

Abstract
Membrane lipid composition is a major determinant of cell excitability. In this study, we assessed the role of membrane cholesterol composition in the distribution and function of Kv1.5-based channels in rat cardiac membranes. In isolated rat atrial myocytes, the application of methyl-β-cyclodextrin (MCD), an agent that depletes membrane cholesterol, caused a delayed increase in the Kv1.5-based sustained component, Ikur, which reached steady state in ∼7 min. This effect was prevented by preloading the MCD with cholesterol. MCD-increased current was inhibited by low 4-aminopyridine concentration. Neonatal rat cardiomyocytes transfected with Green Fluorescent Protein (GFP)-tagged Kv1.5 channels showed a large ultrarapid delayed-rectifier current (IKur), which was also stimulated by MCD. In atrial cryosections, Kv1.5 channels were mainly located at the intercalated disc, whereas caveolin-3 predominated at the cell periphery. A small portion of Kv1.5 floated in the low-density fractions of step sucrose-gradient preparations. In live neonatal cardiomyocytes, GFP-tagged Kv1.5 channels were predominantly organized in clusters at the basal plasma membrane. MCD caused reorganization of Kv1.5 subunits into larger clusters that redistributed throughout the plasma membrane. The MCD effect on clusters was sizable 7 min after its application. We conclude that Kv1.5 subunits are concentrated in cholesterol-enriched membrane microdomains distinct from caveolae, and that redistribution of Kv1.5 subunits by depletion of membrane cholesterol increases their current-carrying capacity.
Outward K+ currents are essential for shaping cardiomyocyte action potentials. These currents are carried by a range of voltage-gated potassium (Kv) channels that are part of multiprotein complexes containing various auxiliary subunits, scaffolding proteins and/or second messengers that regulate channel location and function (Rettig et al. 1994; Kim et al. 1995).
Lipids are other important determinants of channel function. Their composition greatly influence structural and physical properties of the plasma membrane, such as fluidity, curvature and stiffness, which are important regulators of the gating of voltage-gated channels (Elinder et al. 1996; Lundbaek et al. 1996). Some lipids, like phospholipids, can also directly modulate channel function. Arachidonic acids and their amide anandamide introduce rapid inactivation in otherwise non-inactivating Kv channels by binding to channels close to their selectivity filter (Oliver et al. 2004).
Cholesterol and sphingolipids, two major lipids of the plasma membrane, can also pack tightly together to form microdomains called ‘lipid rafts’. Lipid rafts are dynamic platforms important for the delivery of proteins to the membrane and for sequestering proteins in close physical proximity to control their functional interactions (Pike, 2004). An increasing number of channels has been found to be targeted into these cholesterol- and sphingolipid-rich membrane microdomains, including Kv channels (Levitan et al. 2000; Martens et al. 2000; Martens et al. 2001; Yarbrough et al. 2002; Hajdu et al. 2003; Barbuti et al. 2004; Pouvreau et al. 2004; Wong & Schlichter, 2004; Xia et al. 2004; Brainard et al. 2005; Maguy et al. 2006). However, most of the data available have been obtained in heterologous expression systems and evidence of the localization of endogenous Kv channels in cholesterol-rich membrane microdomains of cardiac myocytes and its functional importance are lacking.
Important clues regarding the regulation of channel function by cholesterol have been obtained owing to the use of methyl-β-cyclodextrin (MCD). This molecule removes cholesterol from plasma membranes of live cells. MCD can be added to culture media or applied to single cells via the bath perfusate and is effective at both physiological and room temperatures (Christian et al. 1997; Heino et al. 2000; Slimane et al. 2001; Barbuti et al. 2004). MCD application changes the properties of several Kv channels in both native tissues and heterologous expression systems (Martens et al. 2000, 2001; Hajdu et al. 2003; Xia et al. 2004). In L-cells stably expressing Kv1.5 channels, MCD shifts the activation and inactivation curves of the current (Martens et al. 2001).
In atrial cardiomyocytes, the ultrarapid delayed-rectifier current (IKur) is an important repolarizing current and is believed to be largely the functional expression of Shaker Kv1.5 channels (Fedida et al. 1993, 2003; Wang et al. 1993; Feng et al. 1997). The aim of this study was to examine the effect of membrane cholesterol depletion on the distribution and function of Kv1.5 subunits in rat cardiomyocytes. We show here that MCD-induced cholesterol depletion enhances IKur by modulating the function and clustering of Kv1.5 subunits.
Methods
Cardiac tissue samples and cardiomyocyte isolation
Animal handling was in accordance with the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health. A 1/1 mixture of xylazine (20 mg ml−1) and ketamine (100 mg ml−1) was prepared and Wistar rats were anaesthetized using an intraperitoneal injection (0.1 ml (100 mg body weight)−1). Whole hearts were rapidly excised and thoroughly washed in phosphate-buffered saline (PBS) to eliminate residual blood. The left atria were then isolated, frozen in liquid nitrogen and stored at −80°C for biochemistry and immunohistochemistry. For electrophysiological studies, atrial myocytes were enzymatically isolated as previously described (Boixel et al. 2001). The left atrium was removed, cut up, and washed in Ca2+-free Krebs–Ringer solution containing (mm): 35 NaCl, 4.75 KCl, 1.19 KH2PO4, 16 Na2HPO4, 10 Hepes, 10 glucose, 25 NaHCO3, 134 sucrose, and 30 2,3-butanedione 2-monoxime (BDM) (pH was adjusted to 7.4 with NaOH), gassed with 95% O2–5% CO2, and maintained at 37°C. Pieces were re-incubated in this solution without BDM and containing bovine serum albumin (BSA) (5 mg ml−1, Hoechst-Behring), 200 U ml−1 collagenase (type IV, Sigma Chemical Co.), and 6 U ml−1 protease (type XXIV, Sigma). After 30 min of digestion, the enzyme solution was replaced by the same solution containing only collagenase (400 U ml−1). Isolated myocytes were resuspended in a bicarbonate-buffered Tyrode solution containing 2 mm Ca2+ and incubated at 37°C with continuous gassing with 95% O2–5% CO2 for at least 1 h before use.
One-day-old neonatal Wistar rats were killed by decapitation with sharp scissors and hearts were rapidly excised and washed to remove blood and debris in pre-oxygenated Tyrode solution containing (mm): 135 NaCl, 4 KCl, 2 MgCl2, 10 Hepes, 1 NaH2PO4, 20 glucose, 2.5 pyruvate, adjusted to pH 7.4 with NaOH. The ventricles were carefully minced and dissociated into single cells by proteolytic enzymes in Tyrode solution containing 0.1 mg ml−1 collagenase A (Roche Applied Science) and 1% of bovine serum albumin, during repeated digestions with gentle continuous stirring and aeration with 100% O2 at 37°C for 10 min. Cell were centrifuged at 100 g for 10 min and the pellet resuspended in growth medium containing serum to inhibit proteolytic enzymes. This step was repeated 6 times and pellets were pooled after each digestion. After 1 h of pre-plating to purify the myocyte population from fibroblasts, the cells were counted, adjusted to the desired density (1 × 106 cells per 9.6 cm2) for seeding on laminin-coated (Roche) LabTek borosilicate slides (Nunc) and grown in Dulbecco's modified Eagle's medium (Gibco) supplemented with 10% horse serum (Biowest SAS), 5% fetal bovine serum (Biowest Ltd), 100 U ml−1 penicillin and 100 mg ml−1 streptomycin, in standard conditions (37°C, 5% CO2).
Recombinant proteins and transfection
GFP-tagged human Kv1.5 (HKv1.5) cDNA was generated by RT-PCR and inserted into the multicloning site of the expression vector pcDNA3 as previously described (Godreau et al. 2002, 2003). Twenty-four hours after cell isolation, neonatal ventricular cardiomyocytes (1 × 106 cells per 9.6 cm2) were transfected with 1 μg of GFP-tagged Kv1.5 expression vector using 4 μl of FuGENE 6 Transfection Reagent (Roche Diagnostics) plus OptiMEM (Gibco) that yields ∼10% of transfected cells.
Membrane microdomains separation and Western blot analysis
Protein extraction
Frozen atrial appendages (n = 15) were powdered with a mortar in liquid nitrogen and homogenized on ice with a glass Potter in 2 ml TNE solution (mm: 20 Tris; 150 NaCl; 1 EDTA, pH 7.4) to which we added Complete EDTA-free Protease Inhibitor cocktail (Roche Applied Science). The homogenate was centrifuged at 1000 g for 5 min at 4°C. The pellet was resuspended in extraction buffer, re-homogenized with the Potter and centrifuged again at 1000 g for 5 min at 4°C. This latter step was repeated three times to enhance protein extraction efficiency. All supernatants, corresponding to total protein fraction, were collected and pooled in Eppendorf tubes. Triton X-100 was then added to the total protein fraction with a final concentration of 1% in order to solubilize proteins localized in the bulk plasma membrane. Note that all steps of the protein extraction were performed at 4°C; at this temperature lipid rafts are insoluble in 1% Triton X-100. After 30 min incubation on ice, the protein concentration was determined using a Bio-Rad protein assay (Bio-Rad Laboratories). Then an Eppendorf tube was filled with 17.2 mg of the obtained protein fraction and the volume was completed to 2 ml with cold extraction buffer (4°C).
Sucrose gradient separation and SDS-PAGE
Two millilitres of 80% sucrose solution was placed in a SW41 centrifuge tube (Beckman); 2 ml of the total protein (17.2 mg) was placed on the sucrose solution and the preparation was mixed with a vortex-agitator for 30 s. Four millilitres of 35% sucrose was gently poured onto the mixture, followed by 4 ml of 5% sucrose. The gradient was then centrifuged in a Centrikon T-1170 ultracentrifuge (Kontron Instruments), for 18 h at 178 300 g and 4°C, without braking. Fractions of 1 ml were collected from the top to the bottom of the centrifuge tube (12 fractions kept at −80°C). Each sample fraction was sonicated, its protein concentration measured, and the same protein quantity loaded into each lane of 12.5% polyacrylamide-SDS gels. After separation, proteins were transferred to PVDF membrane (NEN). The membrane was blocked with 5% non-fat milk and incubated with appropriate primary and secondary antibodies. Negative controls consisted in omitting the primary antibody and in incubating membranes with only the secondary antibody. Development was done using Western Lightning Chemoluminescence Reagent Plus (Perkin Elmer Life Science Inc.) and BioFlex Scientific Imaging Films (Clonex Corporation).
Antibodies
Mouse anti-caveolin-3 was from BD Transduction Laboratories, rabbit anti-Kv1.5 was from Alomone Laboratories and rabbit anti-connexin-43 (Zymed Laboratories) was from Chemicon International. Sarcomeric α-actinin was from Sigma. Peroxidase-conjugated goat anti-mouse and FITC-conjugated anti-rabbit IgG were from Jackson ImmunoResearch Laboratory. Texas Red-conjugated anti-mouse IgG and FITC-conjugated anti-rabbit IgG were obtained from Amersham.
Immunohistochemistry–confocal microscopy
Cryostat sections (7 μm) of rat atrial tissue were fixed in 4% formaldehyde for 10 min, permeabilized with 0.2% Triton X-100 for 5 min then pre-incubated for 30 min at room temperature (RT) in PBS containing 5% BSA to block non-specific binding sites. Tissues were rinsed in PBS (pH 7.4) between each step. Then slides were incubated with rabbit anti-Kv1.5 polyclonal antibodies (pAbs) (1: 20), with mouse anti-caveolin-3 monoclonal antibodies (mAbs) (1: 50), with a mixture of mouse anti-caveolin-3 and rabbit anti-connexin-43 (1: 50) antibodies, or with a mixture of mouse anti-α-actinin sarcomeric and rabbit anti-Kv1.5 antibodies for 1 h at RT. After three washes in PBS, binding of primary antibodies was detected with Texas Red-conjugated anti-mouse IgG (1: 40) and FITC-conjugated anti-rabbit IgG (1: 40). All antibodies were prepared in PBS containing 2% BSA. To check for the specificity of the anti-Kv1.5 antibody, the immunostaining was repeated using a primary antibody incubated overnight with the antigen at 4°C. In this condition, no staining could be detected on cryosection of atrial myocardium (data not shown). Sections were finally washed in PBS, mounted in Vectashield medium and observed with a Zeiss Axiovert-200 confocal microscope equipped with an argon laser. Images were acquired and analysed with LSM510 version 3.2 software.
Neonatal cardiomyocytes were observed with the confocal microscope using a ×63 oil immersion objective with pinhole set at 1 Airy. The 488 nm line of an argon laser source (10% of full power) was used for GFP excitation. All experiments on neonatal cardiomyocytes were performed at 5% CO2 and 37°C using a temperature-controlled stage.
The quantification of clusters in live myocytes was performed using the integrated morphometry analysis of the MetaMorph software to measure surface size of clusters. This program uses a threshold tool to set a grey level that discriminates clusters from background light and measures the area of signal above the threshold. Surface area of clusters were expressed in pixels and averaged for each cell. The counting was performed on stack images, conducted in double blind and expressed as number of clusters per myocyte.
Electrophysiology
Whole-cell patch-clamp currents were recorded as previously described (Godreau et al. 2002). Cells were bathed or perfused in solutions containing (mm): 135 NaCl (or choline chloride), 4 KCl, 2 CaCl2, 2 MgCl2, 1 Na2HPO4, 2.5 sodium pyruvate, 10 Hepes, 20 glucose; pH was adjusted to 7.4 with NaOH. Patch pipettes were filled with a solution containing (mm): 115 potassium aspartate, 10 KCl, 2 KH2PO4, 3 MgCl2, 5 EGTA, 5 MgATP, 10 Hepes, 10 glucose and pH was adjusted to 7.2 with KOH. Activation plots were generated by dividing the current by the difference between test and reversal potential and were fitted by the Boltzmann distribution equation: 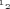 , where G represents the conductance calculated at membrane potential V,
, where G represents the conductance calculated at membrane potential V, 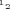 the potential at which half the channels are activated, and k the slope factor. All experiments were carried out at RT (22–24°C). Leak current was not compensated.
the potential at which half the channels are activated, and k the slope factor. All experiments were carried out at RT (22–24°C). Leak current was not compensated.
Drugs
5-Methyl-β-cyclodextrin (MCD), 4-aminopyridine (4-AP) and tetraethylammonium (TEA) were obtained from Sigma and dissolved in the extracellular solution. Cholesterol–MCD (8: 2) complexes were obtained as follows: 100 mg of cholesterol (Sigma) was dissolved in 2 ml of a 1/1 chloroform–methanol solution and 12.8 μl of this solution was added to 10 ml of extracellular solution and left overnight at 37°C.
Statistical analysis
Data are presented as means ±s.e.m. When appropriate, Student's paired or unpaired t tests were used to determine the significance of differences. P values < 0.05 were considered significant.
Results
Methyl-β-cyclodextrin increases IKur-related component in atrial myocytes
We first examined the effect of the extracellular application of 2% MCD on the outward potassium currents of atrial myocytes. In these cells, test pulses from −90 mV to +50 mV elicited an outward current characterized by a fast inactivating (It) and a maintained component, the latter corresponding to IKur. We observed a slow increase in IKur (after ∼400 s of MCD application) to 15.6 ± 1.3 pA pF−1, from a baseline of 8.5 ± 1.2 pA pF−1, n = 20; P << 0.001) (Fig. 1A). It is worth noting that following 15–20 min applications of MCD a leak current was likely to occur. Thus, the application of MCD was limited to 20 min or when such a current was noticeable, the data of this recording were rejected. In around 50% of cells, a rapid inhibition of the fast outward current amplitude was observed just after MCD application (from 2.1 ± 0.7 nA to 1.8 ± 0.6 nA, n = 10, P < 0.05). We first checked that prolonged current recording did not affect the kinetics and amplitude of the outward current by repeating the experiments using a control external solution (n = 5; Fig. 1C and D). In order to determine the specificity of MCD effects on current, the molecule was saturated with cholesterol at a ratio of 8: 2 (ratio previously shown to prevent its capacity to buffer lipids: Christian et al. 1997; Heino et al. 2000; Slimane et al. 2001). As illustrated in Fig. 1B, prolonged cell exposure to the cholesterol–MCD complex caused no increase in the outward K+ current (n = 10) while the fast inhibition was still observed. The MCD-activated current was sensitive to 4-AP as indicated by its inhibition with 50 μm 4-AP (46.3 ± 3.7%, n = 8) and was further suppressed at 2 mm 4-AP (74.6 ± 8.1%, n = 6; Fig. 2A and B). The outward current was poorly sensitive to TEA (4.7 ± 1.2%, n = 5) (Fig. 2C). Following MCD exposure, there was a slight but statistically non-significant (n.s.) change in steady-state activation of the maintained current, as indicated by the values of the slope factor (11.8 ± 5.5 mV in control versus 4.1 ± 1.5 mV in MCD, n = 7, n.s.) and the  (20.9 ± 6.2 mV in control versus 16.9 ± 4.7 mV in MCD; n = 7, n.s.) (Fig. 3). Taken together, these results indicate that prolonged MCD application increases a 4-AP-sensitive component of outward current in atrial myocytes, an effect which appears to be due to the depletion of membrane cholesterol.
(20.9 ± 6.2 mV in control versus 16.9 ± 4.7 mV in MCD; n = 7, n.s.) (Fig. 3). Taken together, these results indicate that prolonged MCD application increases a 4-AP-sensitive component of outward current in atrial myocytes, an effect which appears to be due to the depletion of membrane cholesterol.
Figure 1. Effects of cholesterol depletion on outward currents in native adult atrial myocytes.

A, direct and long period effects of MCD application on the outward current elicited as indicated by the command waveform at 0.2 Hz, shown in the inset. B, effects of MCD–cholesterol application. C, time course changes in Ikur amplitude in control conditions, upon application of MCD and MCD–cholesterol complexes. D, bar graphs summarizing the effects of MCD and MCD–cholesterol complex, measured after 700 s application.
Figure 2. Pharmacological properties of the MCD-activated current in native adult atrial myocytes.

Effects of 50 μm (A) and 2 mm 4-AP (B), and 10 mm TEA (C), on the 650 s MCD-stimulated outward current elicited as indicated by the command waveform at 0.2 Hz.
Figure 3. MCD effects on electrophysiological parameters of the outward K+ current of native adult atrial myocytes.

Current traces elicited during incremental depolarizing test pulses in control (A) conditions and after MCD application (B). Cell capacitance: 75 pF. C, current density–voltage relationships of Ikur recorded in control conditions (•) and following MCD (^) application. D, voltage dependence of current activation in control (•) and MCD (^) conditions. In C and D, each point is the average of 7 cells.
Methyl-β-cyclodextrin increases Kv1.5 subunit-encoded current in neonatal rat cardiomyocytes
To further determine the nature of the 4-AP-sensitive current activated by MCD and because it is difficult to dissect the various components of the K+ outward current in atrial myocytes, we overexpressed the Kv1.5 subunit in neonatal cardiomyocytes which is believed to underlie a large part of the atrial sustained current, IKur. Neonatal myocytes were used because they are easier to transfect and to maintain in culture than adult myocytes. Moreover, they represent a more physiological environment for cardiac ion channels than heterologous expression systems. Cardiomyocytes expressing GFP-tagged Kv1.5 channels showed a large voltage-dependent, rapidly activating and slowly inactivating outward current upon depolarizing voltage steps (Fig. 4A). For instance, at +60 mV, a large current activated in transfected myocytes (183 ± 24.7 pA pF−1, n = 7, versus 8.6 ± 1.6 pA pF−1, in control, n = 6), suggesting that roughly 30 times more functional channels were present in the cell membrane. This current was inhibited by 500 μm 4-AP, confirming that it resulted from the functional expression of GFP-Kv1.5 channels. The application of MCD caused a slow increase in the Kv1.5-based current, which started around 7 min after drug application (average time of 7.4 ± 0.8 min, maximum increase 33 ± 11%, n = 7) (Fig. 4B and C). The MCD-activated outward current in neonatal cardiomyocytes was inhibited by 500 μm 4-AP (n = 5; Fig. 4B). As for adult cardiomyocytes, cholesterol-loaded MCD complexes had no effect on Kv1.5-encoded current (n = 5) (Fig. 4C). The stimulatory effect of MCD was associated with a decrease of the slope factor of the steady-state activation curve of the Kv1.5-encoded current (25.9 ± 3.7 mV in control and 15.2 ± 2.3 mV in MCD, n = 5, P < 0.05) together with a slight but statistically significant shift of its 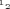 (18.1 ± 1.5 mV in control and 13.5 ± 1.2 mV in MCD; n = 5, P < 0.05) (Fig. 5). These results indicate that membrane cholesterol depletion caused by MCD is associated with increased Kv1.5 subunit channel function.
(18.1 ± 1.5 mV in control and 13.5 ± 1.2 mV in MCD; n = 5, P < 0.05) (Fig. 5). These results indicate that membrane cholesterol depletion caused by MCD is associated with increased Kv1.5 subunit channel function.
Figure 4. MCD enhances Kv1.5-encoded currents in neonatal cardiomyocytes.

A, traces of currents recorded during incremental 10 mV step depolarizations in control and in Kv1.5-transfected cardiomyocytes. B, effects of MCD on the outward current elicited by a test pulse from −80 to +60 mV in control, and after 350, 500 and 600 s MCD applications. Effect of 500 μm 4-AP on 650 s MCD-stimulated current is also shown. C, time course of the MCD and MCD–cholesterol complex effects on the outward current.
Figure 5. Effect of cholesterol depletion on outward current parameters resulting from Kv1.5 subunit overexpression in neonatal cardiomyocytes.

Current density–voltage relationships (A) and voltage dependence (B) of IKur activation under control conditions (•) and following 7 min MCD application (^). In A and B, each point represents average data from 5 cells.
Kv1.5 channel complexes are not localized in caveolae of atrial myocardium
To examine the mechanism of action of cholesterol on Kv1.5 channel properties, we next studied how the lipid regulates channel organization in the plasma membrane. In cardiac myocytes, cholesterol-enriched lipid rafts (caveolae) containing caveolin-3 are abundant. Immunostaining of rat atrial cryosections revealed robust caveolin-3 staining, seen principally at the cell periphery but not at the level of the intercalated disc (Fig. 6B). In contrast, Kv1.5 channel labelling predominated at the level of the intercalated disc, with some staining at the edge of the cell periphery (Fedida et al. 2003) (Fig. 6A). We also co-immunostained cryosections with antibodies directed both against caveolin-3 and connexin-43 (Cx-43), which is another intercalated disc protein (Fig. 6C and D). Again, there was no overlay between caveolin-3 and Cx-43 confirming the lack of caveolin in intercalated discs of the atrial myocardium (Fig. 6E).
Figure 6. Kv1.5 do not co-localized with caveolin-3 in adult atrial tissue.

Immunolocalization of Kv1.5 subunits (A) and caveolin-3 (B) in cryosections of atrial myocardium. Double immunostaining of connexin-43 (FITC, C) and caveolin-3 (Texas Red, D) in cryosections of atrial myocardium. E, merged image of the same area of the section in C and D, showing the lack of overlap between connexin-43 and caveolin-3 stainings.
We also separated detergent-resistant from detergent-soluble membrane proteins of rat atrial myocardium with a step sucrose gradient (n = 4). As expected, caveolin-3 was found predominantly in low density sucrose fractions 4–5, corresponding to lipid rafts (Fig. 7). A single or a doublet band at the expected molecular weight of ∼64 kDa of the Kv1.5 subunits (Dobrzynski et al. 2000) was found predominantly in fractions 9–12 but also in low density fractions 4–5 (Fig. 7). Interestingly, Cx-43 was also detected in fractions 4–5 as a doublet corresponding to the phosphorylated and non-phosphorylated forms of the protein. (Fig. 7). Taken together, these results indicate that Kv1.5 subunits are mainly localized in non-lipid raft microdomains. However, a small fraction of Kv1.5 subunits are found in cholesterol-enriched microdomains distinct from caveolae.
Figure 7. Some Kv1.5 channel subunits are localized in lipid raft fractions in adult rat atrial myocytes.

Western blot analysis of the distribution of Kv1.5 subunits, connexin-43 and caveolin-3 on step sucrose gradient of proteins from atrial myocardium.
The clustering of Kv1.5 channels depends on membrane cholesterol
We then examined how plasma membrane cholesterol regulates the subcellular organization of Kv1.5 subunits. We used live neonatal rat cardiomyocytes transfected with GFP-Kv1.5 subunits. In sharp contrast with the homogeneously distributed free GFP, GFP-tagged Kv1.5 subunits were mainly organized in clusters concentrated at the basal plasma membrane as shown by the Z-axis view in Fig. 8A, lower panel. A similar channel distribution in clusters at the basal membrane was observed in fixed cells immunostained with anti-Kv1.5 antibody (Fig. 8D). Cells positive for the anti-Kv1.5 antibody were also stained with the sarcomeric α-actinin antibody with a typical striated pattern indicating that these cells were cardiac myocytes. The localization of Kv1.5 subunits at the basal membrane could be due to the presence of laminin on the coated dishes as observed for Kir4.1 channels (Guadagno & Moukhles, 2004). The role of cholesterol in the formation of Kv1.5 channel clusters was studied by incubating cells with 2% MCD. After 90 min of MCD application, clusters of GFP-Kv1.5 increased in size (25 ± 5 versus 145 ± 32 pixels; n = 20, P < 0.001) and in number (58 ± 5 versus 78 ± 6; n = 20, P < 0.05) and redistributed throughout the plasma membrane (Fig. 8B). In order to determine the time course of the MCD effect on clusters, the same cardiomyocytes were visualized during the first minutes following drug application in a single Z section in order to prevent excessive photobleaching. After 7 min of MCD application (corresponding to the time necessary for MCD to enhance the current), there was already a significant effect on the size (61 ± 11; n = 21 cells, P < 0.01) and a tendency to increase in number (69 ± 6; n = 21 cells) of GFP-Kv1.5 subunit clusters (Fig. 8C). The same time course of MCD effects on Kv1.5 channel distribution was obtained at room temperature (data not shown). These results indicate that cholesterol is an important determinant of the organization of Kv1.5 subunits in clusters and of their distribution into plasma membranes.
Figure 8. Surface expression of Kv1.5 subunits in neonatal cardiomyocytes.

A, in live cardiomyocytes, transfected GFP-tagged Kv1.5 subunits are clustered at the membrane surface adjacent to the bottom of laminin-coated glass support, as shown in the projection of Z sections in the lower panel. In contrast, GFP alone was homogeneously distributed in cardiomyocytes (inset). B, after the application of 2% MCD, clusters increased in size and were redistributed throughout the plasma membrane. C, bar graphs summarizing changes in cluster size upon MCD exposures; data are from 21 cardiomyocytes in control, and following incubation with 2% MCD for 7 min and 1 h 30 min. **P < 0.01, ***P < 0.001. D, double immunostaining of fixed cardiomyocytes using sarcomeric α-actinin and anti-Kv1.5 antibodies showing that Kv1.5-GFP-transfected cells are cardiomyocytes. A, B and D: scale bars represent 10 μm.
Discussion
In the present study we found that plasma membrane cholesterol is an important determinant of the distribution of Kv1.5 subunits and the properties of the corresponding current IKur in atrial cardiomyocytes. A fraction of these subunits are located in membrane microdomains rich in cholesterol but distinct from caveolae. These effects of membrane cholesterol on Kv1.5 subunit distribution and function could have significant consequences for the regulation of cardiac excitability in both physiological and pathophysiological conditions.
The predominant effect of MCD on the outward potassium current of atrial cardiomyocytes is to induce a slow and progressive increase in IKur. This delayed increase in IKur appears to be related to the capacity of MCD to deplete membrane cholesterol, since it was eliminated when MCD was pre-saturated with cholesterol. The effects of MCD on current observed after several minutes of drug application could not be attributed to a non-specific effect due to prolonged patch-clamp recording, as no current changes were observed over corresponding periods in control external solution. The delay between drug application and increase in IKur corresponds to the time necessary for MCD to deplete membrane cholesterol (Launikonis & Stephenson, 2001; Lam et al. 2004). In addition to the delayed increase in sustained current, in some cells MCD causes a fast and reversible inhibition of the peak outward current also observed with the MCD–cholesterol complex suggesting a direct effect of MCD on channels (Eldstrom et al. 2006).
Several arguments indicate that Kv1.5 subunit-based channels contributed in part to the MCD-activated current. First, Kv1.5-based channels are believed to be the molecular basis of IKur in atrial cardiomyocytes (Fedida et al. 1993; Wang et al. 1993; Feng et al. 1997). Second, the MCD-activated current was sensitive to low concentrations of 4-AP, as are Kv1.5 channels (Wang et al. 1993). Third, in neonatal cardiomyocytes, Kv1.5-encoded current is also stimulated after prolonged (15–20 min) application of MCD, an effect no longer observed in the presence of a saturating concentration of cholesterol. However, it is likely that other channels contribute to the MCD-activated current as suggested by the activation of leak-type current after more than 15 min of MCD application.
Previous studies have demonstrated that membrane cholesterol is an important regulator of ion channel properties (Levitan et al. 2000; Martens et al. 2000, 2001; Yarbrough et al. 2002; Hajdu et al. 2003; Barbuti et al. 2004; Pouvreau et al. 2004; Wong & Schlichter, 2004; Xia et al. 2004; Brainard et al. 2005). As for Kv1.5 subunits in the present study, cholesterol depletion caused by MCD increases the activity of maxi-K+ channels in human myometrium (Brainard et al. 2005), K+ channels of Drosophila Kenyon cells (Gasque et al. 2005) or inwardly rectifying Kir channels (Romanenko et al. 2004; Fang et al. 2006). However, the underlying mechanisms of this cholesterol effect on ionic channels are unclear.
Single-channel studies have eliminated the possibility that the lipid modulates single-channel unitary conductances (Romanenko et al. 2004; Toselli et al. 2005). Rather, it seems that cholesterol affects the number of active channels in the plasma membrane. In our study, the relative short delay between MCD application and IKur increase eliminates the possibility of changes in Kv1.5 subunit synthesis. This is in agreement with the lack of variation of Kir mRNA concentrations in endothelial cells following cholesterol accumulation (Romanenko et al. 2004).
There are several reports suggesting that membrane cholesterol can modulate the equilibrium between active and silent forms of channels. For instance, a reduced open probability (Po) of Kir or N-type single Ca2+ channels was observed following cholesterol membrane accumulation (Romanenko et al. 2004; Toselli et al. 2005). The authors proposed that depletion of membrane cholesterol decreases membrane stiffness and consequently the membrane deformation energy associated with channel opening. Such a process explains the effect of cholesterol on N-type channels, whose gating properties are modulated by membrane tension and stiffness (Toselli et al. 2005). However, the magnitude of the effect of changes in membrane deformation on outward current activation is unknown. Changes in cholesterol can also alter surface charge in the vicinity of channels. However, the lack of major shift of the voltage dependence of IKur activation both in adult and neonatal cardiac myocytes following MCD application suggests that such a mechanism is not predominant (Elinder et al. 1996). In the range of time exposures of MCD (10–15 min) chosen in this study, no change in the whole-cell capacitance was observed with the straightforward standard technique used. However, this technique does not allow measuring the change in membrane capacitance expected from the slight decrease in membrane thickness induced by removing cholesterol (Czub & Baginski, 2006).
In rat atrial myocardium, Kv1.5 channels are not localized in caveolae, the lipid rafts containing caveolin-3. First, Kv1.5 channels are detected predominantly at the level of the intercalated disc, but not at the cell periphery where caveolin-3 is seen. Cx-43, another protein localizing to intercalated discs, similarly does not co-localize with caveolin. These data are in good agreement with published studies showing that caveolin-3 is located at the periphery of atrial and ventricular myocytes in t-tubules and not in intercalated discs (Yarbrough et al. 2002; Locke et al. 2005). Moreover, caveolin-3 and Kv1.5 subunits from rat and canine atrial myocardium fail to co-precipitate and are differentially distributed as seen using electron microscopy and immunogold staining (Eldstrom et al. 2006). However, a small portion of Kv1.5 subunits as well as Cx-43 can be detected in the low density sucrose fraction of the sucrose step gradient suggesting that there are subtypes of cholesterol-enriched membrane microdomains distinct from caveolae. In their study, Eldstrom et al. (2006) did not detect the presence of Kv1.5 subunits in the low density fractions of step gradient sucrose. This discrepancy between the two studies could be explained by differences in the procedure used to extract lipid rafts which can yield different subsets of lipid rafts. This is well known for the compartmentation of connexins in lipid rafts that depend on the procedure used (Locke et al. 2005). Furthermore, lipid rafts are heterogeneous structures, ranging from a highly ordered cholesterol core through less-ordered regions to the disordered structure of the bulk membrane. Proteins could be located in distinct regions of lipid rafts, resulting in distinct detergent solubility as reported for neuronal glycosylphosphatidylinositol-anchored (GPI) proteins (Madore et al. 1999). Proteins of the intercalated disc may also have a low affinity for surrounding lipids, so that they can be solubilized by detergents. For instance, the T cell receptor in Jurkat cells acquires resistance to Triton extraction when cross-linked by an antibody (Janes et al. 1999). However, the Eldstrom et al. (2006) study and our own study agree that the majority of the Kv1.5 subunits are not in the lipid raft fraction. It is possible that the location of a small fraction of Kv1.5 channels in lipid rafts is transient, taking place between channel delivery to the plasma membrane and their recruitment into specialized domains. It was reported that connexins in raft fractions are distinct from those in gap junctions and could be unpaired hemichannels en route to junctional domains (Schubert et al. 2002; Locke et al. 2005). The composition of membrane in cholesterol might regulate the partition of channels in these distinct compartments, and in turn their activity.
In live cardiomyocytes, we found that cholesterol regulates the organization of GFP-Kv1.5 subunits in clusters at the plasma membrane. Clustering appears as a general feature of channel organization and was reported for several K+ and Ca2+ channels (Ianoul et al. 2004) both in heterologous expression systems and in native tissues such as postsynaptic sites on the soma and proximal dendrites for Kv2.1 channels (Muennich & Fyffe, 2004) or the atrial myocardium for Kv1.5 channels using electron microscopy (Dobrzynski et al. 2000). Clustering may depend on the integrity of lipid-rich membrane microdomains, as reported for Kv2.1 in HEK-293 cells incubated with MCD (O'Connell & Tamkun, 2005). Overexpression of the Kv1.5 subunits may result in the accumulation of the protein in lipid rafts. We also found that effect of cholesterol depletion on clusters of Kv1.5 subunits is fast and occurred over a similar time course to MCD effects on IKur. This observation suggests a relationship between the ability of Kv1.5 subunits to function as channels and their spatial organization. This is reminiscent of the observations that the effects of cholesterol on the equilibrium between active and silent Kir 2.1 and 2.2 channels depends on integrity of lipid rafts (Romanenko et al. 2004; Fang et al. 2006). The physical clustering of KcsA potassium channels determines the mode of gating and activation of the channel (Molina et al. 2006). It has been shown, in HEK-293 cells, that clusters of Kv2.1 channels represent a well delineated membrane domain where channels are delivered via trafficking vesicles and trapped by several retention proteins within in the cluster perimeters (O'Connell et al. 2006). It is also possible that the increase in size and number of clusters of Kv1.5 channels following MCD application is due to more channels delivered in the plasma membrane. Thus, one other mechanism underlying the effect of cholesterol depletion on K+ current could be that Kv1.5 subunit clusters seen in the absence of MCD correspond to a lipid-rich compartment with ready access to the plasma membrane. When cholesterol is removed, this pool could release the Kv1.5 subunits and allow them to move rapidly into the membrane where they can form functional channels.
In conclusion, membrane cholesterol content regulates IKur, very likely by modulating the spatial organization and activity of Kv1.5 channels. It has been recently reported that inhibition of Kir channel function by cholesterol could be an important mechanism underlying the endothelial dysfunction caused by hypercholesterolaemia (Fang et al. 2006). There is also clinical evidence that treatment lowering plasma cholesterol is associated with a reduced risk of cardiac arrhythmia, notably atrial arrhythmias (Hanna et al. 2006). Thus, it would be of interest to study the pharmacological consequences of the cholesterol effect on potassium currents.
Acknowledgments
We thank Association Française contre les Myopathies, Fondation de France, the Agence Nationale de la Recherche (ANR-05-PCOD-006-01) and the Canadian Institutes of Health Research for their financial support. A. Maguy was supported by Groupe de Réflexion sur la Recherche Cardiovasculaire and Association Française contre les Myopathies, J. Abi-Char by the Société Française de Pharmacologie and the Association Française contre les Myopathies, and E. Balse by ANR-05-PCOD-006-01.
References
- Barbuti A, Gravante B, Riolfo M, Milanesi R, Terragni B, DiFrancesco D. Localization of pacemaker channels in lipid rafts regulates channel kinetics. Circ Res. 2004;94:1325–1331. doi: 10.1161/01.RES.0000127621.54132.AE. [DOI] [PubMed] [Google Scholar]
- Boixel C, Gonzalez W, Louedec L, Hatem SN. Mechanisms of L-type Ca2+ current downregulation in rat atrial myocytes during heart failure. Circ Res. 2001;89:607–613. doi: 10.1161/hh1901.096702. [DOI] [PubMed] [Google Scholar]
- Brainard AM, Miller AJ, Martens JR, England SK. Maxi-K channels localize to caveolae in human myometrium: a role for an actin-channel-caveolin complex in the regulation of myometrial smooth muscle K+ current. Am J Physiol Cell Physiol. 2005;289:C49–C57. doi: 10.1152/ajpcell.00399.2004. [DOI] [PubMed] [Google Scholar]
- Christian AE, Haynes MP, Phillips MC, Rothblat GH. Use of cyclodextrins for manipulating cellular cholesterol content. J Lipid Res. 1997;38:2264–2272. [PubMed] [Google Scholar]
- Czub J, Baginski M. Comparative molecular dynamics study of lipid membranes containing cholesterol and ergosterol. Biophys J. 2006;90:2368–2382. doi: 10.1529/biophysj.105.072801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobrzynski H, Rothery SM, Marples DD, Coppen SR, Takagishi Y, Honjo H, Tamkun MM, Henderson Z, Kodama I, Severs NJ, Boyett MR. Presence of the Kv1.5 K+ channel in the sinoatrial node. J Histochem Cytochem. 2000;48:769–780. doi: 10.1177/002215540004800606. [DOI] [PubMed] [Google Scholar]
- Eldstrom J, Van Wagoner DR, Moore ED, Fedida D. Localization of Kv1.5 channels in rat and canine myocyte sarcolemma. FEBS Lett. 2006;580:6039–6046. doi: 10.1016/j.febslet.2006.09.069. [DOI] [PubMed] [Google Scholar]
- Elinder F, Madeja M, Arhem P. Surface charges of K channels. Effects of strontium on five cloned channels expressed in Xenopus oocytes. J Gen Physiol. 1996;108:325–332. doi: 10.1085/jgp.108.4.325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang Y, Mohler ER, 3, Hsieh E, Osman H, Hashemi SM, Davies PF, Rothblat GH, Wilensky RL, Levitan I. Hypercholesterolemia suppresses inwardly rectifying K+ channels in aortic endothelium in vitro and in vivo. Circ Res. 2006;98:1064–1071. doi: 10.1161/01.RES.0000218776.87842.43. [DOI] [PubMed] [Google Scholar]
- Fedida D, Eldstrom J, Hesketh JC, Lamorgese M, Castel L, Steele DF, Van Wagoner DR. Kv1.5 is an important component of repolarizing K+ current in canine atrial myocytes. Circ Res. 2003;93:744–751. doi: 10.1161/01.RES.0000096362.60730.AE. [DOI] [PubMed] [Google Scholar]
- Fedida D, Wible B, Wang Z, Fermini B, Faust F, Nattel S, Brown AM. Identity of a novel delayed rectifier current from human heart with a cloned K+ channel current. Circ Res. 1993;73:210–216. doi: 10.1161/01.res.73.1.210. [DOI] [PubMed] [Google Scholar]
- Feng J, Wible B, Li GR, Wang Z, Nattel S. Antisense oligodeoxynucleotides directed against Kv1.5 mRNA specifically inhibit ultrarapid delayed rectifier K+ current in cultured adult human atrial myocytes. Circ Res. 1997;80:572–579. doi: 10.1161/01.res.80.4.572. [DOI] [PubMed] [Google Scholar]
- Gasque G, Labarca P, Darszon A. Cholesterol-depleting compounds modulate K+-currents in Drosophila Kenyon cells. FEBS Lett. 2005;579:5129–5134. doi: 10.1016/j.febslet.2005.08.022. [DOI] [PubMed] [Google Scholar]
- Godreau D, Vranckx R, Maguy A, Goyenvalle C, Hatem SN. Different isoforms of synapse-associated protein, SAP97, are expressed in the heart and have distinct effects on the voltage-gated K+ channel Kv1.5. J Biol Chem. 2003;278:47046–47052. doi: 10.1074/jbc.M308463200. [DOI] [PubMed] [Google Scholar]
- Godreau D, Vranckx R, Maguy A, Rucker-Martin C, Goyenvalle C, Abdelshafy S, Tessier S, Couetil JP, Hatem SN. Expression, regulation and role of the MAGUK protein SAP-97 in human atrial myocardium. Cardiovasc Res. 2002;56:433–442. doi: 10.1016/s0008-6363(02)00602-8. [DOI] [PubMed] [Google Scholar]
- Guadagno E, Moukhles H. Laminin-induced aggregation of the inwardly rectifying potassium channel, Kir4.1, and the water-permeable channel, AQP4, via a dystroglycan-containing complex in astrocytes. Glia. 2004;47:138–149. doi: 10.1002/glia.20039. [DOI] [PubMed] [Google Scholar]
- Hajdu P, Varga Z, Pieri C, Panyi G, Gaspar R., Jr Cholesterol modifies the gating of Kv1.3 in human T lymphocytes. Pflugers Arch. 2003;445:674–682. doi: 10.1007/s00424-002-0974-y. [DOI] [PubMed] [Google Scholar]
- Hanna IR, Heeke B, Bush H, Brosius L, King-Hageman D, Dudley SC, Jr, Beshai JF, Langberg JJ. Lipid-lowering drug use is associated with reduced prevalence of atrial fibrillation in patients with left ventricular systolic dysfunction. Heart Rhythm. 2006;3:881–886. doi: 10.1016/j.hrthm.2006.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heino S, Lusa S, Somerharju P, Ehnholm C, Olkkonen VM, Ikonen E. Dissecting the role of the Golgi complex and lipid rafts in biosynthetic transport of cholesterol to the cell surface. Proc Natl Acad Sci U S A. 2000;97:8375–8380. doi: 10.1073/pnas.140218797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ianoul A, Street M, Grant D, Pezacki J, Taylor RS, Johnston LJ. Near-field scanning fluorescence microscopy study of ion channel clusters in cardiac myocyte membranes. Biophys J. 2004;87:3525–3535. doi: 10.1529/biophysj.104.046383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janes PW, Ley SC, Magee AI. Aggregation of lipid rafts accompanies signaling via the T cell antigen receptor. J Cell Biol. 1999;147:447–461. doi: 10.1083/jcb.147.2.447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim E, Niethammer M, Rothschild A, Jan YN, Sheng M. Clustering of Shaker-type K+ channels by interaction with a family of membrane-associated guanylate kinases. Nature. 1995;378:85–88. doi: 10.1038/378085a0. [DOI] [PubMed] [Google Scholar]
- Lam RS, Shaw AR, Duszyk M. Membrane cholesterol content modulates activation of BK channels in colonic epithelia. Biochim Biophys Acta. 2004;1667:241–248. doi: 10.1016/j.bbamem.2004.11.004. [DOI] [PubMed] [Google Scholar]
- Launikonis BS, Stephenson DG. Effects of membrane cholesterol manipulation on excitation–contraction coupling in skeletal muscle of the toad. J Physiol. 2001;534:71–85. doi: 10.1111/j.1469-7793.2001.00071.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levitan I, Christian AE, Tulenko TN, Rothblat GH. Membrane cholesterol content modulates activation of volume-regulated anion current in bovine endothelial cells. J Gen Physiol. 2000;115:405–416. doi: 10.1085/jgp.115.4.405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Locke D, Liu J, Harris AL. Lipid rafts prepared by different methods contain different connexin channels, but gap junctions are not lipid rafts. Biochemistry. 2005;44:13027–13042. doi: 10.1021/bi050495a. [DOI] [PubMed] [Google Scholar]
- Lundbaek JA, Birn P, Girshman J, Hansen AJ, Andersen OS. Membrane stiffness and channel function. Biochemistry. 1996;35:3825–3830. doi: 10.1021/bi952250b. [DOI] [PubMed] [Google Scholar]
- Madore N, Smith KL, Graham CH, Jen A, Brady K, Hall S, Morris R. Functionally different GPI proteins are organized in different domains on the neuronal surface. EMBO J. 1999;18:6917–6926. doi: 10.1093/emboj/18.24.6917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maguy A, Hebert TE, Nattel S. Involvement of lipid rafts and caveolae in cardiac ion channel function. Cardiovasc Res. 2006;69:798–807. doi: 10.1016/j.cardiores.2005.11.013. [DOI] [PubMed] [Google Scholar]
- Martens JR, Navarro-Polanco R, Coppock EA, Nishiyama A, Parshley L, Grobaski TD, Tamkun MM. Differential targeting of Shaker-like potassium channels to lipid rafts. J Biol Chem. 2000;275:7443–7446. doi: 10.1074/jbc.275.11.7443. [DOI] [PubMed] [Google Scholar]
- Martens JR, Sakamoto N, Sullivan SA, Grobaski TD, Tamkun MM. Isoform-specific localization of voltage-gated K+ channels to distinct lipid raft populations. Targeting of Kv1.5 to caveolae. J Biol Chem. 2001;276:8409–8414. doi: 10.1074/jbc.M009948200. [DOI] [PubMed] [Google Scholar]
- Molina ML, Barrera FN, Fernandez AM, Poveda JA, Renart ML, Encinar JA, Riquelme G, Gonzalez-Ros JM. Clustering and coupled gating modulate the activity in KcsA, a potassium channel model. J Biol Chem. 2006;281:18837–18848. doi: 10.1074/jbc.M600342200. [DOI] [PubMed] [Google Scholar]
- Muennich EA, Fyffe RE. Focal aggregation of voltage-gated, Kv2.1 subunit-containing, potassium channels at synaptic sites in rat spinal motoneurones. J Physiol. 2004;554:673–685. doi: 10.1113/jphysiol.2003.056192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Connell KM, Rolig AS, Whitesell JD, Tamkun MM. Kv2.1 potassium channels are retained within dynamic cell surface microdomains that are defined by a perimeter fence. J Neurosci. 2006;26:9609–9618. doi: 10.1523/JNEUROSCI.1825-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Connell KM, Tamkun MM. Targeting of voltage-gated potassium channel isoforms to distinct cell surface microdomains. J Cell Sci. 2005;118:2155–2166. doi: 10.1242/jcs.02348. [DOI] [PubMed] [Google Scholar]
- Oliver D, Lien CC, Soom M, Baukrowitz T, Jonas P, Fakler B. Functional conversion between A-type and delayed rectifier K+ channels by membrane lipids. Science. 2004;304:265–270. doi: 10.1126/science.1094113. [DOI] [PubMed] [Google Scholar]
- Pike LJ. Lipid rafts: heterogeneity on the high seas. Biochem J. 2004;378:281–292. doi: 10.1042/BJ20031672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pouvreau S, Berthier C, Blaineau S, Amsellem J, Coronado R, Strube C. Membrane cholesterol modulates dihydropyridine receptor function in mice fetal skeletal muscle cells. J Physiol. 2004;555:365–381. doi: 10.1113/jphysiol.2003.055285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rettig J, Heinemann SH, Wunder F, Lorra C, Parcej DN, Dolly JO, Pongs O. Inactivation properties of voltage-gated K+ channels altered by presence of β-subunit. Nature. 1994;369:289–294. doi: 10.1038/369289a0. [DOI] [PubMed] [Google Scholar]
- Romanenko VG, Fang Y, Byfield F, Travis AJ, Vandenberg CA, Rothblat GH, Levitan I. Cholesterol sensitivity and lipid raft targeting of Kir2.1 channels. Biophys J. 2004;87:3850–3861. doi: 10.1529/biophysj.104.043273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schubert AL, Schubert W, Spray DC, Lisanti MP. Connexin family members target to lipid raft domains and interact with caveolin-1. Biochemistry. 2002;41:5754–5764. doi: 10.1021/bi0121656. [DOI] [PubMed] [Google Scholar]
- Slimane TA, Lenoir C, Bello V, Delaunay JL, Goding JW, Chwetzoff S, Maurice M, Fransen JA, Trugnan G. The cytoplasmic/transmembrane domain of dipeptidyl peptidase IV, a type II glycoprotein, contains an apical targeting signal that does not specifically interact with lipid rafts. Exp Cell Res. 2001;270:45–55. doi: 10.1006/excr.2001.5337. [DOI] [PubMed] [Google Scholar]
- Toselli M, Biella G, Taglietti V, Cazzaniga E, Parenti M. Caveolin-1 expression and membrane cholesterol content modulate N-type calcium channel activity in NG108-15 cells. Biophys J. 2005;89:2443–2457. doi: 10.1529/biophysj.105.065623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Fermini B, Nattel S. Sustained depolarization-induced outward current in human atrial myocytes. Evidence for a novel delayed rectifier K+ current similar to Kv1.5 cloned channel currents. Circ Res. 1993;73:1061–1076. doi: 10.1161/01.res.73.6.1061. [DOI] [PubMed] [Google Scholar]
- Wong W, Schlichter LC. Differential recruitment of Kv1.4 and Kv4.2 to lipid rafts by PSD-95. J Biol Chem. 2004;279:444–452. doi: 10.1074/jbc.M304675200. [DOI] [PubMed] [Google Scholar]
- Xia F, Gao X, Kwan E, Lam PP, Chan L, Sy K, Sheu L, Wheeler MB, Gaisano HY, Tsushima RG. Disruption of pancreatic β-cell lipid rafts modifies Kv2.1 channel gating and insulin exocytosis. J Biol Chem. 2004;279:24685–24691. doi: 10.1074/jbc.M314314200. [DOI] [PubMed] [Google Scholar]
- Yarbrough TL, Lu T, Lee HC, Shibata EF. Localization of cardiac sodium channels in caveolin-rich membrane domains: regulation of sodium current amplitude. Circ Res. 2002;90:443–449. doi: 10.1161/hh0402.105177. [DOI] [PubMed] [Google Scholar]