all-points histograms derived from the numbered 30 s recording periods (1, control; 2, 0.5 μm TTX + 50 μm CdCl2) and a 15 s recording period during the perfusion of BMI used to determine the tonic current. The differences between the Gaussian means marked by the dotted lines are indicated. Inset: average sIPSC from the numbered time periods of the recording (1, 1130 events; 2, 919 events). B, cumulative probability of the log scaled interevent intervals recorded during the two numbered time periods (same number of events as used in the inset of A). C, the average tonic current is significantly reduced by blocking action potentials and Ca2+ entry into the terminals (39.1 ± 11.8 pA versus 13.5 ± 2.8 pA, n = 6 cells). *P = 0.031 Wilcoxon matched-pairs signed-ranks test).
The Imean method allows the measurement of phasic current per unit time
Phasic and tonic currents were determined as described in the Methods. Briefly, an all-points histogram was plotted every 1 s, obtaining a distribution skewed to the side of synaptic events (Fig. 2B). The mean of a Gaussian fit to the non-skewed side of the distribution was considered as the mean holding current that formed the basis for the determination of the tonic current (Fig. 2A and B). There was no obvious correlation between the holding current and the magnitudes of the tonic or phasic currents. All the points outside the Gaussian fit were considered to be part of the phasic current (phasic Imean). To test whether the all-point histogram of the tonic current without the phasic current can be described by an unskewed Gaussian distribution, we simulated the activation of high-affinity non-desensitizing GABAARs (α1β3δ) using an established model (Haas & MacDonald, 1999). The openings of a single simulated channel activated by 1 mm GABA show a high similarity to the experimentally obtained recordings (compare Fig. 3A with Fig. 5A of Haas & MacDonald, 1999). To generate a tonic current of approximately 25 pA we simulated the activation of 1250 channels by 5 μm GABA (Vh=−70 mV, GABA reversed potential (EGABA) = 0 mV; Fig. 3B) The all-points histogram of this current, as well as that of this current summed with a real current recorded without the contribution of GABAARs (Fig. 3B) both showed unskewed Gaussian distributions. Based on these realistic simulations, the deviation from the Gaussian distribution in the Imean method when phasic currents are present (Fig. 2B) must be due to the presence of phasic events.
Figure 3. Tonic current without phasic current follows a Gaussian distribution in the Imean method.

A, simulation of single channel openings of an α1β3δ GABAA channel activated by 1 mm GABA based on the kinetic model of Haas & MacDonald (1999) (compare this simulated trace with the experimental trace shown in Fig. 5A of that publication). B, upper panels: simulated current generated by the activation of 1250 α1β3δ GABAA channels by 5 μm GABA (Vh=−70 mV, EGABA= 0 mV) using the kinetic model as in A. The current trace recorded in the presence of BMI is the same as that shown in Fig. 2B. Total current was determined as the sum of the simulated current and the current recorded during BMI. Lower panels: Gaussian fits to right sides of the all-points histograms of the indicated traces. The peak of the Gaussian denotes the mean holding current. Note on the insets the lack of deviation from the Gaussian distribution (i.e. the absence of a skew) compared with when phasic currents are present (insets on Fig. 2B).
We next investigated how well the Imean method can resolve changes in phasic currents following a predicted decrease in the size of IPSCs by altering the holding potential. We recorded sIPSCs in a CA1 PC at −60 mV and then changed the holding potential to −13 mV. This decrease in holding potential should have reduced the average sIPSC by 78%. The averages of all sIPSCs recorded under the two conditions yielded a gross underestimate of the predicted change (Fig. 4A) (Stell & Mody, 2002).
Figure 4. Accuracy of the Imean method for detecting changes in the phasic current.

CA1 PC recorded in the presence of 3 mm kynurenic acid and 5 μm GABA at two separate holding potentials of −60 and −13 mV. A, left panel: average sIPSC (of the indicated number of events) recorded at the two potentials. The change in driving force is expected to reduce the average IPSCs at −13 mV by 78% compared with that recorded at −60 mV. Middle panel: count-matching the number of events recorded at −13 mV with the largest amplitude sIPSCs recorded at −60 mV gives a more accurate value of 72% reduction. Right panel: using the Imean method gives a similar decrease (84%). B, left panel: average sIPSC recorded under control conditions in a CA1 PC and in another cell in the presence of 200 nm gabazine (SR; averages of the largest 100 events). Middle panel: the phasic current calculated by the Imean method shows a similar reduction without the need for event detection and count-matching. Right panel: sample all-point histograms measured over 1 s epochs recorded in two different cells in different conditions. Note the decrease in the skewed part of the histogram corresponding to the phasic current when the cell is exposed to gabazine (SR). In A and B, the Imean was obtained as the average of 60 or 30 1-s segments, respectively.
This is due to the smallest events recorded at −60 mV being buried in the noise when recorded at −13 mV (Stell & Mody, 2002). This can be circumvented by using the count-matching method (Stell & Mody, 2002), i.e. by selecting an equal number of the largest events recorded at −60 mV to the total number of events recorded at −13 mV, the decrease of the average IPSCs became a more realistic 72% (from 53.2 pA at −60 mV to −15.1 pA at −13 mV). Using the Imean method during the two recording conditions, with no need for any event detection or count-matching, we calculated a similar decrease in the phasic current (−3.49 ± 0.17 pA at −60 mV compared with 0.54 ± 0.05 pA at −13 mV), corresponding to a 84 ± 3% decrease that very closely matches the one predicted from the smaller driving force (Fig. 4A).
We next tested the accuracy of the Imean method under conditions of selective reduction of the phasic current. We have previously shown that low concentrations of gabazine (200 nm) selectively reduce the phasic current without affecting the tonic current, due to the higher GABA affinity of the extrasynaptic receptors (Stell & Mody, 2002). We recorded inhibitory currents in CA1 PCs in aCSF containing 3 mm kynurenic acid and 5 μm GABA with or without 200 nm gabazine present. This low concentration of gabazine statistically decreased Imean phasic from −6.5 ± 1.4 to −1.7 ± 0.4 pA (P = 0.02, n = 5, unpaired t test; Fig. 4B). This corresponds to a 74% reduction, close to the value determined by count-matching the 100 largest sIPSCs recorded with or without gabazine present that detected a 64% reduction (control: 89.6 ± 10.7 pA; 200 nm gabazine: 30 ± 3.96 pA; n = 5; Fig. 4B). The small (∼10%) difference between the two methods is probably due to our present experimental conditions where recordings in the absence of gabazine were compared with recordings in gabazine in different neurons. Nevertheless, the 74% reduction produced by gabazine as found by the Imean method compares very well to the 71% reduction found in experiments where the IPSCs were compared before and after perfusion of gabazine in the same cells (Stell & Mody, 2002). Therefore, the Imean method appears to be accurate even when very small phasic events, normally beyond the detection threshold of conventional methods, contribute to the all-points histograms. As expected (Stell & Mody, 2002), tonic currents determined by the Imean method were not statistically different between the two conditions (26.2 ± 3.9 pA in control and 23.1 ± 2.4 pA in 200 nm gabazine; P = 0.51, n = 5, unpaired t test). Thus, the Imean method accurately detected a selective reduction in phasic inhibition. In addition to not requiring detection and count-matching of the events, the phasic Imean should be sensitive to the combined effects of sIPSCs frequency, amplitude and decay, which do not have to be measured separately. Small amplitude or slow rise time events missed by traditional detection programs are not overlooked by this new measurement method because no threshold-crossing is set as a prerequisite for detection. The method can also be used for sEPSC analysis or for outward currents (Klaassen et al. 2006).
Small changes in sIPSC frequency, amplitude and decay are better detected by the Imean method then by measuring them individually
The traditional analysis of sIPSCs involves their detection based on some threshold-crossing or template-matching algorithm and then analysing them individually for amplitude, decay and their overall frequency. We wanted to ascertain that our newly devised phasic Imean measurement was as sensitive to changes in IPSCs as the analysis of each individual variable. To control changes in the parameters of sIPSCs, we simulated whole-cell voltage-clamped sIPSCs and systematically changed their frequencies, amplitudes and decay time constants. We developed an IPSC simulator running in IgorPro where frequency, peak amplitude, 10–90% rise time (RT10-90), weighted decay (τw) as well as baseline noise standard deviation (σBL) could be individually modified (see Methods for details). Analysis of a 15 s period of simulated sIPSCs, generated using frequency and event properties recorded from several CA1 PCs, yielded an excellent correspondence between the properties of the simulated and recorded sIPSCs, with no statistically significant differences in frequency, peak amplitude, RT10-90 or τw (experiment: frequency, 26.8 ± 2.15 Hz; peak amplitude, 47.7 ± 2.93 pA, RT10-90, 0.53 ± 0.01 ms, τw, 6.36 ± 0.17 ms, n = 7; simulation: frequency, 26.4 ± 2.20 Hz; peak amplitude, 46.9 ± 2.95 pA, RT10-90, 0.53 ± 0.01 ms, τw 6.42 ± 0.19 ms; n = 7, P > 0.05 for each parameter, unpaired t test; Fig. 5). Next, we systematically varied individual IPSC parameters in the simulator and compared them to the simulated control parameters. When IPSC peak amplitude was increased by 25%, both phasic Imean (control, −7.96 ± 0.65 pA versus+25%, −10.6 ± 0.87 pA; n = 7, P = 0.034 unpaired t test) and the individual peak amplitude analysis showed statistically significant differences (from 46.9 ± 2.95 to 58.5 ± 4.01 pA; n = 7; P = 0.040, unpaired t test). Smaller increases in peak amplitude failed to produce a statistically significant increase in Imean. However, phasic Imean showed a statistical difference when the frequency was increased by ∼25% (to −10.2 ± 0.68 pA, n = 7, P = 0.036, unpaired t test) but the individual frequency analysis did not yield a significant difference (from 26.4 ± 2.20 to 33.3 ± 3.06 Hz, n = 7; P = 0.095, unpaired t test). This was in contrast to the analysis of individual τw where a ∼15% increase was statistically different (from 6.42 ± 0.19 to 7.36 ± 0.23 ms, n = 7, P = 0.009, unpaired t test) yet phasic Imean was unable to resolve a statistically significant difference (−9.45 ± 0.80 pA, n = 7, P = 0.176, unpaired t test). A difference of ∼25% in τw was needed to statistically significantly change phasic Imean (to −9.89 ± 0.53 pA, n = 7, P = 0.041, unpaired t test). This lack of sensitivity for τw was due to a higher coefficient of variance of phasic Imean compared with the variance of τw (phasic Imean CV was 22.36% compared with the τw CV of 8.28% for an increase in τw by 15%).
Figure 5. Comparison of experimental and simulated events.

A, whole-cell voltage-clamp recording from a CA1 PC (Vh=−70 mV). Left inset: higher magnification of a 1 s segment denoted by dashed lines. Right inset: single sIPSC (indicated by asterisks) overlaid from an actual experiment and from a simulation using the parameters obtained from the actual recording. B, a simulated 15 s trace using the parameters obtained from the recording in A. Inset: higher magnification of a 1 s segment marked by dashed lines. C, distribution of interevent intervals (corresponding to the frequency of 27.2 Hz of the events in A) used to generate the trace in B. D, plot of phasic Imean (P-Imean) cumulative average (calculated every second) showing no difference between experiment and simulation. E, peak, 10–90% rise time and τw distributions used to generate the trace in B, which were obtained by using in the simulation the mean and standard deviations of the respective parameters from the recording in A. F, average simulated sIPSC showing the difference when the peak and τw are both increased by 15% and 10%, respectively (grey line). G, when peak amplitude and τw are compared between two sets of simulations, they do not show statistical differences (peak: P = 0.174, n = 7; τw: P = 0.064, n = 7, unpaired t tests). The combined effect of the simultaneous, albeit non-significant, changes in the two parameters results in a significantly different phasic Imean(from −8.08 ± 0.53 to −10.0 ± 0.60 pA, n = 7, P = 0.031, unpaired t test).
However, when two sIPSCs parameters increased concomitantly, phasic Imean showed a much higher sensitivity than the analysis of the individual parameters. In a new set of simulations, when the peak amplitude and frequency were increased, albeit non-significantly, by ∼15% (from 47.6 ± 3.13 to 55.8 ± 3.96 pA, n = 7, P = 0.131, unpaired t test) and by ∼10% (from 26.2 ± 2.14 to 29.3 ± 2.16 Hz, n = 7, P = 0.326 unpaired t test), respectively, phasic Imean increased to reach statistical significance (from −8.08 ± 0.53 to −10.4 ± 0.73 pA, n = 7, P = 0.024, unpaired t test). Significantly different phasic Imean values were also obtained, for example, when the peak amplitude was increased by ∼10% and the frequency by ∼15% (phasic Imean: from −8.08 ± 0.53 to −10.3 ± 0.77 pA, n = 7, P = 0.039, unpaired t test), or when the peak was increased by ∼15% and the τw by ∼10% (phasic Imean: from −8.08 ± 0.53 to −10.0 ± 0.60 pA, n = 7, P = 0.031, unpaired t test; Fig. 5F and G). Notably, in any of the simulations none of the individual parameters were significantly different from each other.
Imean detected even smaller differences when all three sIPSCs parameters were increased simultaneously by insignificant amounts. When the peak amplitude, frequency and τw were slightly increased by ∼5% (to 50.3 ± 3.35 pA, n = 7, P = 0.560, unpaired t test), by ∼10% (to 30.0 ± 2.77 Hz, n = 7, P = 0.303, unpaired t test) and by ∼5% (to 6.68 ± 0.20 ms, n = 7, P = 0.444, unpaired t test), respectively, the phasic Imean increased significantly (from −8.08 ± 0.53 to −10.2 ± 0.78 pA, n = 7, P = 0.044, unpaired t test).
We also studied how opposite changes in two parameters influences phasic Imean. A significant 25% increase in peak amplitude (to 58.3 ± 3.91 pA, n = 7, P = 0.040) and a simultaneous 30% significant decrease in frequency (to 18.71 ± 1.44 Hz, n = 7, P = 0.015) did not modify phasic Imean (from −7.96 ± 0.65 to −7.03 ± 0.54 pA, n = 7, P = 0.290). Thus, as sIPSC properties change in opposite directions, the total current mediated by the synaptic events may remain constant. These simulations show that the Imean method can reliably detect small combined differences in IPSC parameters, and, when used in combination with conventional event detection, can predict when two opposing changes cancel out each other. At some combination of the parameters the individual events may merge to create a shifted baseline current that may be difficult to separate from the tonic current. However, as shown in Figs 6 and 7, in our recordings this condition was not reached even at IPSC frequencies of ∼100 Hz.
Figure 6. Increased tonic inhibitory current during spontaneous bursts of sIPSCs.

A, whole-cell voltage-clamp recording from a molecular layer interneuron in the presence of 3 mm kynurenic acid and 5 μm GABA (Vh=−70 mV) showing a spontaneous burst of sIPSCs (•). Lower panel shows expanded 5 s segments from the burst (•, 52 Hz) and control (○, 9 Hz) conditions. The dashed line indicates the baseline current in the control condition. B, simulated traces using the recorded parameters (frequency, peak amplitude, 10–90% rise time, τw and baseline noise) from the experimental condition. The dashed line indicates a baseline current of 0 pA. C, left panel: Gaussian fits to all-points histograms of the simulated traces using the parameters obtained from the experimental condition at 1-times and 1.5-times the maximum experimentally obtained event frequency during the burst. Shifts in baseline currents are as follows during the respective sIPSC frequencies: 10.4 Hz (−1.31 pA), 50.9 Hz (−2.69 pA) and at 75.7 Hz (−4.46 pA). Right panel: the difference in the tonic current measured in the experiment between the control condition and the spontaneous burst. GABA spillover must account for 13.4 pA of the increase in tonic current (16.1 pA − 2.69 pA).
Figure 7. Tonic inhibition increases during induced sIPSCs bursts.

A, left panel: whole-cell voltage-clamp recording from a CA1 PC in the presence of 3 mm kynurenic acid and 5 μm GABA (Vh=−70 mV) where sucrose (300 mm, black line) was applied by low pressure from a pipette positioned close to the soma to induce a burst of IPSCs (•). Lower panel shows expanded 5 s segments from the burst (•, 68 Hz) and control (○, 19 Hz) conditions. The dotted line indicates the baseline current in the control condition. Right panel: Gaussian fits to the all-points histograms (dashed lines) of the two traces on the left indicating the shift in baseline current produced by application of sucrose (s.d. of the Gaussian fits: control 4.54 pA; burst 15.1 pA). B, left panel: simulated traces using the experimentally recorded sIPSC parameters. The dotted line indicates a baseline current of 0 pA. Right panel: Gaussian fits to all-points histograms of the simulated traces using the parameters obtained from the experimental condition at 1-times and 1.5-times the maximum experimentally obtained event frequency during the burst. The shifts in baseline current at the indicated frequencies are as follows: 21 Hz (−1.06 pA), 66 Hz (−2.93 pA) and at 102 Hz (−4.77 pA). GABA spillover should account for 20.1 pA of the increases in tonic current (23 pA – 2.93 pA). C, same as A but recorded from a Gabra5/Gabrd−/− double knockout mouse CA1 PC (Vh=−70 mV) that lacks tonic inhibition altogether. Note the absence of a tonic inhibitory current during the induced sIPSC burst (s.d. of the Gaussian fits: control 5.26 pA; burst 6.14 pA). D, linear regression between the burst-induced change in tonic and phasic currents indicating a high degree of correlation during a condition of increased vesicular GABA release (n = 14). Line represents the linear fit while dashed lines represent 95% confidence intervals. E, histogram plots of phasic and tonic current measured during induced bursts in wild type (WT) and in Gabra5/Gabrd−/− double knockout mice that lack tonic currents. (n = 14; *P < 0.001, unpaired t test).
Spontaneous bursts of sIPSCs increase the tonic inhibitory current
Some dentate gyrus interneurons show a bursting pattern of firing (Han et al. 1993). We recorded spontaneous bursts of IPSCs in dentate gyrus molecular layer interneurons in the presence of 3 mm kynurenic acid and 5 μm GABA without blocking GABA uptake with NO-711 (n = 4; Fig. 6A). During the bursts, the IPSCs in the cell shown in Fig. 6A had a frequency of 52.1 Hz which was ∼6 times higher than during baseline conditions (9.1 Hz). There was also an increase in peak amplitude, τw and σBL during the burst compared with the control baseline (burst: peak amplitude 118 ± 3.68 pA, τw 9.09 ± 0.43 ms, σBL 5.21 pA; control: peak amplitude 32.7 ± 1.77 pA, τw 6.25 ± 0.27 ms, σBL 3.88 pA; Fig. 6A). Besides an increase in phasic current there was a shift in the holding current during the burst compared with the baseline of 16.1 pA (Fig. 6C). This difference in holding current could be a result of either a summation of sIPSCs due to their high frequency or of GABA spillover from synapses to extra- and peri-synaptic receptors. Therefore, by using the frequency and kinetics of the sIPSCs as well as the σBL recorded in the control and burst segments we generated simulated traces of 15 s. The events were added to a baseline with a mean current of 0 pA plus the σBL measured from the recording. Next, an all point histogram of the whole 15 s was plotted and a Gaussian was fitted to obtain the baseline current (see Methods for details). A simulated control period (with an event frequency of 10.4 Hz) had a holding baseline of −1.31 pA. This increased to −2.69 pA when a burst was simulated (events at 50.9 Hz) and to a mere −4.46 pA when the frequency was increased 1.5 times, an overshoot, just in case some of the sIPSCs were not detected in the actual recording (75.7 Hz; Fig. 6B and C). This clearly indicates that the increase in frequency and altered kinetics of the sIPSCs accounts for only 2.69 pA of the baseline shift whereas the rest of the 13.4 pA should represent the activation of extrasynaptic receptors due to GABA spillover.
Increased tonic inhibitory current during induced bursts of sIPSCs
We next wanted to confirm the results of our simulations by inducing sIPSCs burst in a recorded cell. This was done in CA1 PCs recorded in whole-cell voltage-clamp in the presence of 3 mm kynurenic acid and 5 μm GABA. After a stable baseline recording was obtained, a short puff of sucrose (300 mm) was applied by a picospritzer to the soma of the recorded cell (Mody et al. 1994) (Fig. 7A) to induce a burst of sIPSCs (control: frequency 20.5 ± 0.62 Hz, peak amplitude 38.4 ± 1.67 pA, RT10-90 0.61 ± 0.02 ms, τw 6.54 ± 0.27 ms; burst: frequency 58.1 ± 3.36 Hz, peak amplitude 42.9 ± 1.80 pA, RT10-90 0.51 ± 0.02 ms, τw not determined due to the high number of overlapping events; n = 5 bursts periods with their adjacent control period). The bursts induced a 24.3 ± 1.48 pA increase in holding current and an increase from 6.95 ± 0.65 to 17.3 ± 0.8 pA in phasic current (see Methods under Burst analysis; n = 14). To determine if this increase in holding current was a consequence of the summation of high frequency of events or of GABA spillover, we simulated one of the bursts (Fig. 7B). There was a −1.06 pA shift in baseline when the events were simulated at a control frequency (21 Hz) while at 65.6 Hz (burst event frequency) this shift increased to −2.93 pA, and to −4.77 pA when the frequency of events was increased 1.5 times (102.4 Hz; Fig. 7C). However, when the change in holding current was determined in the actual CA1 PC, the baseline shifted by 23 pA (Fig. 7A, right) indicating that 20.1 pA of the baseline shift was due to activation of extrasynaptic receptors following GABA spillover. In addition, the changes in phasic and tonic currents produced by the enhanced vesicular release triggered by the sucrose puffs were highly correlated (r = 0.86, P < 0.0001; n = 14, Fig. 7D).
The GABAAR α5 subunit is mainly distributed extrasynaptically in CA1 and CA3 PCs of the hippocampus (Sur et al. 1999b; Pirker et al. 2000; Brunig et al. 2002; Serwanski et al. 2006). Mice lacking the GABAAR α5 subunit (Gabra5−/−) show a residual amount of tonic current in CA1 PCs mediated by an up-regulation of the GABAAR δ subunit without change of phasic current (Glykys & Mody, 2006). Recently, we generated Gabra5/Gabrd−/− double mutant mice which lack altogether tonic inhibitory current in CA1 PCs (1.47 ± 0.68 pA, n = 6). These mice can be used to determine if summation of high-frequency sIPSCs or the activation of extrasynaptic receptors is responsible for the change in baseline current during the bursts. We repeated the experiment in slices obtained from these mice where brief sucrose puffs were applied to CA1 PCs which increased the sIPSCs frequency and kinetics (control: frequency 25.4 ± 1.29 Hz, peak amplitude 41.9 ± 2.06 pA, RT10-90 0.49 ± 0.02 ms, τw 5.46 ± 0.15 ms; burst: frequency 61 ± 2.66 Hz, peak amplitude 53.3 ± 2.36 pA, RT10-90 0.47 ± 0.03 pA, τw 4.64 ± 0.11; n = 6). As expected, in a neuron where there is no tonic current, the shift in baseline current during the sucrose puff only increased by 3.93 ± 0.89 pA while phasic current reached 19.5 ± 0.91 pA during the burst (n = 14; Fig. 7C and E). Notably, there were no statistical differences between wildtype and Gabra5/Gabrd−/− phasic currents in either burst or control conditions (P > 0.05, unpaired t test, Fig. 7E). These results strongly support the idea that excessive GABA released from vesicles triggered by the sucrose puffs spills over onto extrasynaptic receptors during sIPSC bursts and causes the increase in the baseline current during the bursts, i.e. tonic current. As expected, this increase is highly correlated with the increase in phasic current during the burst (Fig. 7D).
A decrease in phasic current is correlated with a decrease in tonic current
The use of Imean allowed us to calculate the phasic and tonic current in any desired epoch (e.g. 1 s) of the traces recorded in the presence of TTX and CdCl2 (Fig. 1) to determine if a correlation exists between the two currents (Fig. 8A). The phasic Imean decreased from −9.56 ± 1.68 pA in the control condition to −5.31 ± 0.72 pA in the presence of TTX and CdCl2 (P = 0.032, n = 6, paired t test). The cross-correlation between phasic and tonic inhibitory current showed a positive peak at t = 0 s (0.40 ± 0.11, n = 6; Fig. 8B) indicating that at this time resolution (1 s) the two currents are correlated to each other with no phase lag. In reality, the change in phasic current is expected to precede that in the tonic current due to the time required for diffusion of GABA to extrasynaptic sites. In all recorded neurons the two types of inhibition had a positive covariance (21.5 ± 10.9, n = 6) and a significant linear regression (P < 0.0001; n = 5; Fig. 8C for an example). These results indicate that a large fraction of the tonic inhibitory current is mediated by action potential-dependent GABA release. It should be noted, however, that as mIPSCs are frequent, vesicular GABA release is still present in the absence of action potentials which may be responsible for the tonic current persisting in the presence of TTX + CdCl2.
Figure 8. Correlation between phasic and tonic inhibitory currents following blockade of action potential-dependent GABA release.

A, whole-cell voltage-clamp recording from the same CA1 PC depicted in Fig. 1A showing phasic and tonic Imean simultaneously measured every second. The current in the presence of BMI was subtracted to yield a mean of 0 pA. Lines above the recordings indicate when TTX (5 μm) + CdCl2 (50 μm) and BMI (100 μm) were applied. B, normalized cross-correlation of phasic and tonic currents from the recording in A showing a positive correlation at 0 s. All neurons analysed (n = 6) showed a mean positive cross-correlation at 0 s of 0.40 ± 0.11. C, linear regression displaying 292 continuous seconds from the neuron depicted in A showing that phasic and tonic currents are correlated. Line represents the linear fit while dashed lines represent 95% confidence intervals
Thus far, we have shown that phasic and tonic inhibitory currents are both reduced when Ca2+ entry into the terminals and action potentials are blocked. Although this treatment reduces, but does not abolish, vesicular GABA release, it provides only an indirect proof for the vesicular origin of the source of GABA responsible for the two types of inhibition. Therefore, we wanted to examine the direct effect of reducing vesicular GABA content on the phasic and tonic inhibitory currents. Concanamycin A (folimycin) blocks the vacuolar-type ATPase, thus reducing the neurotransmitter concentration in vesicles (Drose & Altendorf, 1997; Harrison & Jahr, 2003). We used concanamycin A to reduce vesicular GABA content. CA1 PCs were incubated in either DMSO or concanamycin A (100 nm or 3.8 μm) for > 1 h and recorded in the presence of 3 mm kynurenic acid and 7.5 μm NO-711. When compared with control conditions, 100 nm concanamycin A decreases both phasic and tonic currents (control: phasic Imean 4.28 ± 0.55 pA, tonic 33.5 ± 4.34 pA, n = 6; 100 nm concanamycin A: phasic Imean 3.63 ± 0.27 pA, tonic 21.2 ± 6.58 pA, n = 5; Fig. 9A and D). In the presence of 3.8 μm concanamycin A the frequency and amplitudes of sIPSCs were reduced to 6.7 ± 1.14 Hz, and 18.7 ± 1.44 pA, respectively (from 22.8 ± 2.27 Hz and 32 ± 1.28 pA under control conditions), as expected after reduction of vesicular filling. This produced a corresponding decrease in phasic Imean (0.59 ± 0.11 pA) and a correlated decrease in the tonic current (14.5 ± 2.95 pA, n = 6; Fig. 9A and D). One-way ANOVA for all three groups showed significance for both phasic (P < 0.0001) and tonic currents (P = 0.029) while Dunnett's post hoc test showed a significant difference between the control condition and 3.8 μm concanamycin A (phasic ImeanP < 0.0001, tonic P = 0.018; Fig. 9C). A linear regression between phasic and tonic currents using the values obtained in all recorded neurons (control and concanamycin A) showed a significant correlation (r = 0.493, P = 0.044; Fig. 9D) with a large positive covariance (11.6) of the two types of inhibition. These results are consistent with both the phasic and tonic currents being strongly dependent on the amount of GABA contained in the vesicles.
Figure 9. Correlated decrease in tonic and phasic inhibitions following a reduction in vesicular GABA content.

A, whole-cell voltage-clamp recording from a CA1 PC in the presence of 3 mm kynurenic acid and 5 μm GABA (Vh=−70 mV) showing a 44.3 pA tonic current. Lower panel: 5 s expanded segment of the trace shown above (phasic Imean=−4.21 pA) B, same as A but in a CA1 PC recorded in a slice pre-incubated in 3.8 μm concanamycin A for 1 h. The tonic current is 14.55 pA. Lower panel: 5 s expanded segment of the trace shown above (phasic Imean=−0.50 pA). C, bar graphs of tonic (left) and phasic (right) currents recorded under three different conditions: control (▪), 100 nm (•) and 3.8 μm concanamycin A (▴). *P < 0.05 (one-way ANOVA, Dunnett's post hoc test). D, linear regression between the normalized phasic and tonic currents. Symbols correspond to the experimental conditions in C; open symbols are individual experiments, filled symbols are mean ±s.e.m. Continuous line represents the linear fit to the data points while the dashed lines represent the 95% confidence intervals (r = 0.493, P = 0.044).
Discussion
A novel approach to measuring inhibition (Imean) enabled us to simultaneously determine phasic and tonic inhibitory currents during short epochs in mouse hippocampal neurons. Using a variety of experimental approaches to decrease or increase the amount of GABA released from synaptic vesicles, we have shown a strong positive correlation between the two types of inhibitions. We conclude that vesicular GABA release is responsible for most of the tonic inhibitory current seen in hippocampal neurons under our recording conditions.
The Imean method computes a global inhibitory current over a chosen time period and can account for the combined effects of changes in frequency and in various properties of the recorded synaptic currents. Previous methods calculated the overall synaptic currents over various time periods (charge) based on measuring the area of the average detected event and multiplying it by the mean frequency of the events over the period of interest. This method fails at high event frequencies when events overlap or at small event amplitudes when events cannot be detected. In contrast, the Imean method does not rely on the use of detected events. It is highly sensitive to small changes in frequency and event properties even when each of these properties does not change significantly in the detected events. Moreover, as the Imean reflects the compound effect of all alterations in event properties, changes in opposite directions may cancel each other, producing no net change in the global current. Calculating Imean can be automated without the need for sophisticated detection algorithms. In our study, the major advantage of the Imean method was to simultaneously measure tonic and phasic inhibitory currents during short epochs thus allowing us for the first time to quantify correlations between the two types of inhibition.
Tonic inhibition is mediated by various extrasynaptic receptors expressed in different areas of the brain (Semyanov et al. 2004; Farrant & Nusser, 2005). These receptors are especially well-suited to sense low concentrations of GABA due to their specific subunit composition with high affinity for GABA which includes either δ or the α5 subunits (Saxena & MacDonald, 1994; Burgard et al. 1996; Fisher & MacDonald, 1997). The potential sources of extracellular GABA responsible for the activation of neuronal peri- and extra-synaptic receptors have received great attention because drugs modulating interstitial GABA levels, especially those that block GABA transporters, are of great therapeutic potential (Eckstein-Ludwig et al. 1999; Dalby, 2003; Perucca, 2005). Several sources of extracellular GABA have been proposed, including the reversal of GABA transporters (Richerson & Wu, 2003), non-vesicular and action potential-independent leakage mechanisms (Attwell et al. 1993; Wall & Usowicz, 1997; Rossi et al. 2003), action potential-dependent mechanisms (Kaneda et al. 1995; Brickley et al. 1996; Bright et al. 2007) as well as release from astrocytes (Liu et al. 2000; Kozlov et al. 2006).
In the present study, we have shown that tonic and phasic inhibitory currents are correlated under conditions of both enhanced and decreased vesicular GABA release. However, not every condition resulted in a perfect ‘one-to-one’ correlation between phasic and tonic inhibitory currents. For example, when vesicular GABA content was reduced, and thus the GABA released from the vesicles was diminished, phasic current was affected more than tonic current. This is consistent with the different affinities for GABA of the receptors that mediate the two types of inhibition. It was previously shown that low concentrations of gabazine (200 nm) significantly reduce phasic current without altering tonic inhibitory current, demonstrating that the GABAAR assemblies mediating tonic current have a higher affinity for GABA (Stell & Mody, 2002) as shown in expression systems (Saxena & MacDonald, 1994; Brown et al. 2002). Therefore, in the presence of low extracellular GABA levels after reduced vesicular filling, tonic inhibitory current would still be present, while the lower affinity GABAARs responsible for phasic inhibition in the recorded cell would not be active. As vesicles from synapses onto neighbouring cells also release GABA, albeit at reduced levels, the ambient GABA levels may not be depleted by the same amount as the reduction of phasic current recorded in a single neuron. This also means that in pathological processes where synaptic vesicle filling might be impaired, tonic inhibitory current would become the predominant force in maintaining inhibitory control. Conversely, the high affinity of the GABA receptors responsible for generating the tonic conductance may also be responsible for the deviation from linearity of the correlation between the phasic and tonic currents shown in Fig. 8C. As phasic inhibition continues to increase, the levels of GABA may reach a concentration that can no longer enhance the tonic conductance. This property might impose a natural limitation on the size of the tonic conductance that can be generated under a given condition.
The correlation between tonic and phasic current was also evident during enhancement of vesicular GABA release. During bursts of sIPSCs (spontaneously occurring or induced by enhancing vesicular GABA release by applying sucrose puffs) there was a simultaneous and highly correlated increase in tonic current. We have shown that the increased steady current during the bursts is due to spillover of GABA onto nearby peri- and extra-synaptic receptors producing tonic inhibitory current. This idea was unequivocally confirmed by recordings from Gabra5/Gabrd−/− mice in CA1 PCs that are devoid of any tonic current.
Our results are in contrast to studies in adult CGCs, where the magnitude of tonic inhibitory current was not dependent on action potential-mediated GABA release (Wall & Usowicz, 1997; Rossi et al. 2003), but are in accordance with recordings from CGCs early on during development where tonic and phasic inhibitions are both action potential dependent (Kaneda et al. 1995; Brickley et al. 1996) and in thalamic relay neurons of the dorsal lateral geniculate nucleus (Bright et al. 2007). It has to be noted, however, that CGCs have specialized inhibitory/excitatory synapses which are ensheathed by an astrocytic glomerulus (Eccles et al. 1967; Jakab & Hamori, 1988) most probably preventing long distance diffusion of GABA. Most inhibitory synapses in the CNS are devoid of such specialized synaptic structure thus allowing the free diffusion of GABA to neighbouring cells. This more frequent synaptic arrangement allows diffusional transmission by vesicular GABA through spillover from the synaptic cleft to nearby extra- and peri-synaptic receptors before being transported back into neurons and astrocytes (Sykova, 2004).
Our results do not completely rule out the contribution of astrocytes, reversed GABA transporters or action potential-independent mechanisms to the maintenance of ambient GABA levels, particularly during specific physiological or pathological conditions (Attwell et al. 1993; Liu et al. 2000; Richerson & Wu, 2003; Rossi et al. 2003; Wu et al. 2003; Sykova, 2004). For example, in lactating animals there is a retraction of astrocytic processes in hypothalamic magnocellular neurons which results in an enhanced diffusion of glutamate needed for synaptic cross-talk (Piet et al. 2004). Reversal of the GABA transporter has been demonstrated in the presence of high extracellular K+ as well as in association with a decrease in extracellular Na+ concentration, which resemble conditions of high-frequency neuronal firing (Gaspary et al. 1998). Nevertheless, our results under conditions designed to restore ambient GABA concentrations to mimic those expected in vivo, demonstrate that the main component of tonic inhibitory currents in hippocampal neurons is mediated by spillover subsequent to the release of GABA-containing vesicles. Until vesicular release can be entirely abolished in our slices, it will be difficult to assign a numerical value to just how large this main component is.
The GABA concentration released into the synaptic cleft can reach the millimolar range (Nusser et al. 2001; Mozrzymas et al. 2003). In the absence of a barrier but depending on several diffusional parameters (Sykova, 2004) and the number of vesicles released, GABA molecules will freely diffuse over some distance resulting in a sufficient concentration to activate the high-affinity GABAARs responsible for tonic current. An activity-dependent GABA spillover from presynaptic terminals will be most effective on adjacent extra- and peri-synaptic receptors. Further away from the releasing boutons, GABA transporters could decrease the GABA concentration in a distance- and region-specific manner (Engel et al. 1998). Other factors affecting the diffusion of GABA in the extracellular space may also play a role as it is seen with glutamate diffusion (Rusakov & Kullmann, 1998; Sykova, 2004). In contrast to a glia-dependent GABA release or uptake that may influence a basal tone of inhibition in a wider area, synaptic GABA spillover should have more direct and local influence on the postsynaptic neuron's firing and integrative properties (Mitchell & Silver, 2003; Chadderton et al. 2004). Thus interneurons can influence other neurons not just by intersynaptic diffusion (Scimemi et al. 2004) but also by activating extrasynaptic receptors on neighbouring cells that do not receive synapses from the given interneuron. In this manner, burst of sIPSCs mediated by specific classes of interneurons like fast spiking cells onto PCs (Somogyi & Klausberger, 2005) could readily influence the amount of tonic inhibition of their postsynaptic target cells as well as that of nearby but not synaptically connected neurons.
The close involvement of tonic inhibition in various physiological (e.g. ovarian cycle (Maguire et al. 2005)) and pathological (e.g. ethanol action (Wei et al. 2004; Glykys et al. 2007), epilepsy (Houser & Esclapez, 2003; Peng et al. 2004)) events in the brain makes it critical to understand the origin of the transmitter activating this type of inhibition. Based on our study, the main source for tonic inhibition under physiological conditions is the GABA contained in the same vesicles that are responsible for generating phasic inhibition.
Acknowledgments
We thank Guido Faas for stimulating discussions, M. Rafizadeh and M. Lazaro for technical assistance. Supported by the Gonda Fellowship (J.G.), The Coelho Endowment and NIH grant NS30549 (I.M.).
References
- Attwell D, Barbour B, Szatkowski M. Nonvesicular release of neurotransmitter. Neuron. 1993;11:401–407. doi: 10.1016/0896-6273(93)90145-h. [DOI] [PubMed] [Google Scholar]
- Brickley SG, Cull-Candy SG, Farrant M. Development of a tonic form of synaptic inhibition in rat cerebellar granule cells resulting from persistent activation of GABAA receptors. J Physiol. 1996;497:753–759. doi: 10.1113/jphysiol.1996.sp021806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bright DP, Aller MI, Brickley SG. Synaptic release generates a tonic GABAA receptor-mediated conductance that modulates burst precision in thalamic relay neurons. J Neurosci. 2007;27:2560–2569. doi: 10.1523/JNEUROSCI.5100-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown N, Kerby J, Bonnert TP, Whiting PJ, Wafford KA. Pharmacological characterization of a novel cell line expressing human α4β3δ GABAA receptors. Br J Pharmacol. 2002;136:965–974. doi: 10.1038/sj.bjp.0704795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunig I, Scotti E, Sidler C, Fritschy JM. Intact sorting, targeting, and clustering of gamma-aminobutyric acid A receptor subtypes in hippocampal neurons in vitro. J Comp Neurol. 2002;443:43–55. doi: 10.1002/cne.10102. [DOI] [PubMed] [Google Scholar]
- Burgard EC, Tietz EI, Neelands TR, MacDonald RL. Properties of recombinant gamma-aminobutyric acid A receptor isoforms containing the α5 subunit subtype. Mol Pharmacol. 1996;50:119–127. [PubMed] [Google Scholar]
- Caraiscos VB, Elliott EM, You T, Cheng VY, Belelli D, Newell JG, Jackson MF, Lambert JJ, Rosahl TW, Wafford KA, MacDonald JF, Orser BA. Tonic inhibition in mouse hippocampal CA1 pyramidal neurons is mediated by α5 subunit-containing gamma-aminobutyric acid type A receptors. Proc Natl Acad Sci U S A. 2004;101:3662–3667. doi: 10.1073/pnas.0307231101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chadderton P, Margrie TW, Hausser M. Integration of quanta in cerebellar granule cells during sensory processing. Nature. 2004;428:856–860. doi: 10.1038/nature02442. [DOI] [PubMed] [Google Scholar]
- Cope DW, Hughes SW, Crunelli V. GABAA receptor-mediated tonic inhibition in thalamic neurons. J Neurosci. 2005;25:11553–11563. doi: 10.1523/JNEUROSCI.3362-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalby NO. Inhibition of gamma-aminobutyric acid uptake: anatomy, physiology and effects against epileptic seizures. Eur J Pharmacol. 2003;479:127–137. doi: 10.1016/j.ejphar.2003.08.063. [DOI] [PubMed] [Google Scholar]
- Ding R, Asada H, Obata K. Changes in extracellular glutamate and GABA levels in the hippocampal CA3 and CA1 areas and the induction of glutamic acid decarboxylase-67 in dentate granule cells of rats treated with kainic acid. Brain Res. 1998;800:105–113. doi: 10.1016/s0006-8993(98)00507-1. [DOI] [PubMed] [Google Scholar]
- Drasbek KR, Jensen K. THIP, a hypnotic and antinociceptive drug, enhances an extrasynaptic GABAA receptor-mediated conductance in mouse neocortex. Cereb Cortex. 2006;16:1134–1141. doi: 10.1093/cercor/bhj055. [DOI] [PubMed] [Google Scholar]
- Drose S, Altendorf K. Bafilomycins and concanamycins as inhibitors of V-ATPases and P-ATPases. J Exp Biol. 1997;200:1–8. doi: 10.1242/jeb.200.1.1. [DOI] [PubMed] [Google Scholar]
- Eccles JC, Ito M, Szentagothai J. The Cerebellum as a Neuronal Machine. Heidelberg: Springer; 1967. [Google Scholar]
- Eckstein-Ludwig U, Fei J, Schwarz W. Inhibition of uptake, steady-state currents, and transient charge movements generated by the neuronal GABA transporter by various anticonvulsant drugs. Br J Pharmacol. 1999;128:92–102. doi: 10.1038/sj.bjp.0702794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engel D, Schmitz D, Gloveli T, Frahm C, Heinemann U, Draguhn A. Laminar difference in GABA uptake and GAT-1 expression in rat CA1. J Physiol. 1998;512:643–649. doi: 10.1111/j.1469-7793.1998.643bd.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrant M, Nusser Z. Variations on an inhibitory theme: phasic and tonic activation of GABAA receptors. Nat Rev Neurosci. 2005;6:215–229. doi: 10.1038/nrn1625. [DOI] [PubMed] [Google Scholar]
- Fisher JL, MacDonald RL. Functional properties of recombinant GABAA receptors composed of single or multiple β subunit subtypes. Neuropharmacology. 1997;36:1601–1610. doi: 10.1016/s0028-3908(97)00133-0. [DOI] [PubMed] [Google Scholar]
- Gaspary HL, Wang W, Richerson GB. Carrier-mediated GABA release activates GABA receptors on hippocampal neurons. J Neurophysiol. 1998;80:270–281. doi: 10.1152/jn.1998.80.1.270. [DOI] [PubMed] [Google Scholar]
- Glykys J, Mody I. Hippocampal network hyperactivity after selective reduction of tonic inhibition in GABAA receptor α5 subunit-deficient mice. J Neurophysiol. 2006;95:2796–2807. doi: 10.1152/jn.01122.2005. [DOI] [PubMed] [Google Scholar]
- Glykys J, Peng Z, Chandra D, Homanics GE, Houser CR, Mody I. A new naturally occurring GABAA receptor subunit partnership with high sensitivity to ethanol. Nat Neurosci. 2007;10:40–48. doi: 10.1038/nn1813. [DOI] [PubMed] [Google Scholar]
- Haas KF, MacDonald RL. GABAA receptor subunit γ2 and δ subtypes confer unique kinetic properties on recombinant GABAA receptor currents in mouse fibroblasts. J Physiol. 1999;514:27–45. doi: 10.1111/j.1469-7793.1999.027af.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han ZS, Buhl EH, Lorinczi Z, Somogyi P. A high degree of spatial selectivity in the axonal and dendritic domains of physiologically identified local-circuit neurons in the dentate gyrus of the rat hippocampus. Eur J Neurosci. 1993;5:395–410. doi: 10.1111/j.1460-9568.1993.tb00507.x. [DOI] [PubMed] [Google Scholar]
- Harrison J, Jahr CE. Receptor occupancy limits synaptic depression at climbing fiber synapses. J Neurosci. 2003;23:377–383. doi: 10.1523/JNEUROSCI.23-02-00377.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houser CR, Esclapez M. Downregulation of the α5 subunit of the GABAA receptor in the pilocarpine model of temporal lobe epilepsy. Hippocampus. 2003;13:633–645. doi: 10.1002/hipo.10108. [DOI] [PubMed] [Google Scholar]
- Jakab RL, Hamori J. Quantitative morphology and synaptology of cerebellar glomeruli in the rat. Anat Embryol Berl. 1988;179:81–88. doi: 10.1007/BF00305102. [DOI] [PubMed] [Google Scholar]
- Jensen K, Chiu CS, Sokolova I, Lester HA, Mody I. GABA transporter-1 (GAT1)-deficient mice: differential tonic activation of GABAA versus GABAB receptors in the hippocampus. J Neurophysiol. 2003;90:2690–2701. doi: 10.1152/jn.00240.2003. [DOI] [PubMed] [Google Scholar]
- Jia F, Pignataro L, Schofield CM, Yue M, Harrison NL, Goldstein PA. An extrasynaptic GABAA receptor mediates tonic inhibition in thalamic VB neurons. J Neurophysiol. 2005;94:4491–4501. doi: 10.1152/jn.00421.2005. [DOI] [PubMed] [Google Scholar]
- Kaneda M, Farrant M, Cull-Candy SG. Whole-cell and single-channel currents activated by GABA and glycine in granule cells of the rat cerebellum. J Physiol. 1995;485:419–435. doi: 10.1113/jphysiol.1995.sp020739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klaassen A, Glykys J, Maguire J, Labarca C, Mody I, Boulter J. Seizures and enhanced cortical GABAergic inhibition in two mouse models of human autosomal dominant nocturnal frontal lobe epilepsy. Proc Natl Acad Sci U S A. 2006;103:19152–19157. doi: 10.1073/pnas.0608215103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozlov AS, Angulo MC, Audinat E, Charpak S. Target cell-specific modulation of neuronal activity by astrocytes. Proc Natl Acad Sci U S A. 2006;103:10058–10063. doi: 10.1073/pnas.0603741103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuntz A, Clement HW, Lehnert W, van Calker D, Hennighausen K, Gerlach M, Schulz E. Effects of secretin on extracellular amino acid concentrations in rat hippocampus. J Neural Transm. 2004;111:931–939. doi: 10.1007/s00702-003-0082-y. [DOI] [PubMed] [Google Scholar]
- Lerma J, Herranz AS, Herreras O, Abraira V, Martin del Rio R. In vivo determination of extracellular concentration of amino acids in the rat hippocampus. A method based on brain dialysis and computerized analysis. Brain Res. 1986;384:145–155. doi: 10.1016/0006-8993(86)91230-8. [DOI] [PubMed] [Google Scholar]
- Liu QY, Schaffner AE, Chang YH, Maric D, Barker JL. Persistent activation of GABAA receptor/Cl− channels by astrocyte-derived GABA in cultured embryonic rat hippocampal neurons. J Neurophysiol. 2000;84:1392–1403. doi: 10.1152/jn.2000.84.3.1392. [DOI] [PubMed] [Google Scholar]
- Maguire JL, Stell BM, Rafizadeh M, Mody I. Ovarian cycle-linked changes in GABAA receptors mediating tonic inhibition alter seizure susceptibility and anxiety. Nat Neurosci. 2005;8:797–804. doi: 10.1038/nn1469. [DOI] [PubMed] [Google Scholar]
- Mitchell SJ, Silver RA. GABA spillover from single inhibitory axons suppresses low-frequency excitatory transmission at the cerebellar glomerulus. J Neurosci. 2000;20:8651–8658. doi: 10.1523/JNEUROSCI.20-23-08651.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell SJ, Silver RA. Shunting inhibition modulates neuronal gain during synaptic excitation. Neuron. 2003;38:433–445. doi: 10.1016/s0896-6273(03)00200-9. [DOI] [PubMed] [Google Scholar]
- Mody I. Distinguishing between GABAA receptors responsible for tonic and phasic conductances. Neurochem Res. 2001;26:907–913. doi: 10.1023/a:1012376215967. [DOI] [PubMed] [Google Scholar]
- Mody I, De Koninck Y, Otis TS, Soltesz I. Bridging the cleft at GABA synapses in the brain. Trends Neurosci. 1994;17:517–525. doi: 10.1016/0166-2236(94)90155-4. [DOI] [PubMed] [Google Scholar]
- Mozrzymas JW, Zarnowska ED, Pytel M, Mercik K. Modulation of GABAA receptors by hydrogen ions reveals synaptic GABA transient and a crucial role of the desensitization process. J Neurosci. 2003;23:7981–7992. doi: 10.1523/JNEUROSCI.23-22-07981.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nusser Z, Mody I. Selective modulation of tonic and phasic inhibitions in dentate gyrus granule cells. J Neurophysiol. 2002;87:2624–2628. doi: 10.1152/jn.2002.87.5.2624. [DOI] [PubMed] [Google Scholar]
- Nusser Z, Naylor D, Mody I. Synapse-specific contribution of the variation of transmitter concentration to the decay of inhibitory postsynaptic currents. Biophys J. 2001;80:1251–1261. doi: 10.1016/S0006-3495(01)76101-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nusser Z, Sieghart W, Somogyi P. Segregation of different GABAA receptors to synaptic and extrasynaptic membranes of cerebellar granule cells. J Neurosci. 1998;18:1693–1703. doi: 10.1523/JNEUROSCI.18-05-01693.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng Z, Huang CS, Stell BM, Mody I, Houser CR. Altered expression of the δ subunit of the GABAA receptor in a mouse model of temporal lobe epilepsy. J Neurosci. 2004;24:8629–8639. doi: 10.1523/JNEUROSCI.2877-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perucca E. An introduction to antiepileptic drugs. Epilepsia. 2005;46:31–37. doi: 10.1111/j.1528-1167.2005.463007.x. [DOI] [PubMed] [Google Scholar]
- Piet R, Vargova L, Sykova E, Poulain DA, Oliet SH. Physiological contribution of the astrocytic environment of neurons to intersynaptic crosstalk. Proc Natl Acad Sci U S A. 2004;101:2151–2155. doi: 10.1073/pnas.0308408100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pirker S, Schwarzer C, Wieselthaler A, Sieghart W, Sperk G. GABAA receptors: immunocytochemical distribution of 13 subunits in the adult rat brain. Neuroscience. 2000;101:815–850. doi: 10.1016/s0306-4522(00)00442-5. [DOI] [PubMed] [Google Scholar]
- Richerson GB, Wu YM. Dynamic equilibrium of neurotransmitter transporters: Not just for reuptake anymore. J Neurophysiol. 2003;90:1363–1374. doi: 10.1152/jn.00317.2003. [DOI] [PubMed] [Google Scholar]
- Rossi DJ, Hamann M. Spillover-mediated transmission at inhibitory synapses promoted by high affinity α6 subunit GABAA receptors and glomerular geometry. Neuron. 1998;20:783–795. doi: 10.1016/s0896-6273(00)81016-8. [DOI] [PubMed] [Google Scholar]
- Rossi DJ, Hamann M, Attwell D. Multiple modes of GABAergic inhibition of rat cerebellar granule cells. J Physiol. 2003;548:97–110. doi: 10.1113/jphysiol.2002.036459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rusakov DA, Kullmann DM. Extrasynaptic glutamate diffusion in the hippocampus: ultrastructural constraints, uptake, and receptor activation. J Neurosci. 1998;18:3158–3170. doi: 10.1523/JNEUROSCI.18-09-03158.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxena NC, MacDonald RL. Assembly of GABAA receptor subunits: role of the δ subunit. J Neurosci. 1994;14:7077–7086. doi: 10.1523/JNEUROSCI.14-11-07077.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scimemi A, Fine A, Kullmann DM, Rusakov DA. NR2B-containing receptors mediate cross talk among hippocampal synapses. J Neurosci. 2004;24:4767–4777. doi: 10.1523/JNEUROSCI.0364-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semyanov A, Walker MC, Kullmann DM, Silver RA. Tonically active GABAA receptors: modulating gain and maintaining the tone. Trends Neurosci. 2004;27:262–269. doi: 10.1016/j.tins.2004.03.005. [DOI] [PubMed] [Google Scholar]
- Serwanski DR, Miralles CP, Christie SB, Mehta AK, Li X, De Blas AL. Synaptic and nonsynaptic localization of GABAA receptors containing the α5 subunit in the rat brain. J Comp Neurol. 2006;499:458–470. doi: 10.1002/cne.21115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somogyi P, Klausberger T. Defined types of cortical interneurone structure space and spike timing in the hippocampus. J Physiol. 2005;562:9–26. doi: 10.1113/jphysiol.2004.078915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sperk G, Schwarzer C, Tsunashima K, Fuchs K, Sieghart W. GABAA receptor subunits in the rat hippocampus I: immunocytochemical distribution of 13 subunits. Neuroscience. 1997;80:987–1000. doi: 10.1016/s0306-4522(97)00146-2. [DOI] [PubMed] [Google Scholar]
- Stell BM, Brickley SG, Tang CY, Farrant M, Mody I. Neuroactive steroids reduce neuronal excitability by selectively enhancing tonic inhibition mediated by δ subunit-containing GABAA receptors. Proc Natl Acad Sci U S A. 2003;100:14439–14444. doi: 10.1073/pnas.2435457100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stell BM, Mody I. Receptors with different affinities mediate phasic and tonic GABAA conductances in hippocampal neurons. J Neurosci. 2002;22:RC223. doi: 10.1523/JNEUROSCI.22-10-j0003.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun C, Sieghart W, Kapur J. Distribution of α1, α4, γ2, and δ subunits of GABAA receptors in hippocampal granule cells. Brain Res. 2004;1029:207–216. doi: 10.1016/j.brainres.2004.09.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sur C, Farrar SJ, Kerby J, Whiting PJ, Atack JR, McKernan RM. Preferential coassembly of α4 and δ subunits of the gamma-aminobutyric acid A receptor in rat thalamus. Mol Pharmacol. 1999a;56:110–115. doi: 10.1124/mol.56.1.110. [DOI] [PubMed] [Google Scholar]
- Sur C, Fresu L, Howell O, McKernan RM, Atack JR. Autoradiographic localization of α5 subunit-containing GABAA receptors in rat brain. Brain Res. 1999b;822:265–270. doi: 10.1016/s0006-8993(99)01152-x. [DOI] [PubMed] [Google Scholar]
- Sykova E. Extrasynaptic volume transmission and diffusion parameters of the extracellular space. Neuroscience. 2004;129:861–876. doi: 10.1016/j.neuroscience.2004.06.077. [DOI] [PubMed] [Google Scholar]
- Tossman U, Jonsson G, Ungerstedt U. Regional distribution and extracellular levels of amino acids in rat central nervous system. Acta Physiol Scand. 1986;127:533–545. doi: 10.1111/j.1748-1716.1986.tb07938.x. [DOI] [PubMed] [Google Scholar]
- Wall MJ, Usowicz MM. Development of action potential-dependent and independent spontaneous GABAA receptor-mediated currents in granule cells of postnatal rat cerebellum. Eur J Neurosci. 1997;9:533–548. doi: 10.1111/j.1460-9568.1997.tb01630.x. [DOI] [PubMed] [Google Scholar]
- Wei W, Faria LC, Mody I. Low ethanol concentrations selectively augment the tonic inhibition mediated by delta subunit-containing GABAA receptors in hippocampal neurons. J Neurosci. 2004;24:8379–8382. doi: 10.1523/JNEUROSCI.2040-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu YM, Wang WG, Richerson GB. Vigabatrin induces tonic inhibition via GABA transporter reversal without increasing vesicular GABA release. J Neurophysiol. 2003;89:2021–2034. doi: 10.1152/jn.00856.2002. [DOI] [PubMed] [Google Scholar]
- Zoli M, Jansson A, Sykova E, Agnati LF, Fuxe K. Volume transmission in the CNS and its relevance for neuropsychopharmacology. Trends Pharmacol Sci. 1999;20:142–150. doi: 10.1016/s0165-6147(99)01343-7. [DOI] [PubMed] [Google Scholar]