

Abstract
The transport of long-chain fatty acids (LCFAs) across mitochondrial membranes is regulated by carnitine palmitoyltransferase I (CPTI) activity. However, it appears that additional fatty acid transport proteins, such as fatty acid translocase (FAT)/CD36, influence not only LCFA transport across the plasma membrane, but also LCFA transport into mitochondria. Plasma membrane-associated fatty acid binding protein (FABPpm) is also known to be involved in sacrolemmal LCFA transport, and it is also present on the mitochondria. At this location, it has been identified as mitochondrial aspartate amino transferase (mAspAT), despite being structurally identical to FABPpm. Whether this protein is also involved in mitochondrial LCFA transport and oxidation remains unknown. Therefore, we have examined the ability of FABPpm/mAspAT to alter mitochondrial fatty acid oxidation. Muscle contraction increased (P < 0.05) the mitochondrial FAT/CD36 content in rat (+22%) and human skeletal muscle (+33%). By contrast, muscle contraction did not alter the content of mitochondrial FABPpm/mAspAT protein in either rat or human muscles. Electrotransfecting rat soleus muscles, in vivo, with FABPpm cDNA increased FABPpm protein in whole muscle (+150%; P < 0.05), at the plasma membrane (+117%; P < 0.05) and in mitochondria (+80%; P < 0.05). In these FABPpm-transfected muscles, palmitate transport into giant vesicles was increased by +73% (P < 0.05), and fatty acid oxidation in intact muscle was increased by +18% (P < 0.05). By contrast, despite the marked increase in mitochondrial FABPpm/mAspAT protein content (+80%), the rate of mitochondrial palmitate oxidation was not altered (P > 0.05). However, electrotransfection increased mAspAT activity by +70% (P < 0.05), and the mitochondrial FABPpm/mAspAT protein content was significantly correlated with mAspAT activity (r= 0.75). It is concluded that FABPpm has two distinct functions depending on its subcellular location: (a) it contributes to increasing sarcolemmal LCFA transport while not contributing directly to LCFA transport into mitochondria; and (b) its primary role at the mitochondria level is to transport reducing equivalents into the matrix.
The cellular transport of long-chain fatty acids (LCFAs) across the plasma membrane has long been thought to occur via passive diffusion. However, in recent years a substantial body of literature has emerged indicating that LCFA uptake into muscle cells occurs via a protein-mediated process (for review see Bonen et al. 2002; Luiken et al. 2004; Koonen et al. 2005). A number of proteins have been shown to facilitate the uptake of LCFAs into parenchymal cells, including fatty acid translocase, the homologue of human CD36 (FAT/CD36, 88 kDa) (Abumrad et al. 1993), a family of fatty acid transport proteins (FATP1–6, 63–70 kDa) (Schaffer & Lodish, 1994; Hirsch et al. 1998; Gimeno et al. 2003) and plasma membrane-associated fatty acid binding protein (FABPpm, 43 kDa) (Stremmel et al. 1985; Schwieterman et al. 1988; Isola et al. 1995). Whereas little is known about the regulation of FATP1–6 in muscle tissue, considerable evidence has accumulated to indicate that FAT/CD36 and FABPpm are important in regulating the uptake of LCFAs into cardiac and skeletal muscle.
Recently, FAT/CD36 was also found to be present in mitochondrial membranes of skeletal muscle (Campbell et al. 2004; Bezaire et al. 2006). In rodents, electrically induced muscle contraction (30 min) increased the mitochondrial content of FAT/CD36 (Campbell et al. 2004). Similar results have been observed in mitochondria isolated from human muscle after 2 h of moderate-intensity exercise (∼60% maximal O2 consumption rate) (Holloway et al. 2006). In both rodents and humans, the muscle contraction-induced increases in muscle mitochondrial FAT/CD36 content were associated with increases in fatty acid oxidation in isolated mitochondria (Campbell et al. 2004; Holloway et al. 2006). In addition, when FAT/CD36 was blocked by sulfo-N-succinimidyloleate (SSO), a specific inhibitor of FAT/CD36, mitochondrial fatty acid oxidation was almost completely inhibited (∼90%) in mitochondria obtained from either resting or exercised muscle (Campbell et al. 2004; Bezaire et al. 2006; Holloway et al. 2006). These studies do not negate the well-known role of carnitine palmitoyltransferase (CPTI) activity in mitochondrial fatty acid oxidation. However, it appears that FAT/CD36 works in conjunction with CPTI, as together these two proteins predict rates of mitochondrial fatty acid oxidation (r= 0.90; Bezaire et al. 2006), and FAT/CD36 and CPTI are colocalized in mitochondria (Campbell et al. 2004; Schenk & Horowitz, 2006).
Whether FABPpm, another LCFA transport protein, also contributes to regulating mitochondrial LCFA oxidation is not known. However a series of studies have shown that FABPpm and mitochondrial aspartate aminotransferase (mAspAt) are identical proteins (Berk et al. 1990; Stump et al. 1993; Bradbury & Berk, 2000), and are found at the plasma membrane and on mitochondria, respectively (Cechetto et al. 2002). To date it is known that plasma membrane FABPpm contributes to regulating fatty acid transport (Schwieterman et al. 1988; Stump et al. 1993; Zhou et al. 1995) while in mitochondria, mAspAt catalyses the following reversible reaction: glutamate + oxaloacetate ⇌ aspartate + 2-oxoglutarate (Lehninger et al. 1993). Transfecting 3T3 fibroblasts (Isola et al. 1995) or skeletal muscle (Clarke et al. 2004) with mAspAt cDNA increased plasmalemmal FABPpm content and increased the rates of LCFA transport (Isola et al. 1995; Clarke et al. 2004), as well as the rates of LCFA oxidation in muscle (Clarke et al. 2004). Whether the up-regulation of LCFA oxidation was due to the increased influx of LCFA into the muscle and/or to an increase in FABPpm/mAspAt content at the mitochondria was not determined. Furthermore, there is strong evidence suggesting that FABPpm collaborates with FAT/CD36 to transport LCFAs across the plasma membrane (Luiken et al. 1999), and there may also be a similar role for FABPpm/mASpAT in mitochondria where FAT/CD36 is also present. Thus, it is important to resolve whether FABPpm/mAspAt has a role in both plasma membrane LCFA transport and in mitochondrial LCFA oxidation.
Because we have previously shown that up-regulation of LCFA oxidation in contracting muscles involves the translocation of FAT/CD36 to the mitochondria (Campbell et al. 2004; Holloway et al. 2006), we first examined whether FABPpm/mAspAt was also translocated to the mitochondria during electrically induced muscle contraction in a rat muscle and after 2 h of cycling exercise in human muscle. In addition, we also transfected rat muscle with mAspAt cDNA to examine the effects of FABPpm/mAspAt up-regulation on the rates of plasma membrane LCFA transport, and on the rates of LCFA oxidation in intact muscle and in isolated mitochondria. Finally, we also examined the effects of FABPpm/mAspAt up-regulation on mAspAT activity in whole muscle and isolated mitochondria.
Methods
Electrical stimulation of rat muscle
Female Sprague–Dawley rats (n= 5, weighing ∼175 g) were used in the contraction experiments. Animals were housed in a climate- and temperature-controlled room, on a 12–12 h reverse light–dark cycle. Rat chow and water were provided ad libitum. The study was approved by the University of Guelph Animal Ethics Committee.
To ascertain whether mitochondrial FABPpm/mAspAT content was altered in response to muscle contraction in a similar manner to FAT/CD36, rat hindlimbs were acutely (30 min) stimulated as we have previously described (Bonen et al. 2000; Campbell et al. 2004). Briefly, while under anaesthesia (intraperitoneal injections of sodium pentobarbital, 6 mg (100 g body weight)−1; MTC Pharmaceuticals, Cambridge, ON, Canada), a small incision was made in one hindlimb, and stimulating electrodes placed on the exposed sciatic nerve. The contralateral muscle in the same animal acted as a resting control. Muscle contraction consisted of stimulating the sciatic nerve in one hindlimb (10 V, 100 Hz, 100 ms trains, 20 tetani min−1 for 30 min; Grass S48 Stimulator, Astro-Medical Inc., Longueuil, QC, Canada), as we have reported previously (Campbell et al. 2004). Immediately after stimulation, the contracting soleus and the contralateral resting soleus were removed for mitochondrial isolation as described below, and used for Western blotting analysis.
Human exercise experiments
Twelve healthy, recreationally active, individuals volunteered for this study (n= 9 males; n= 3 females; age, 22 ± 1 year; weight, 77 ± 4 kg; body mass index, 24 ± 1 kg m−2; peak O2 consumption rate 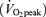 , 49 ± 2 ml min−1 (kg body mass)−1. Subjects were fully informed of the purpose of the experiments and of any possible risk before giving written consent to participate. The study was approved by the University of Guelph Ethics Committee.
, 49 ± 2 ml min−1 (kg body mass)−1. Subjects were fully informed of the purpose of the experiments and of any possible risk before giving written consent to participate. The study was approved by the University of Guelph Ethics Committee.
Participants cycled for 2 h at ∼60%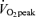 on a cycle ergometer (LODE Instrument, Groningen, the Netherlands), following a 12 h overnight fast. Pulmonary O2 uptake was measured with a metabolic cart (SensorMedics Vmax model, CA, USA). Prior to exercising, both legs were prepared for muscle biopsies of the vastus lateralis muscle. Ventilatory and muscle samples were obtained before and after 2 h of cycling. Muscle samples were obtained under local anaesthesia (2% lignocaine (lidocaine) without adrenaline (epinephrine)) using the percutaneous needle biopsy technique described by Bergstrom (1975). Immediately following tissue sampling, visible fat and connective tissue were dissected free from the muscle and the samples were blotted to remove excess blood. The tissue was used for the immediate isolation of mitochondria and for the determination of selected proteins with Western blotting.
on a cycle ergometer (LODE Instrument, Groningen, the Netherlands), following a 12 h overnight fast. Pulmonary O2 uptake was measured with a metabolic cart (SensorMedics Vmax model, CA, USA). Prior to exercising, both legs were prepared for muscle biopsies of the vastus lateralis muscle. Ventilatory and muscle samples were obtained before and after 2 h of cycling. Muscle samples were obtained under local anaesthesia (2% lignocaine (lidocaine) without adrenaline (epinephrine)) using the percutaneous needle biopsy technique described by Bergstrom (1975). Immediately following tissue sampling, visible fat and connective tissue were dissected free from the muscle and the samples were blotted to remove excess blood. The tissue was used for the immediate isolation of mitochondria and for the determination of selected proteins with Western blotting.
Rat solei electrotransfection
Female Sprague–Dawley rats (weighing ∼175 g) were used in these experiments.
During surgery, rats were maintained unconscious by inhalation of the anaesthetic isoflorane (AErane; Baxter, Deerfield, IL, USA) at 2–2.5% in pure O2 at a flow rate of 2 l min−1. Electrotransfection of FABPpm/mAspAt was performed as we have previously described (Clarke et al. 2004). Briefly, the control limb was left ‘intact’ and the experimental lower hindlimb was shaved and sterilized with iodine solution. A 1.5 cm incision was made laterally along the left hindlimb parallel to the tibia and extending to the Achilles tendon. The soleus muscle was exposed and 100 μl 0.45% saline solution containing 250 μg plasmid cDNA (provided by Dr A. Iriarte, University of Missouri, USA) was injected using a 27 gauge needle. The plasmid contained the open reading frame of FABPpm under the control of the CMV promotor as we have previously described (Clarke et al. 2004), with the addition of a FLAG epitope (Sigma, St Louis, MO, USA), inserted at the C-terminus by PCR methods. The modified FABPpm–FLAG was subcloned into pcDNA3.1 between EcoRI and XbaI sites. Immediately following the cDNA injection, electroporation of the intact soleus muscle was performed as we have previously described (Clarke et al. 2004) (eight electric pulses, 200 V cm−1, 1 Hz, 20 ms in duration) (ECM 830 Square Wave Electroporator; BTX, Holliston, MA, USA) using Tweezertrodes (BTX). After the pulse was delivered, the overlying superficial muscle was sutured, and the skin incision was closed. The animals were randomly divided into three groups 7 days after this procedure. The first group was used for the analysis of whole-muscle measurements (Western blot analysis, palmitate transport, oxidation and incorporation into diacylglycerol (DAG) and triacylglycerol (TAG) pools) (n= 12). The second group was used to isolate vesicles (n= 12). Because of tissue requirements, the soleus muscles of eight animals were pooled to create one sample for each independent fatty acid transport assay. The third group was used for isolating mitochondria (n= 5). Because of tissue requirements, the soleus muscles of two animals were pooled to create one sample for mitochondrial analysis (i.e. n= 1 is two rats), thus 10 rats were used to create five data points. However, prior to pooling for mitochondrial isolations, a small section of muscle (∼10 mg) was removed for determining the enzymatic characteristics of each animal (see below). Additional experiments were conducted with an empty vector to ensure that the transfection procedures did not alter any of the measured parameters.
Preparation of giant vesicles
Giant vesicles from pooled control and electrotransfected solei muscles were generated as previously described (Bonen et al. 1998, 2000, 2004; Luiken et al. 2001; Luiken et al. 2002b; Koonen et al. 2002; Steinberg et al. 2002). Briefly, the tissues were cut into thin layers (1–3 mm thick) and incubated for 1 h at 34°C in 140 mm KCl/10 mm MOPS (pH 7.4), aprotinin (30 μg ml−1, Sigma) and collagenase (type VII, 150 units ml−1) in a shaking water bath. At the end of the incubation, the supernatant fraction was collected and the remaining tissue was washed with KCl/MOPS and 10 mm EDTA which resulted in a second supernatant fraction. Both supernatant fractions were pooled, and Percoll (G.E. Healthcare, Aurora, OH, USA), and aprotinin were added to final concentrations of 3.5% (v/v) and 10 μg ml−1, respectively. The resulting suspension was placed at the bottom of a density gradient consisting of a 3 ml middle layer of 4% Nycodenz (w/v) and a 1 ml KCl/MOPS upper layer. This sample was centrifuged at 60 g for 45 min at room temperature (25°C). Subsequently, the vesicles were harvested from the interface of the upper and middle layer, diluted in KCl/MOPS and re-centrifuged at 12 000 g for 5 min. The pellet was resuspended in KCl/MOPS.
Palmitate uptake by giant vesicles
Palmitate uptake studies were performed as we have previously described (Bonen et al. 1998, 2004; Luiken et al. 2001, 2002c; Koonen et al. 2002; Steinberg et al. 2002). Fatty acid-free bovine serum albumin (BSA; MP Biomedicals, Solon, OH, USA), [9,10-3H]palmitate (Perkin Elmer, Wellesley, MA, USA) and [14C]mannitol (Perkin Elmer) were used in the experiments. For palmitate transport measurements, 40 μl 0.1% BSA in KCl/MOPS, containing unlabelled (15 μm) and radiolabelled [3H]palmitate (0.3 μCi) and [14C]mannitol (0.06 μCi), was added to 40 μl vesicle suspension. The incubation was carried out for 15 s. Palmitate uptake was terminated by addition of 1.4 ml ice-cold KCl/MOPS, 2.5 mm HgCl2 and 0.1% BSA. The sample was then centrifuged in a microfuge at 12 000 r.p.m. (13 000g) for 2 min. The supernatant was discarded, and radioactivity was determined in the tip of the tube.
Whole-muscle palmitate metabolism
In control and electrotransfected soleus muscle we examined the metabolism of palmitate. For these measurements we used our previously published methods (Dyck et al. 1997, 2000, 2001). Briefly, soleus muscle strips were preincubated for 20 min (30°C), followed by a 60 min incubation period (30°C). The incubation medium contained 0.5 mm palmitate, and 5 mm glucose, as well as 2 μCi [14C]palmitate (G.E. Healthcare). Palmitate oxidation was determined by acidifying the incubation medium to release the 14CO2, which was trapped by benzethonium hydroxide (Sigma). To determine the incorporation of palmitate into DAG and TAG pools, muscle was homogenized, spotted on silica gel plates (Silica Gel GF, 250 mm; Analtech, Newark, DE, USA), and resolved in solvent (60: 40: 4, heptane/isopopylether/acetic acid) for 45 min. Plates were air-dried, sprayed with dichlorofluorescein dye and visualized under long-wave ultraviolet light. Individual lipid bands were quantified against known standards, and scraped into vials for liquid scintillation counting.
Isolation of mitochondria from skeletal muscle
Differential centrifugation was used to obtain pure and intact mitochondria containing both intermyofibrillar (IMF) and subsarcelommal (SS) fractions (Campbell et al. 2004). All procedures were identical to those that we have previously published (Bezaire et al. 2006; Holloway et al. 2006). Briefly, muscle (∼300 mg) was homogenized with a tight-fitting Teflon pestle. The homogenate was centrifuged at 800 g for 10 min, to separate the SS and IMF mitochondria. The IMF mitochondria were treated with a protease (0.025 ml g−1; Sigma) for exactly 5 min to digest the myofibrils. Further centrifugation was used to remove the myofibrils, and recombine the IMF with the SS mitochondria. The combined samples were centrifuged twice at 10 000 g for 10 min. The pellet was resuspended in 1 μl buffer per milligram of tissue. Following the oxidation measurements, the remaining mitochondria were further purified using a Percoll gradient for Western blotting analysis. Samples were centrifuged at 20 000 g for 1 h and the mitochondrial layer was removed. The Percoll was removed from the sample by further centrifuging at 20 000 g for 5 h.
Mitochondrial palmitate oxidation
Labelled CO2 production from palmitate oxidation and acid-soluble trapped 14C were measured during state 4 respiration following a 30 min incubation of viable mitochondria in a sealed system, as previously described by us (Campbell et al. 2004; Bezaire et al. 2006; Holloway et al. 2006). Briefly, viable mitochondria (100 μl) were added to a system containing a pre-gassed modified Krebs–Ringer solution supplemented with 5 mm ATP, 1 mm NAD+, 0.5 mmdl-carnitine, 0.1 mm coenzyme A, 25 μm cytochrome C and 0.5 mm malate. A microcentrifuge tube containing 500 μl 1 m benzethonium hydroxide inserted into a 1.5 ml centrifuge tube, was placed in the system to capture 14CO2 produced during the oxidation reaction. The system was then sealed with a rubber cap and further sealed with parafilm. The reaction was initiated by the addition of a 6: 1 palmitate–BSA complex (containing 10 μCi [1-14C]palmitate, for a final palmitate concentration of 77 μm) administered by syringe through the rubber cap. The reaction continued for 30 min at 37°C and was terminated with the addition of ice-cold 12 N perchloric acid by syringe through the rubber cap.
A fraction of the reaction medium was removed through the cap and analysed for isotopic fixation. Gaseous CO2 produced from oxidation of [1-14C]palmitate was measured by acidifying the remaining reaction mixture. Liberated 14CO2 was trapped by the benzethonium hydroxide over a 90 min incubation period at room temperature. The microcentrifuge tube containing the 14CO2 was put in a scintillation vial, and radioactivity was determined.
Mitochondrial viability
Electrotransfection was performed on five rats (weighing ∼250 g), and mitochondria isolated from both electrotransfected and contralateral control limbs as outlined above. Using a Clark-type electrode, mitochondrial respiration rates were measured in state 3 and state 4 conditions, and the respiratory control ratios (RCRs) were calculated for both control and electrotransfected limbs to ensure the isolation procedure yielded intact mitochondria, and that electrotransfection did not alter the viability. Briefly, following the isolation procedure outlined above, 500 μg (∼50 μl) mitochondria was added to 1500 μl oxygraph medium containing (mm): mannitol 225, sucrose 75, Tris-base 10, K2HPO4 10, EDTA 0.1 and MgCl2 0.8; pH 7.0. State 3 respiration was measured in the presence of 5 mm pyruvate, 2 mm malate and 300 μm ADP, and subsequently, state 4 respiration was measured following the consumption of the ADP. The RCR was calculated as the ratio between state 3 and state 4 respiration rates (Chavez et al. 1995; Mogensen & Sahlin, 2005; Sahlin et al. 2007).
β-Hydroxyacyl-CoA dehydrogenase, citrate synthase and aspartate aminotransferase activities
A portion of the soleus muscle (∼10 mg) was immediately homogenized in 100 μl/mg of a 100 mm potassium phosphate buffer (Bergmeyer, 1974b) and used for the measurements of β-hydroxyacyl-CoA dehydrogenase (β-HAD), citrate synthase (CS) and mAspAT activities. Total muscle β-HAD activity was measured in Tris-HCl buffer (50 mm Tris-HCl, 2 mm EDTA, 250 μm NADH, pH 7.0) and 0.04% Triton X-100. The reaction was started by addition of 100 μm acetoacetyl-CoA and absorbance was measured at 340 nm over a 2 min period (37°C) (Bergmeyer, 1974b).
CS activity was determined in isolated mitochondria as well as in aliquots of homogenized whole muscle. CS activity in intact mitochondria was determined by first assaying the extramitochondrial fraction in the suspension (1: 20 dilution) and then assaying the total CS activity of the suspension (1: 20 dilution) after lysing the mitochondria with 0.04% Triton X-100 and repeated freeze-thawing. The net difference provided a measure of the viability of the mitochondria, and when compared to the total muscle CS activity provided a measure of the mitochondria recovered during our isolation procedure. The CS activity was assayed spectrophotometrically at 37°C by measuring the disappearance of NADH at 412 nm (Bergmeyer, 1974b).
Mitochondrial aspartate aminotransferase activity was also measured in isolated mitochondria and in aliquots of homogenized whole muscle. Mitochondria and homogenate were repeatedly freeze-thawed prior to analysis. Maximal measurements were determined in a 75 mm phosphate buffer (75 mm phosphate, 60 mm 2-oxoglutarate, 6 U malate dehydrogenase, 0.25% Triton X-100, and excess NADH; adjusted to optimize spectrophotometer sensitivity range). The reaction was started by addition of 400 mm aspartate and absorbance was measured at 340 nm over a 2 min period (25°C) (Bergmeyer, 1974a).
Western blotting
Whole-muscle crude membranes were generated as we have previously described (Bonen et al. 1998, 2000; Luiken et al. 2001), analysed for total protein (bicinchoninic (BCA) protein assay), and 25 μg denatured protein was loaded for Western blotting. The plasma membrane content of fatty acid transporters was determined by generating vesicles (as described above), analysing for total protein (BCA protein assay), and loading 10 μg denatured protein for Western blotting. Purified isolated mitochondrial fractions were analysed for total protein (BCA protein assay) and 25 μg denatured protein from each sample was loaded for Western blotting. All proteins were separated by electrophoresis on an 8% SDS-polyacrylamide gel and transferred to a polyvinylidene difluoride (PVDF) membrane. The FABPpm polyclonal antibody used was produced in our laboratories and has been used previously in our work (Bonen et al. 1998, 2000; Luiken et al. 2001). The MO-25 antibody used to detect FAT/CD36 was produced by N. N. Tandon (Thrombosis Research Laboratory, Rockville, Maryland, USA). Commercially available antibodies were used to detect cytochrome c oxidase IV (Cox-IV; Invitrogen, Burlington, ON, Canada), sarco(endo)plasmic reticulum Ca2+ ATPase (SERCA2) (Sigma) and Glut-4 (Chemicon International Inc., Temecula, CA, USA). In addition, an epitopic FLAG-tag (Sigma), specific to a six amino acid sequence inserted at the N-terminus of FABPpm, was used to detect de novo synthesis as a result of the cDNA electrotransfected into soleus muscles. An internal control of previously extracted crude membranes from rat was used in each gel. Blots were quantified using chemiluminescence and the Syngene 2 ChemiGenius bioimaging system (Syngene, Cambridge, UK).
Statistics
All data are presented as the means ±s.e.m. All data were compared using a paired t test. Associations between variables were investigated using Pearson correlation analyses. Statistical significance was accepted at P < 0.05.
Results
Effects of contraction on mitochondrial FABPpm content
Initially we examined whether muscle contraction or exercise increased the mitochondrial content of FABPpm/mAspAt, as we have previously shown for FAT/CD36 in both rat (Campbell et al. 2004) and human muscle (Holloway et al. 2006). In accordance with our previous work (Campbell et al. 2004), electrical stimulation increased FAT/CD36 content on rat mitochondria by +22% (P < 0.05, Fig. 1C). By contrast, electrical stimulation did not alter rat mitochondrial FABPpm content (Fig. 1A). Similarly, 2 h of cycling at ∼60%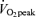 , increased mitochondrial FAT/CD36 content in human muscle by +33% (P < 0.05, Fig. 1D), but did not alter mitochondrial FABPpm/mAspAt content (Fig. 1B).
, increased mitochondrial FAT/CD36 content in human muscle by +33% (P < 0.05, Fig. 1D), but did not alter mitochondrial FABPpm/mAspAt content (Fig. 1B).
Figure 1. Effects of muscle contraction on the content of mitochondrial FABPpm/mAspAT and FAT/CD36.

Values are means ±s.e.m., expressed in arbitrary units. A, mitochondrial FABPpm/mAspAT content in rat soleus muscle at rest, and following 30 min of electrical stimulation (n= 5). B, mitochondrial FABPpm/mAspAT content in human vastus lateralis muscle at rest, and following 2 h of cycling at ∼60% peak O2 consumption rate 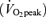 (n= 12). C, mitochondrial FAT/CD36 content in rat soleus muscle at rest, and following 30 min of electrical stimulation (n= 5). D, mitochondrial FAT/CD36 content in human vastus lateralis muscle at rest, and following 2 h of cycling at ∼60%
(n= 12). C, mitochondrial FAT/CD36 content in rat soleus muscle at rest, and following 30 min of electrical stimulation (n= 5). D, mitochondrial FAT/CD36 content in human vastus lateralis muscle at rest, and following 2 h of cycling at ∼60%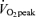 (n= 7). *Significantly different (P < 0.05) from rest.
(n= 7). *Significantly different (P < 0.05) from rest.
Effects of FABPpm/mAspAT electrotransfection on plasma membrane proteins, palmitate transport into giant vesicles and palmitate oxidation in intact soleus muscle
While the contraction-induced up-regulation of FAT/CD36 has been linked to increased rates of mitochondrial LCFA oxidation (Campbell et al. 2004; Holloway et al. 2006), the lack of change in mitochondrial FABPpm/mAspAt during muscle contraction suggests that this protein is not involved with fatty acid oxidation in mitochondria. To examine this further we investigated the effects of up-regulating FABPpm/mAspAt on rates of LCFA transport and fatty acid oxidation in intact muscle and in isolated mitochondria.
When FABPpm/mAspAt was transfected into soleus muscles, there was a +150% increase in muscle FABPpm/mAspAT content (P < 0.05, Fig. 2A), and a +117% increase in the plasmalemmal FABPpm content (P < 0.05, Fig. 2B). The content of FAT/CD36, another fatty acid transporter, was not altered with transfection in either whole muscle or at the plasma membrane (data not shown). Transfection increased (P < 0.05) the rate of palmitate transport into giant sarcolemmal vesicles by +73% (Fig. 2C).
Figure 2. Effects of electrotransfection with FABPpm cDNA on soleus muscle.

Values are means ±s.e.m.; protein contents expressed in arbitrary units, and transport expressed in pmol (mg wet weight)−1 15 s−1 (n= 12). A, whole-muscle FABPpm protein content. B, plasmalemmal FABPpm protein content. C, palmitate transport into giant sarcolemmal vesicles. ww, wet weight. *Significantly different (P < 0.05) from control.
In intact soleus muscle, there was a somewhat smaller increase in the rate of palmitate oxidation following transfection (+18%, P < 0.05, Fig. 3A), while the rates of palmitate incorporation into DAG (Fig. 3B) or TAG pools were not altered (Fig. 3C).
Figure 3. Effects of electrotransfection with FABPpm cDNA on the fate of palmitate in soleus muscle.

Values are means ±s.e.m., expressed in nmol mg wet weight−1 hour−1 (n= 12). A, palmitate incorporation into the diacylglycerol pool. B, palmitate incorporation into triacylglycerol pool. C, palmitate oxidation. ww, wet weight. *Significantly different (P < 0.05) from control.
Electrotransfecting FABPpm/mAspAT in mitochondria
Next we determined whether the electrotransfection of FABPpm/mAspAt into muscle increased FABPpm/mAspAt content in mitochondria, and whether this altered the rates of palmitate oxidation and/or aspartate aminotransferase activities in these organelles. The mitochondrial isolation procedure yielded intact mitochondria as the RCR values were similar those to previously published (Chavez et al. 1995; Mogensen & Sahlin, 2005; Sahlin et al. 2007), as were the mitochondrial recovery and viability as calculated from CS values (see Methods) (Bezaire et al. 2006; Bruce et al. 2006; Holloway et al. 2006). In addition, control and electrotransfection RCR values were not different (10.4 ± 1.9 versus 10.0 ± 1.4, respectively; P= 0.84). The absence of SERCA (110 kDa) and Glut-4 (45 kDa) proteins, and the presence of Cox-IV (22 kDa), FAT/CD36 (88 kDa) and FABPpm (43 kDa) (Fig. 4) indicated that the isolation procedures successfully yielded highly purified mitochondria without contamination from other sources.
Figure 4. Representative Western blots performed on mitochondria isolated from control and electrotransfected solei.

A FLAG-tag specific for the exogenous expression of FABPpm (43 kDa) was only found on the purified mitochondrial extracts obtained from the FABPpm/mAspAT electrotransfected muscles (Fig. 4). Electrotransfection of FABPpm/mAspAT into soleus muscle increased the mitochondrial FABPpm/mAspAT protein content by +80% (P < 0.05, Fig. 5A), without altering mitochondrial FAT/CD36 (Fig. 5B) or COX-IV (Fig. 5C) contents. In addition, control experiments with an empty vector did not alter mitochondrial FABPpm/mAspAT, FAT/CD36 or Cox-IV contents (data not shown).
Figure 5. Effects of electrotransfection with FABPpm cDNA on the content of soleus muscle mitochondrial proteins, and the ability of mitochondria to oxidize palmitate.

Values are means ±s.e.m., expressed in arbitrary units (n= 5). A, mitochondrial FABPpm/mAspAT protein content. B, mitochondrial FAT/CD36 protein content. C, Cox-IV protein content. D, isolated mitochondrial palmitate oxidation, expressed in nmol mg protein−1 hour−1. *Significantly different (P < 0.05) from control.
Mitochondrial palmitate oxidation and enzymatic activity of FABPpm/mAspAT
Although electrotransfection increased FABPpm/mAspAT content on mitochondria by +80%, isolated mitochondrial palmitate oxidation rates were not altered (Fig. 5D). By contrast, FABPpm/mAspAT electrotransfection resulted in a +90% increase (P < 0.001) in whole-muscle mAspAT activity (Fig. 6A), and a +70% increase (P < 0.001) in isolated mitochondrial mAspAT activity (Fig. 6B). Whole-muscle β-HAD activity (Fig. 6C) and whole-muscle CS activity (Fig. 6D) were not altered. In control experiments with an empty vector, mAspAT activity was not altered (data not shown). There was a significant correlation (r= 0.75) between mAspAT activity and mitochondrial FABPpm protein levels (Fig. 7).
Figure 6. Effects of electrotransfection with FABPpm cDNA on mitochondrial enzymatic activity.

Values are means ±s.e.m., expressed in μmol min−1 g wet weight−1. A, mitochondrial aspartate aminotransferase (mAspAT) activity in homogenates (n= 10). B, mitochondrial aspartate aminotransferase activity (mAspAT) in isolated mitochondria (n= 5). C, homogenate β-hydroxyacyl-CoA dehydrogenase (β-HAD) activity (n= 10). D, homogenate citrate synthase (CS) activity (n= 10). ww, wet weight. *Significantly different (P < 0.05) from control.
Figure 7. Pearson correlation calculated between mitochondrial FABPpm/mAspAT content and mAspAT activity in homogenates.

n= 20 (10 control and 10 transfected) muscles.
Discussion
Studies by Berk and colleagues demonstrated that FABPpm and mAspAt are identical proteins (Berk et al. 1990; Stump et al. 1993; Bradbury & Berk, 2000), that are located at the plasma membrane and the mitochondria, respectively. We previously reported that FABPpm/mAspAT overexpression increased not only the rates of LCFA transport into muscle but it also increased the rates of palmitate oxidation in whole muscle (Clarke et al. 2004). This raised the spectre that FABPpm might have a role in regulating fatty acid oxidation at the mitochondrial level, as we have repeatedly demonstrated for FAT/CD36 (Campbell et al. 2004; Bezaire et al. 2006; Holloway et al. 2006). The present work has shown that (i) unlike for FAT/CD36, muscle contraction in rats and exercise in humans do not increase mitochondrial FABPpm/mAspAT content, whereas (ii) the up-regulation of this protein in muscle stimulates the rate of fatty acid transport but (iii) fails to alter the rate of fatty acid oxidation in isolated mitochondria. Hence (iv) the FABPpm-induced up-regulation of fatty acid oxidation in whole muscle would seem to be solely related to the increased rate of plasmalemmal fatty acid transport. Finally (v) overexpression of FABPpm/mAspAT markedly increased the activity of aspartate aminotransferase. Thus, these studies have shown that despite the fact that FABPpm and mAspAt are identical proteins (Berk et al. 1990; Stump et al. 1993), they have quite distinct metabolic functions depending on their subcellular location. Specifically, the protein appears to function as a LCFA transport protein on the plasma membrane, and as an enzyme associated with the mitochondrial membranes involved in the transport of reducing equivalents into mitochondria.
Although LCFA transport was originally viewed as a purely passive process, it has become increasingly apparent that LCFA transport proteins are involved in the regulation of cellular fatty acid uptake and metabolism. Research at the level of the plasma membrane, specifically pertaining to FAT/CD36 and FABPpm proteins, has provided insight into the regulation of LCFA transport. It has been shown that FAT/CD36 and FABPpm proteins are up-regulated whenever fatty acid oxidation is chronically increased, such as during 7-day low-frequency muscle stimulation in rodents (Bonen et al. 1999; Koonen et al. 2004) and exercise training in humans (Tunstall et al. 2002), or in insulin-deficient animals (Luiken et al. 2002a). In these studies, the rates of fatty acid transport are concomitantly altered as a result of changes in FAT/CD36 and FABPpm protein expression and their appearance at the plasma membrane (Bonen et al. 1999; Luiken et al. 2002a; Koonen et al. 2004). In addition, rates of fatty acid transport can also be altered when these transporter proteins are redistributed to the plasma membrane in the absence of any change in their protein expression, which not only occurs acutely but also chronically. A translocation of these LCFA transporter proteins from a low-density microsomal pool to the plasma membrane has been observed following muscle contraction (Bonen et al. 2000) and AMP kinase activation (Luiken et al. 2003; Chabowski et al. 2005), thereby facilitating a very rapid up-regulation of fatty acid transport into muscle cells. A similar chronic translocation (‘relocation’) was found in muscle from obese Zucker rats (Luiken et al. 2001) and in obese and type 2 diabetic humans (Bonen et al. 2004). Taken altogether it is apparent that FAT/CD36 and FABPpm can influence fatty acid metabolism both acutely and chronically by altering the rate of LCFA entry into parenchymal cells.
Recently, FAT/CD36 has been shown to translocate to the mitochondria during exercise, in both rat and human skeletal muscle, increasing the ability of mitochondria to oxidize palmitate (Campbell et al. 2004; Holloway et al. 2006), and hence providing another level of regulation in fatty acid oxidation. At the level of the plasma membrane, the acute regulation of FAT/CD36 and FABPpm has in the past appeared similar (Chabowski et al. 2005; Chabowski et al. 2006). However, the results of the current work suggest that the acute regulation of these proteins may be different at the level of the mitochondria. These results support the hypothesis that FAT/CD36 content on mitochondria increases following muscle contraction (Campbell et al. 2004; Holloway et al. 2006), while contraction does not alter mitochondrial FABPpm protein content, in either rats or in human skeletal muscle, and represents a much less flexible system.
As fatty acid oxidation appeared to increase during exercise without alterations in mitochondrial FABPpm/mAspAT content, a series of studies were designed to determine the role of FABPpm/mAspAT at the mitochondrial level. Electrotransfection of FABPpm cDNA into soleus muscle has previously been shown to increase rates of plasma membrane palmitate transport and whole-muscle palmitate oxidation, independent of changes in FAT/CD36 content (Clarke et al. 2004). In the present study we have replicated these findings, and investigated the role of mitochondrial FABPpm/mAspAT. Because FABPpm is located both on the plasma membrane and mitochondria (Stump et al. 1993; Cechetto et al. 2002), it was not known whether the increase in palmitate oxidation following electrotransfection of FABPpm resulted solely from enhanced rates of fatty acid transport across the plasma membrane, or whether FABPpm/mAspAT also played a role in fatty acid oxidation in mitochondria. The novel finding in the current study is that a +80% increase in mitochondrial FABPpm/mAspAT protein content failed to alter mitochondrial palmitate oxidation. This indicates that FABPpm/mAspAT, unlike FAT/CD36 (Campbell et al. 2004; Bezaire et al. 2006; Holloway et al. 2006; Schenk & Horowitz, 2006), is not involved in regulating fatty acid oxidation in mitochondria. Therefore, as whole-muscle oxidation was increased, without an increase in the ability of mitochondria to oxidize palmitate, increases in plasma membrane fatty acid transport and mass action are responsible for the observed increase in whole-muscle oxidation.
While others have suggested that FABPpm and mAspAT are structurally identical proteins, but responsible for different functions as a result of their subcellular compartmentation (Stump et al. 1993; Isola et al. 1995), we are the first to directly establish this. The increase in both the content of FABPpm at the plasma membrane and rate of fatty acid transport across the plasma membrane following electrotransfection, further supports the belief that FABPpm functions as a fatty acid transporter at this level (Isola et al. 1995; Luiken et al. 1999; Turcotte et al. 1999; Turcotte et al. 2000; Clarke et al. 2004). Altering the mitochondrial FABPpm/mAspAT protein presumably did not affect transport into mitochondria, because oxidation remained constant. However, increasing the mitochondrial content of FABPpm/mAspAT increased mAspAT enzymatic activity. This evidence strongly suggests that FABPpm contributes to transporting fatty acids across plasma membranes, as well as enzymatically regulating the rate of the following reaction (glutamate + oxaloacetate ⇌ aspartate + 2-oxoglutarate) in mitochondria (Lehninger et al. 1993). These different functions for the same protein may be explained by a differential post-translational modification of the protein depending on the subcellular location, or a co-functioning with other proteins (such as FAT/CD36 at the plasma membrane as previously suggested; Luiken et al. 1999).
In summary, we measured mitochondrial FABPpm/mAspAT content in contracting rat and human skeletal muscle, and have demonstrated an inability of FABPpm to translocate to mitochondria during contraction, suggesting that it does not regulate mitochondrial fatty acid oxidation. In addition, we have used electrotransfection to independently alter the content of FABPpm in rat soleus muscle on both the plasma membrane and mitochondria. We propose that the FABPpm-mediated up-regulation of fatty acid oxidation occurs via an increased rate of fatty acid transport across the plasma membrane, and thus mass action accounts for their increased mitochondrial oxidation. It is well known that another fatty acid transport protein, FAT/CD36, can have different functions in different tissues (for review see Febbraio et al. 2001), and the current novel finding is that FABPpm has diverse metabolic functions depending on its subcellular compartmentalization. Functionally, in muscle, it appears that FABPpm is a fatty acid transport protein on the plasma membrane, and FABPpm/mAspAT is a functional enzyme at the level of the mitochondria, facilitating the transport of reducing equivalents into the mitochondrial matrix.
Acknowledgments
This research was funded by the Natural Sciences and Engineering Research Council of Canada (NSERC) (A.B. and L.L.S.), the Canadian Institutes of Health Research (CIHR) (A.B., G.J.F.H. and L.L.S.), the Netherlands Heart Foundation grant 2002.T049 (J.F.C.G.), the Netherlands Organization for Health Research and Development (NWO-ZonMw grant 40–00812-98–03075) (J.J.F.P.L. and J.F.C.G.), and the European Commission (Integrated Project LSHM-CT-2004–005272, Exgenesis) (J.J.F.P.L. and J.F.C.G.). J.J.F.P.L. is the recipient of a VIDI-Innovational Research Grant from the Netherlands Organization of Scientific Research (NWO-ZonMw Grant 016.036.305), J.F.C.G. is the Netherlands Heart Foundation Professor of Cardiac Metabolism, A.B. is a recipient of a Canada Research Chair in Metabolism and Health and G.P.H. is a recipient of an NSERC Canadian Graduate Scholarship.
References
- Abumrad NA, El-Maghrabi MR, Amri EZ, Lopez E, Grimaldi PA. Cloning of a rat adipocyte membrane protein implicated in binding or transport of long-chain fatty acids that is induced during preadipocyte differentiation. Homology with human CD36. J Biol Chem. 1993;268:17665–17668. [PubMed] [Google Scholar]
- Bergmeyer HU. Methods of Enzymatic Analysis. Vol. 2. New York: Verlag Chemie Weinheim; 1974a. [Google Scholar]
- Bergmeyer HU. Methods of Enzymatic Analysis. Vol. 4. New York: Verlag Chemie Weinheim; 1974b. [Google Scholar]
- Bergstrom J. Percutaneous needle biopsy of skeletal muscle in physiological and clinical research. Scand J Clin Lab Invest. 1975;35:609–616. [PubMed] [Google Scholar]
- Berk PD, Wada H, Horio Y, Potter BJ, Sorrentino D, Zhou SL, Isola LM, Stump D, Kiang CL, Thung S. Plasma membrane fatty acid-binding protein and mitochondrial glutamic-oxaloacetic transaminase of rat liver are related. Proc Natl Acad Sci U S A. 1990;87:3484–3488. doi: 10.1073/pnas.87.9.3484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bezaire V, Bruce CR, Heigenhauser GJ, Tandon NN, Glatz JF, Luiken JJ, Bonen A, Spriet LL. Identification of fatty acid translocase on human skeletal muscle mitochondrial membranes: essential role in fatty acid oxidation. Am J Physiol Endocrinol Metab. 2006;290:E509–E515. doi: 10.1152/ajpendo.00312.2005. [DOI] [PubMed] [Google Scholar]
- Bonen A, Dyck DJ, Ibrahimi A, Abumrad NA. Muscle contractile activity increases fatty acid metabolism and transport and FAT/CD36. Am J Physiol Endocrinol Metab. 1999;276:E642–E649. doi: 10.1152/ajpendo.1999.276.4.E642. [DOI] [PubMed] [Google Scholar]
- Bonen A, Luiken JJ, Arumugam Y, Glatz JF, Tandon NN. Acute regulation of fatty acid uptake involves the cellular redistribution of fatty acid translocase. J Biol Chem. 2000;275:14501–14508. doi: 10.1074/jbc.275.19.14501. [DOI] [PubMed] [Google Scholar]
- Bonen A, Luiken JJ, Glatz JF. Regulation of fatty acid transport and membrane transporters in health and disease. Mol Cell Biochem. 2002;239:181–192. [PubMed] [Google Scholar]
- Bonen A, Luiken JJ, Liu S, Dyck DJ, Kiens B, Kristiansen S, Turcotte LP, Van Der Vusse GJ, Glatz JF. Palmitate transport and fatty acid transporters in red and white muscles. Am J Physiol Endocrinol Metab. 1998;275:E471–E478. doi: 10.1152/ajpendo.1998.275.3.E471. [DOI] [PubMed] [Google Scholar]
- Bonen A, Parolin ML, Steinberg GR, Calles-Escandon J, Tandon NN, Glatz JF, Luiken JJ, Heigenhauser GJ, Dyck DJ. Triacylglycerol accumulation in human obesity and type 2 diabetes is associated with increased rates of skeletal muscle fatty acid transport and increased sarcolemmal FAT/CD36. FASEB J. 2004;18:1144–1146. doi: 10.1096/fj.03-1065fje. [DOI] [PubMed] [Google Scholar]
- Bradbury MW, Berk PD. Mitochondrial aspartate aminotransferase: direction of a single protein with two distinct functions to two subcellular sites does not require alternative splicing of the mRNA. Biochem J. 2000;345:423–427. [PMC free article] [PubMed] [Google Scholar]
- Bruce CR, Thrush AB, Mertz VA, Bezaire V, Chabowski A, Heigenhauser GJ, Dyck DJ. Endurance training in obese humans improves glucose tolerance and mitochondrial fatty acid oxidation and alters muscle lipid content. Am J Physiol Endocrinol Metab. 2006;291:E99–E107. doi: 10.1152/ajpendo.00587.2005. [DOI] [PubMed] [Google Scholar]
- Campbell SE, Tandon NN, Woldegiorgis G, Luiken JJ, Glatz JF, Bonen A. A novel function for fatty acid translocase (FAT)/CD36: involvement in long chain fatty acid transfer into the mitochondria. J Biol Chem. 2004;279:36235–36241. doi: 10.1074/jbc.M400566200. [DOI] [PubMed] [Google Scholar]
- Cechetto JD, Sadacharan SK, Berk PD, Gupta RS. Immunogold localization of mitochondrial aspartate aminotransferase in mitochondria and on the cell surface in normal rat tissues. Histol Histopathol. 2002;17:353–364. doi: 10.14670/HH-17.353. [DOI] [PubMed] [Google Scholar]
- Chabowski A, Coort SL, Calles-Escandon J, Tandon NN, Glatz JF, Luiken JJ, Bonen A. The subcellular compartmentation of fatty acid transporters is regulated differently by insulin and by AICAR. FEBS Lett. 2005;579:2428–2432. doi: 10.1016/j.febslet.2004.11.118. [DOI] [PubMed] [Google Scholar]
- Chabowski A, Momken I, Coort SL, Calles-Escandon J, Tandon NN, Glatz JF, Luiken JJ, Bonen A. Prolonged AMPK activation increases the expression of fatty acid transporters in cardiac myocytes and perfused hearts. Mol Cell Biochem. 2006;288:201–212. doi: 10.1007/s11010-006-9140-8. [DOI] [PubMed] [Google Scholar]
- Chavez JC, Pichiule P, Boero J, Arregui A. Reduced mitochondrial respiration in mouse cerebral cortex during chronic hypoxia. Neurosci Lett. 1995;193:169–172. doi: 10.1016/0304-3940(95)11692-p. [DOI] [PubMed] [Google Scholar]
- Clarke DC, Miskovic D, Han XX, Calles-Escandon J, Glatz JF, Luiken JJ, Heikkila JJ, Bonen A. Overexpression of membrane-associated fatty acid binding protein (FABPpm) in vivo increases fatty acid sarcolemmal transport and metabolism. Physiol Genomics. 2004;17:31–37. doi: 10.1152/physiolgenomics.00190.2003. [DOI] [PubMed] [Google Scholar]
- Dyck DJ, Miskovic D, Code L, Luiken JJ, Bonen A. Endurance training increases FFA oxidation and reduces triacylglycerol utilization in contracting rat soleus. Am J Physiol Endocrinol Metab. 2000;278:E778–E785. doi: 10.1152/ajpendo.2000.278.5.E778. [DOI] [PubMed] [Google Scholar]
- Dyck DJ, Peters SJ, Glatz J, Gorski J, Keizer H, Kiens B, Liu S, Richter EA, Spriet LL, Van Der Vusse GJ, Bonen A. Functional differences in lipid metabolism in resting skeletal muscle of various fiber types. Am J Physiol Endocrinol Metab. 1997;272:E340–E351. doi: 10.1152/ajpendo.1997.272.3.E340. [DOI] [PubMed] [Google Scholar]
- Dyck DJ, Steinberg G, Bonen A. Insulin increases FA uptake and esterification but reduces lipid utilization in isolated contracting muscle. Am J Physiol Endocrinol Metab. 2001;281:E600–E607. doi: 10.1152/ajpendo.2001.281.3.E600. [DOI] [PubMed] [Google Scholar]
- Febbraio M, Hajjar DP, Silverstein RL. CD36: a class B scavenger receptor involved in angiogenesis, atherosclerosis, inflammation, and lipid metabolism. J Clin Invest. 2001;108:785–791. doi: 10.1172/JCI14006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gimeno RE, Ortegon AM, Patel S, Punreddy S, Ge P, Sun Y, Lodish HF, Stahl A. Characterization of a heart-specific fatty acid transport protein. J Biol Chem. 2003;278:16039–16044. doi: 10.1074/jbc.M211412200. [DOI] [PubMed] [Google Scholar]
- Hirsch D, Stahl A, Lodish HF. A family of fatty acid transporters conserved from mycobacterium to man. Proc Natl Acad Sci U S A. 1998;95:8625–8629. doi: 10.1073/pnas.95.15.8625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holloway GP, Bezaire V, Heigenhauser GJ, Tandon NN, Glatz JF, Luiken JJ, Bonen A, Spriet LL. Mitochondrial long chain fatty acid oxidation, fatty acid translocase/CD36 content and carnitine palmitoyltransferase I activity in human skeletal muscle during aerobic exercise. J Physiol. 2006;571:201–210. doi: 10.1113/jphysiol.2005.102178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isola LM, Zhou SL, Kiang CL, Stump DD, Bradbury MW, Berk PD. 3T3 fibroblasts transfected with a cDNA for mitochondrial aspartate aminotransferase express plasma membrane fatty acid-binding protein and saturable fatty acid uptake. Proc Natl Acad Sci U S A. 1995;92:9866–9870. doi: 10.1073/pnas.92.21.9866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koonen DP, Benton CR, Arumugam Y, Tandon NN, Calles-Escandon J, Glatz JF, Luiken JJ, Bonen A. Different mechanisms can alter fatty acid transport when muscle contractile activity is chronically altered. Am J Physiol Endocrinol Metab. 2004;286:E1042–E1049. doi: 10.1152/ajpendo.00531.2003. [DOI] [PubMed] [Google Scholar]
- Koonen DP, Coumans WA, Arumugam Y, Bonen A, Glatz JF, Luiken JJ. Giant membrane vesicles as a model to study cellular substrate uptake dissected from metabolism. Mol Cell Biochem. 2002;239:121–130. [PubMed] [Google Scholar]
- Koonen DP, Glatz JF, Bonen A, Luiken JJ. Long-chain fatty acid uptake and FAT/CD36 translocation in heart and skeletal muscle. Biochim Biophys Acta. 2005;1736:163–180. doi: 10.1016/j.bbalip.2005.08.018. [DOI] [PubMed] [Google Scholar]
- Lehninger AL, Nelson DL, Cox MM. Principles of Biochemistry. New York: Worth; 1993. [Google Scholar]
- Luiken JJ, Arumugam Y, Bell RC, Calles-Escandon J, Tandon NN, Glatz JF, Bonen A. Changes in fatty acid transport and transporters are related to the severity of insulin deficiency. Am J Physiol Endocrinol Metab. 2002a;283:E612–E621. doi: 10.1152/ajpendo.00011.2002. [DOI] [PubMed] [Google Scholar]
- Luiken JJ, Arumugam Y, Dyck DJ, Bell RC, Pelsers MM, Turcotte LP, Tandon NN, Glatz JF, Bonen A. Increased rates of fatty acid uptake and plasmalemmal fatty acid transporters in obese Zucker rats. J Biol Chem. 2001;276:40567–40573. doi: 10.1074/jbc.M100052200. [DOI] [PubMed] [Google Scholar]
- Luiken JJ, Bonen A, Glatz JF. Cellular fatty acid uptake is acutely regulated by membrane-associated fatty acid-binding proteins. Prostaglandins Leukot Essent Fatty Acids. 2002b;67:73–78. doi: 10.1054/plef.2002.0401. [DOI] [PubMed] [Google Scholar]
- Luiken JJ, Coort SL, Koonen DP, Van Der Horst DJ, Bonen A, Zorzano A, Glatz JF. Regulation of cardiac long-chain fatty acid and glucose uptake by translocation of substrate transporters. Pflugers Arch. 2004;448:1–15. doi: 10.1007/s00424-003-1199-4. [DOI] [PubMed] [Google Scholar]
- Luiken JJ, Coort SL, Willems J, Coumans WA, Bonen A, Van Der Vusse GJ, Glatz JF. Contraction-induced fatty acid translocase/CD36 translocation in rat cardiac myocytes is mediated through AMP-activated protein kinase signaling. Diabetes. 2003;52:1627–1634. doi: 10.2337/diabetes.52.7.1627. [DOI] [PubMed] [Google Scholar]
- Luiken JJ, Koonen DP, Willems J, Zorzano A, Becker C, Fischer Y, Tandon NN, Van Der Vusse GJ, Bonen A, Glatz JF. Insulin stimulates long-chain fatty acid utilization by rat cardiac myocytes through cellular redistribution of FAT/CD36. Diabetes. 2002c;51:3113–3119. doi: 10.2337/diabetes.51.10.3113. [DOI] [PubMed] [Google Scholar]
- Luiken JJ, Turcotte LP, Bonen A. Protein-mediated palmitate uptake and expression of fatty acid transport proteins in heart giant vesicles. J Lipid Res. 1999;40:1007–1016. [PubMed] [Google Scholar]
- Mogensen M, Sahlin K. Mitochondrial efficiency in rat skeletal muscle: influence of respiration rate, substrate and muscle type. Acta Physiol Scand. 2005;185:229–236. doi: 10.1111/j.1365-201X.2005.01488.x. [DOI] [PubMed] [Google Scholar]
- Sahlin K, Mogensen M, Bagger M, Fernstrom M, Pedersen PK. The potential for mitochondrial fat oxidation in human skeletal muscle influences whole body fat oxidation during low-intensity exercise. Am J Physiol Endocrinol Metab. 2007;292:E223–E230. doi: 10.1152/ajpendo.00266.2006. [DOI] [PubMed] [Google Scholar]
- Schaffer JE, Lodish HF. Expression cloning and characterization of a novel adipocyte long chain fatty acid transport protein. Cell. 1994;79:427–436. doi: 10.1016/0092-8674(94)90252-6. [DOI] [PubMed] [Google Scholar]
- Schenk S, Horowitz JF. Coimmunoprecipitation of FAT/CD36 and CPT I in skeletal muscle increases proportionally with fat oxidation after endurance exercise training. Am J Physiol Endocrinol Metab. 2006;291:E254–E260. doi: 10.1152/ajpendo.00051.2006. [DOI] [PubMed] [Google Scholar]
- Schwieterman W, Sorrentino D, Potter BJ, Rand J, Kiang CL, Stump D, Berk PD. Uptake of oleate by isolated rat adipocytes is mediated by a 40-kDa plasma membrane fatty acid binding protein closely related to that in liver and gut. Proc Natl Acad Sci U S A. 1988;85:359–363. doi: 10.1073/pnas.85.2.359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinberg GR, Dyck DJ, Calles-Escandon J, Tandon NN, Luiken JJ, Glatz JF, Bonen A. Chronic leptin administration decreases fatty acid uptake and fatty acid transporters in rat skeletal muscle. J Biol Chem. 2002;277:8854–8860. doi: 10.1074/jbc.M107683200. [DOI] [PubMed] [Google Scholar]
- Stremmel W, Lotz G, Strohmeyer G, Berk PD. Identification, isolation, and partial characterization of a fatty acid binding protein from rat jejunal microvillous membranes. J Clin Invest. 1985;75:1068–1076. doi: 10.1172/JCI111769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stump DD, Zhou SL, Berk PD. Comparison of plasma membrane FABP and mitochondrial isoform of aspartate aminotransferase from rat liver. Am J Physiol Gastrointest Liver Physiol. 1993;265:G894–G902. doi: 10.1152/ajpgi.1993.265.5.G894. [DOI] [PubMed] [Google Scholar]
- Tunstall RJ, Mehan KA, Wadley GD, Collier GR, Bonen A, Hargreaves M, Cameron-Smith D. Exercise training increases lipid metabolism gene expression in human skeletal muscle. Am J Physiol Endocrinol Metab. 2002;283:E66–E72. doi: 10.1152/ajpendo.00475.2001. [DOI] [PubMed] [Google Scholar]
- Turcotte LP, Swenberger JR, Tucker MZ, Yee AJ. Training-induced elevation in FABPPM is associated with increased palmitate use in contracting muscle. J Appl Physiol. 1999;87:285–293. doi: 10.1152/jappl.1999.87.1.285. [DOI] [PubMed] [Google Scholar]
- Turcotte LP, Swenberger JR, Tucker MZ, Yee AJ, Trump G, Luiken JJ, Bonen A. Muscle palmitate uptake and binding are saturable and inhibited by antibodies to FABPPM. Mol Cell Biochem. 2000;210:53–63. doi: 10.1023/a:1007046929776. [DOI] [PubMed] [Google Scholar]
- Zhou SL, Stump D, Kiang CL, Isola LM, Berk PD. Mitochondrial aspartate aminotransferase expressed on the surface of 3T3-L1 adipocytes mediates saturable fatty acid uptake. Proc Soc Exp Biol Med. 1995;208:263–270. doi: 10.3181/00379727-208-43854. [DOI] [PubMed] [Google Scholar]