

Abstract
The functional significance of shear stress-induced vasodilatation in large conduit arteries is unclear since changes in the diameter have little effect on the resistance to blood flow. However, changes in diameter have a relatively large effect on wall shear stress which suggests that the function of flow-mediated dilatation is to reduce wall shear stress. The mean and pulsatile components of shear stress vary widely throughout the arterial system and areas of low mean and high amplitude of wall shear stress are prone to the development of atheroma. In this study, using an in vivo model with the ability to control flow rate and amplitude of flow independently, we investigated the characteristics of the response of the iliac artery to variations in both the mean and amplitude of wall shear stress. The results of this study confirm that increases in mean wall shear stress are an important stimulus for the release of nitric oxide by the endothelium as indicated by changes in arterial diameter and show for the first time, in vivo, that increases in the amplitude of the pulsatile component of shear stress have a small but significant inhibitory effect on this response. A negative feedback mechanism was identified whereby increases in shear stress brought about by increases in blood flow are reduced by the release of nitric oxide from the endothelium causing dilatation of the artery, thus decreasing the stimulus to cell adhesion and, through a direct action of nitric oxide, inhibiting the process of cell adhesion. The results also provide an explanation for the uneven distribution of atheroma throughout the arterial system, which is related to the ratio of pulsatile to mean shear stress and consequent variability in the production of NO.
Flow-mediated dilatation of large conduit arteries was first demonstrated in vivo by Schretzenmayr (1933) and was later shown to be caused by the action of an increase in wall shear stress on the endothelium (Melkumyants et al. 1989) which causes vasodilatation by production of nitric oxide (NO) (Griffith et al. 1984). The functional significance of this dilatation is unclear since changes in the diameter of conduit arteries (e. g. aorta, iliac and femoral) have little effect on the resistance to blood flow, which is determined by the diameter of small-resistance arteries, in particular the arterioles. However, changes in diameter do have a relatively large effect on wall shear stress, which is inversely proportional to the diameter cubed at constant flow and viscosity, thus suggesting the existence of a negative feedback system whereby increases in wall shear stress, brought about, for example, by increases in flow, are reduced by dilatation of the conduit artery. Flow-mediated dilatation has also been directly observed in isolated small-resistance arteries (Sorop et al. 2003) and by implication from perfusion experiments in vivo (Kartamyshev et al. 2007). In these small arteries increases in diameter at constant pressure cause an increase in flow and the net effect of an increase in flow is, in contrast to the situation in large-conduit arteries, an increase in shear stress. However, the diameter of small arteries is determined much more by the physical and metabolic environment of the surrounding tissue making it difficult to detect the precise role of flow-mediated dilatation in vivo (Poucher, 1995).
Arterial wall shear stress can be divided into two components, pulsatile and mean, associated with mean and pulsatile blood flow. These two components of wall shear stress vary widely throughout the arterial system and areas of low mean and high amplitude of wall shear stress are prone to the development of atheroma (Caro et al. 1971; Glagov et al. 1988; van den Berg et al. 2006b). It has been suggested that the mechanisms responsible for this uneven distribution are, firstly, the ability of the endothelium to produce nitric oxide (which is known to be atheroprotective) in response to increases in mean shear stress and, secondly, the ability of high amplitude shear stress to reduce the production of nitric oxide and also activate platelet and neutrophil adhesion.
Flow-mediated arterial dilatation is a measure of the ability of the endothelium to produce nitric oxide and is now routinely used as a diagnostic test of endothelial function in man. In these tests, changes in the characteristics of the shear stress stimulus to the endothelium are uncontrolled, because of changes in blood flow, pressure and arterial diameter, making the interpretation of results difficult. A recent review (Pyke & Tschakovsky, 2005), stated that ‘the quantification that best describes the stimulus responsible for FMD (flow-mediated dilatation) has not been precisely defined’. The methodology in our previous studies in vivo (Snow et al. 1994, 2001) did not allow precise and independent control of both the duration and magnitude of changes in the mean and amplitude of blood flow and pressure. Therefore, the conclusion that the amplitude of shear stress caused arterial constriction was not entirely justified mainly because of concomitant changes in pulse pressure.
The aim of this present investigation, using a preparation in which the characteristics of blood flow could be precisely controlled, was to test the hypothesis for the existence of a negative feedback system regulating wall shear stress and to obtain quantitative data on the characteristics of the stimulus and response involved in order to assess their physiological significance. A preliminary communication of some of the results has been given to the British Pharmacological Society (Kelly & Snow, 2005).
Methods
This investigation was carried out under licenses issued by the Department of Health Ireland as directed by the Cruelty to Animals Act Ireland (1876) and EU Statutory Instructions (2002). Local ethical committee approval was obtained. Twelve female landrace pigs of weight range 24–35 kg were sedated with ketamine (30 mg kg−1i. m.) and xylazine (2.7 mg kg−1i. m.) and then anaesthetized with sodium pentobarbitone (induction, 30 mg kg−1i.v.; maintenance 6 mg kg−1 h−1i.v.). The depth of anaesthesia was assessed by monitoring changes in heart rate and blood pressure in response to a nose pinch. At the end of experimental procedures, animals were killed using a lethal intravenous injection of pentobarbitone.
End tidal 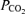 was measured using an infra red CO2 analyser (P. K. Morgan Ltd, Gillingham, Kent, UK) and maintained between 34 and 40 mmHg by positive pressure ventilation using a Palmer pump via a tracheotomy; hypoxia was prevented by adding 40% oxygen to inspired air. Rectal temperature was recorded and maintained at 37 ± 1°C using a heated table and blanket.
was measured using an infra red CO2 analyser (P. K. Morgan Ltd, Gillingham, Kent, UK) and maintained between 34 and 40 mmHg by positive pressure ventilation using a Palmer pump via a tracheotomy; hypoxia was prevented by adding 40% oxygen to inspired air. Rectal temperature was recorded and maintained at 37 ± 1°C using a heated table and blanket.
The ECG lead II was recorded and used to calculate heart rate. Systemic arterial pressure was measured using a 7 F catheter tip manometer (Millar Instruments Inc., Houston, TX, USA) inserted through the right external iliac or the inferior mesenteric artery into the base of the aorta. Blood flow was measured using an ultrasonic flowmeter (Transonic Inc., Ithaca, NY, USA). The diameter of the iliac artery was measured using disc shaped piezo electric crystals attached to a Sonomicrometer 120.0 (Triton Technology Inc., San Diego, CA, USA). The recorded distance between the crystals was corrected for the crystal lens effect (−0.7 mm) and arterial wall thickness (−0.8 mm) to obtain Di the internal diameter. Flow, arterial pressure, iliac artery diameter, ECG and heart rate, were recorded using Power lab preamplifiers and Chart 5 software connected to a Compaq PC(AD Instruments).
Surgical procedures
The abdominal aorta and iliac arteries and left iliac vein were exposed through a left retroperitoneal flank incision. The inferior mesenteric artery, right external and the internal iliac arteries and any small branches between the origin of the left iliac artery at the aorta and the deep femoral artery were ligated. The pig was heparinized (500 i.u. kg−1i.v., maintenance 100 i.u. kg−1 (30 min)−1). The left iliac artery and vein were connected by a shunt which incorporated a variable screw clip resistance and capacitor (Fig. 1). The screw clip allowed control of mean flow and by varying the amount of air in the capacitor the amplitude of the flow pulse was varied without causing any changes in mean flow. A pair of piezo electric crystals was placed on diametrically opposite sides of the artery through a small incision in the connective tissue surrounding the artery. An ultrasonic blood flow transducer was placed around the iliac artery distal to the crystals.
Figure 1. Diagram of the shunt preparation.

The iliac artery and iliac vein were connected by a shunt incorporating a screw clip resistance to control mean flow and a variable capacitor to allow independent control of the amplitude of pulsatile flow. A catheter tip pressure manometer was inserted via the inferior mesenteric artery into the abdominal aorta. A pair of piezo electric crystals was used to measure iliac artery diameter at D. An ultrasonic flow probe was used to measure iliac artery flow.
Calculation of mean and amplitude of pulsatile wall shear stress
The mean and amplitude of wall shear stress were calculated from the mean and amplitude of the volume flow (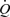 ) using methods fully described by Hunter (2003). In summary, the frequency response of the Transonic flow probe is 100 Hz and the analog output was digitally sampled at 200 s−1 and divided by the cross sectional area of the artery to give the velocity (U) by the Powerlab system. The digitized flow data were tabulated over one to four complete cardiac cycles, depending on the heart rate, such that the number of digitized points was close to but less than 2n, zeros were then added to make up to 2n as required for the fast Fourier algorithm (Fast Fourier algorithm, Excel Microsoft Windows 2000) used to derive the moduli and phases of the possible 2n−1 harmonics. In practice the number of data points per cardiac cycle was in the region of 60 or 120. For a heart rate of 3 Hz, 60 data points per cycle is sufficient to derive the 30th harmonic which has a frequency of 90 Hz, a frequency just below the 100 Hz maximum of the flow probe. For pressure and flow pulses in arteries of large mammals, apart from the coronary arteries, accurate recording of the 10th harmonic is usually regarded as sufficient (Pedley, 1980). However the modulus of wall shear stress is a function of α, the Womersley frequency parameter (α=a(2πfn/υ1/2), where a is radius fn frequency and υ is the kinematic viscosity of blood); α varies as fn1/2 thus higher frequency harmonics contribute significantly more to the wall shear stress waveform than they do to the corresponding flow, velocity and pressure waveforms. The mean wall shear stress (τw, N m−2) was calculated continuously on-line by Powerlab software using the equation:
) using methods fully described by Hunter (2003). In summary, the frequency response of the Transonic flow probe is 100 Hz and the analog output was digitally sampled at 200 s−1 and divided by the cross sectional area of the artery to give the velocity (U) by the Powerlab system. The digitized flow data were tabulated over one to four complete cardiac cycles, depending on the heart rate, such that the number of digitized points was close to but less than 2n, zeros were then added to make up to 2n as required for the fast Fourier algorithm (Fast Fourier algorithm, Excel Microsoft Windows 2000) used to derive the moduli and phases of the possible 2n−1 harmonics. In practice the number of data points per cardiac cycle was in the region of 60 or 120. For a heart rate of 3 Hz, 60 data points per cycle is sufficient to derive the 30th harmonic which has a frequency of 90 Hz, a frequency just below the 100 Hz maximum of the flow probe. For pressure and flow pulses in arteries of large mammals, apart from the coronary arteries, accurate recording of the 10th harmonic is usually regarded as sufficient (Pedley, 1980). However the modulus of wall shear stress is a function of α, the Womersley frequency parameter (α=a(2πfn/υ1/2), where a is radius fn frequency and υ is the kinematic viscosity of blood); α varies as fn1/2 thus higher frequency harmonics contribute significantly more to the wall shear stress waveform than they do to the corresponding flow, velocity and pressure waveforms. The mean wall shear stress (τw, N m−2) was calculated continuously on-line by Powerlab software using the equation:
| (1) |
where 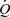 is flow (ml s−1), Di is the internal diameter of the artery (mm) and μ is the viscosity of blood (0.004 N m−2 s).
is flow (ml s−1), Di is the internal diameter of the artery (mm) and μ is the viscosity of blood (0.004 N m−2 s).
The amplitude component of wall shear stress cannot be calculated on-line, as it requires knowledge of the moduli and phases of the harmonics of the wave form of either the pressure gradient (G), or average velocity (U) in the artery. Pedley (1980) derived expressions for both average velocity and wall shear stress (τw) in terms of the mean pressure gradient (Go) and the moduli (Gn) and phases (θn) of the harmonics of the pulsatile component of the pressure gradient. The equations for U and τw in terms of G were combined to give τw in terms U as described by Hunter (2003).
| (2) |
where F(αn) = 2J1(i3/2αn)/i3/2αnJo(i3/2αn) and Jo, J1 are Bessel functions. The term 4 μU0/a is the mean wall shear stress. The moduli (τwn) and phases (θn) of each harmonic of the shear stress waveform were calculated from:
| (3) |
where 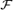 eiΨ=α2 F(α)/[1 −F(α)] and U=
eiΨ=α2 F(α)/[1 −F(α)] and U=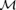 eiΦ
eiΦ
The quantities 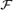 and
and 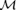 are real moduli and Φ and Ψ are real phases.
are real moduli and Φ and Ψ are real phases. 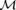 and Φ are known from experimental data (i.e. they are the moduli and phase obtained from the Fourier analysis of the velocity waveform).
and Φ are known from experimental data (i.e. they are the moduli and phase obtained from the Fourier analysis of the velocity waveform).
Examples of the velocity and wall shear stress wave forms throughout one cardiac cycle are shown in Fig. 2. The heart rate was 188 beats min−1, the mean flow 542 ml min−1, Di 3.12 mm, mean velocity Um 1.16 m s−1 and mean wall shear stress τw 6.75 N m−2. The values for τw at each time point were calculated using the moduli and phases of the harmonics of the velocity wave form up to the 10th (31.3 Hz) and the 32nd (100.2 Hz). It can be seen that there are only small differences in the two calculated waveforms of τw and that to estimate the overall amplitude of τw the first 10 harmonics with a frequency up to 31.3 Hz are sufficient. This frequency is well within the frequency response of the flow probe (100 Hz).
Figure 2. Velocity (U) and wall shear stress (τw) wave forms over one cardiac cycle at a heart rate of 188 beats min−1.

Velocity wave form (□), τw calculated using the first 10 harmonics (^) and using 32 harmonics (▵).
Results
When recording began about 2 h after the induction of anaesthesia, the average arterial blood pressure and heart rate in the 10 pigs were 130 ± 20.1 mmHg systolic, 98.1 ± 16.1 mmHg diastolic and 150.7 ± 28.9 beats min−1, respectively.
Effects of a maintained increase in flow on wall shear stress and diameter in the iliac artery
An example of records showing the effect of a maximum increase (shunt fully open) in blood flow (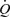 ) in the left iliac artery, maintained for a period of 2 min, on diameter (D), wall shear stress (τw) and arterial pressure (AP) is shown in Fig. 3. Shunt resistance was reduced causing an increase in flow and wall shear stress. In response to this increase in shear stress the artery dilates with a consequent reduction in shear stress until a new equilibrium between shear stress and diameter is achieved. The effect of this increase in diameter, in the presence of a maintained flow, on both the mean and amplitude of pulsatile wall shear stress is shown in Fig. 3). When the shunt resistance is increased flow returned to control values and wall shear stress is now below control. The artery then gradually constricts and diameter and shear stress return to their control values over a period of 30 min. This decay of the response follows a single exponential curve with a time constant (t1/2) of 5 min 55 s. It should be noted that as the artery dilates (b to c; Fig. 3) in the presence of the fixed resistance provided by the screwclip, no further detectable changes in arterial pressure and iliac artery blood flow take place. Therefore, in this preparation the greater part of the resistance to blood flow is distal to the iliac artery.
) in the left iliac artery, maintained for a period of 2 min, on diameter (D), wall shear stress (τw) and arterial pressure (AP) is shown in Fig. 3. Shunt resistance was reduced causing an increase in flow and wall shear stress. In response to this increase in shear stress the artery dilates with a consequent reduction in shear stress until a new equilibrium between shear stress and diameter is achieved. The effect of this increase in diameter, in the presence of a maintained flow, on both the mean and amplitude of pulsatile wall shear stress is shown in Fig. 3). When the shunt resistance is increased flow returned to control values and wall shear stress is now below control. The artery then gradually constricts and diameter and shear stress return to their control values over a period of 30 min. This decay of the response follows a single exponential curve with a time constant (t1/2) of 5 min 55 s. It should be noted that as the artery dilates (b to c; Fig. 3) in the presence of the fixed resistance provided by the screwclip, no further detectable changes in arterial pressure and iliac artery blood flow take place. Therefore, in this preparation the greater part of the resistance to blood flow is distal to the iliac artery.
Figure 3.

A, records of flow (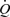 ), calculated mean wall shear stress (τw), external diameter (D) and pressure (AP) in the left iliac artery showing effects of a maintained increase in flow (
), calculated mean wall shear stress (τw), external diameter (D) and pressure (AP) in the left iliac artery showing effects of a maintained increase in flow (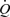 ). When the shunt was fully opened at a, mean blood flow increased from 111 to 754 ml min−1, causing an initial increase in mean wall shear stress from 2.2 to 15.1 N m−2. This reduction in resistance caused a fall in mean pressure from 97 to 86 mmHg and a small decrease from 4.06 to 4.03 mm in the diameter of the artery. The high shear stress was maintained for approximately 20 s until the artery began to dilate (b). The time from the initial increase in shear stress for the artery to reach 50% of the maximum increase in diameter was 33 s. At constant flow, wall shear stress is inversely proportional to D3. Therefore, as the artery dilates, mean wall shear stress is reduced despite the maintained increase in flow, until a new equilibrium between shear stress and diameter is reached at c. B, the effect of an increase in diameter (from b to c above) on the mean and pulsatile wall shear stress (τw) over two cardiac cycles at the same mean and pulsatile flow. The increase in diameter from 4.03 to 5.14 mm reduced mean wall shear stress from 14.4 to 6.5 N m−2, the amplitude from 4.7 to 2.5 N m−2 and the peak from 17.1 to 8.0 N m−2.
). When the shunt was fully opened at a, mean blood flow increased from 111 to 754 ml min−1, causing an initial increase in mean wall shear stress from 2.2 to 15.1 N m−2. This reduction in resistance caused a fall in mean pressure from 97 to 86 mmHg and a small decrease from 4.06 to 4.03 mm in the diameter of the artery. The high shear stress was maintained for approximately 20 s until the artery began to dilate (b). The time from the initial increase in shear stress for the artery to reach 50% of the maximum increase in diameter was 33 s. At constant flow, wall shear stress is inversely proportional to D3. Therefore, as the artery dilates, mean wall shear stress is reduced despite the maintained increase in flow, until a new equilibrium between shear stress and diameter is reached at c. B, the effect of an increase in diameter (from b to c above) on the mean and pulsatile wall shear stress (τw) over two cardiac cycles at the same mean and pulsatile flow. The increase in diameter from 4.03 to 5.14 mm reduced mean wall shear stress from 14.4 to 6.5 N m−2, the amplitude from 4.7 to 2.5 N m−2 and the peak from 17.1 to 8.0 N m−2.
The effects of wall shear stress on arterial diameter were completely abolished by the nitric oxide synthase (NOS) inhibitor, l-NAME (20 mg kg−1i.v.), confirming that the above effects of wall shear stress on diameter were mediated through the production of NO by the endothelium(Fig. 4). In the presence of l-NAME, opening and closing the shunt caused an immediate large increase in wall shear stress and a small reduction in arterial pressure and diameter, as in Fig. 3, but in contrast the dilatation of the artery was abolished.
Figure 4. Records of flow (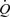 ), calculated mean wall shear stress (τw), external diameter (D) and pressure (AP) in the left iliac artery showing effects of a maintained increase in flow in the presence of l-NAME (20 mg kg−1i.v.).
), calculated mean wall shear stress (τw), external diameter (D) and pressure (AP) in the left iliac artery showing effects of a maintained increase in flow in the presence of l-NAME (20 mg kg−1i.v.).

Shunt resistance was reduced at a causing an increase in flow and shear stress with no resultant dilatation of the artery. The shunt resistance was increased at b and flow and shear stress returned to their control values.
The threshold and sensitivity of the response of the artery to wall shear stress were determined by measuring the increase in diameter in response to stepwise increases in flow (Fig. 5A). At each step, flow and wall shear stress increase together until wall shear stress exceeds the threshold value at which point the artery dilates (a in Fig. 5A). Consequently, the wall shear stress, which stimulates the dilatation, is then reduced and a new equilibrium between wall shear stress and diameter is reached. This process was repeated until either flow reached a maximum (shunt fully open) or further increases in shear stress did not cause an increase in the diameter. The relationship between wall shear stress and arterial diameter in one pig is shown in Fig. 5B, where the diameter of the artery (D) is plotted against the initial stimulating value of wall shear stress (τw). The relationship is not linear over the whole range and tends to plateau at the higher values of wall shear stress. The value of the threshold wall shear stress was taken as the intersection of the two regression lines b and c and the sensitivity of the response as the slope of the initial response, line c.
Figure 5. Records obtained in one pig showing effects of stepwise increases in flow (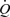 ), maintained for 2 min, on calculated mean wall shear stress (τw), external diameter (D) and arterial pressure (AP).
), maintained for 2 min, on calculated mean wall shear stress (τw), external diameter (D) and arterial pressure (AP).

A, at a, the threshold stimulus of wall shear stress is exceeded and the artery begins to dilate. B, effect of graded increases in wall shear stress (τw) on the external diameter (D) of the iliac artery, where baseline (b) is where there was no detectable response to wall shear stress and c is the response. The threshold value of the wall shear stress (5.97 N m−2) was taken at the intersection of the two regression lines b and c, and the sensitivity of the response (0.036 mm N−1 m−2) as the slope of regression line c.
The effects of a maximum increase in flow, maintained for at least 2 min, on diameter and shear stress were obtained in 10 pigs. An increase in flow of 513% led to an increase in equilibrium wall shear stress of 215%. In every pig the diameter of the iliac artery increased in response to an increase in mean wall shear stress. Overall there was a significant 20.8 ± 3.0% (mean ±s.e.m.,P < 0.001, paired t test) dilatation of the artery. If no increase in Di had taken place, both flow and wall shear stress would have increased by the same 513%. Therefore the effect of the 20.8% increase in Di is to markedly reduce the increase in mean wall shear stress (τw) from 513% to 215%.
The above investigations of the effects of mean flow on wall shear stress and artery diameter were carried out using periods of maintained flow of a minimum of 2 min, by which time an equilibrium had been reached between mean wall shear stress and artery diameter, e.g. c in Fig. 3A. The effects of shorter periods of maintained flow were also investigated and the interaction between the magnitude and duration of wall shear stress on the threshold response measured. The shunt was opened and closed for different combinations of time and flow (Fig. 6A and B). Similar results were obtained in two pigs and the relationship between wall shear stress and duration which caused a threshold response is shown in Fig. 6C. The observed points suggest that the data may be fitted to an hyperbola of the form:
where τw (N m−2) is the wall shear stress stimulus, T is the duration (in s) and a, b and k are constants, i.e. the threshold stimulus is a product of the intensity and the duration. The threshold value of 4.49 N m−2 for a long duration stimulus of 120 s was taken as the value of a, and b and k derived from regression analysis of the linear form 1/(τw− a) =T/k − b/k. The predicted minimum duration (b) of an infinitely large stimulus is 0.8 s. However, the maximum stimulus that was possible in these two experiments was 35 N m−2 (flow 600 ml min−1) and the minimum duration was 2 s. From these results it may be concluded that even transient large increases in shear stress must have a duration in excess of 1–2 s to cause a detectable increase in arterial diameter and that a shear stress below 4.5 N m−2 maintained for 2 min is not likely to be effective.
Figure 6. Records of flow (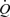 ), mean wall shear stress (τw), external diameter (D) and arterial pressure (AP).
), mean wall shear stress (τw), external diameter (D) and arterial pressure (AP).

Effects of an increase in mean wall shear stress to: A, 9.50 N m−2 maintained for increasing periods of time: 6.5 s (a), 10.3 s (b) and 13.6 s (c); and B, 22.5 N m−2 maintained for increasing periods of time: 2.7 s (a), 4.1 s (b) and 6.1 s (c). In both there is a small detectable increase in arterial diameter after b and a definite increase after c. C, relationship between magnitude of stimulus (τw) and duration (T) to cause a threshold response. τw= (24/T– 0.8) + 4.49.
Results from previous experiments in the dog (Snow et al. 2001) have indicated that the two components of wall shear stress associated with the mean and amplitude of pulsatile flow interact such that the amplitude component of wall shear stress reduces the effect of the mean on the resultant dilatation of the artery. Using the variable capacitor in the shunt circuit, which allowed changes in the amplitude of flow to be varied independently of changes in both mean flow and arterial pressure, an experiment was carried out to verify this effect of amplitude shear stress (Fig. 7). Firstly, mean blood flow is increased with no change in amplitude causing an increase in mean wall shear stress and resultant increase in arterial diameter. When equilibrium between mean wall shear stress and arterial diameter is reached the amplitude of wall shear stress is increased (at constant mean shear stress) in three stages. These increases cause no further changes in arterial diameter. Amplitude is then reduced to control level, with no effect on arterial diameter and finally to show that the artery was still capable of dilating, mean wall shear stress is increased with a consequent increase in diameter. Similar results were obtained in four pigs where increases in the amplitude of wall shear stress (8.0 ± 3.0 to 28.3 ± 1.3 N m−2; mean ±s.e.m.) had no detectable effect on arterial diameter. However, when the initial response of the artery to increases in mean shear stress at low and high amplitude of wall shear stress was analysed, a reduction in the sensitivity of the artery to mean shear stresss was observed (e.g. Fig. 8). This effect of a high amplitude of wall shear stress on the response was investigated in five pigs and in every pig a reduction (40%) in the sensitivity of the response to mean wall shear stress was observed. No significant effects of increasing the amplitude of wall shear stress on the threshold response to mean shear stress were detected.
Figure 7. Records obtained in one pig showing the effects of independent changes in the mean and amplitude of flow (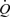 ) on the mean (τw) and calculated amplitude (horizontal bars) of wall shear stress and arterial diameter (D).
) on the mean (τw) and calculated amplitude (horizontal bars) of wall shear stress and arterial diameter (D).

At a mean flow was increased from 183 to 318 ml min−1 with no change in amplitude (100 ml min−1), mean shear stress increased from 9.9 to 16.8 N m−2 followed by dilatation of the artery from 3.85 to 4.1 mm. Equilibrium between mean wall shear stress (12.5 N m−2) and diameter (4.10 mm) was achieved at b; from a to b the amplitude of wall shear stress was constant (7.4 N m−2). The amplitude of wall shear stress was then increased, with no change in mean, to 13.7 N m−2 (c), 17.2 N m−2 (d) and 18.3 N m−2 (e). These increases in the amplitude of wall shear stress at constant mean had no detectable effects on arterial diameter. At f the amplitude of flow was reduced to control level (a) with no effect on arterial diameter. At g mean wall shear stress was increased to 24.6 N m−2 causing a further increase in diameter from 4.1 to 4.5 mm.
Figure 8. Effect of increasing the amplitude of wall shear stress (τw) on the sensitivity of the response of the artery to mean wall shear stress.

Results obtained at low amplitude of wall shear stress (3.27 N m−2, ♦) and high amplitude of wall shear stress 20.58 Nm−2, ⋄). Increasing the amplitude of wall shear stress reduced the sensitivity of the response from 0.398 mm N−1 m−2 to 0.125 mm N−1 m−2 (P < 0.001, t test).
Summary of results
The overall results obtained in 10 pigs are summarized in Table 1 (values are mean ±s.e.m.).
Table 1.
Summary of results shown as mean ±s.e.m.
| Control | Maximum flow | |
|---|---|---|
| Blood flow (ml min−1) | 132 ± 14.8 | 676 ± 73 |
| Mean arterial blood pressure (mmHg) | 92.6 ± 7.5 | 88.3 ± 8.3 |
| Internal diameter (mm) | 2.788 ± 0.171 | 3.359 ± 0.201 |
| τw before artery dilatation (N m−2) | 4.49 ± 0.7 | 26.04 ± 2.53 |
| τw after artery dilatation (N m−2) | — | 14.89 ± 1.58 |
| Time of response to 50% increase in diameter (s) | 47.4 ± 3.3 | |
| Decay of response, T1/2 (s) | 481 ± 72 | |
| Sensitivity of response mm Hg.ml−1.s | ||
| At low amp. τw (7.0 ± 2.4 N m−2) | 0.2 ± 0.003 | |
| At high amp. τw (24.0 ± 4.4 N m−2) | 0.121 ± 0.016 | |
Fully opening the shunt caused an approximately 5-fold increase in both mean blood flow and mean wall shear stress in the iliac artery with no significant change in mean arterial pressure. The increase in mean blood flow was maintained but the consequent 20% increase in the internal diameter of the iliac artery reduced the increase in wall shear stress from 5- to 3-fold. Changes in the amplitude of wall shear stress at constant mean had no effect upon arterial diameter but did reduce the initial sensitivity of the response to mean wall shear stress.
Discussion
The above results (Table 1) were obtained using an in vivo preparation which allowed independent control of the mean and amplitude of blood flow in a conduit artery and confirm the existence of a negative feedback system regulating wall shear stress through changes in arterial diameter. They define the threshold stimulus in terms of the intensity and duration of wall shear stress, the sensitivity of the system (change in arterial diameter/change in wall shear stress) and the duration of the on and off responses. In the presence of a maintained steady mean shear stress, changes in the amplitude of shear stress appear to have no effect upon arterial diameter. However, high amplitude shear stress decreases the sensitivity of, the response of the artery to mean shear stress. The effects of mean wall shear stress were abolished by l-NAME and can be attributed to the release of NO by the endothelium. In these experiments, the iliac artery was isolated from the periphery by the arterio-venous shunt therefore excluding any antidromic signalling from endothelium-derived hyperpolarizing factor (EDHF). Even in the intact pig iliac artery, it has been shown that any EDHF released either by increases in flow or distal intra-arterial injection of acetycholine had no effect on the diameter of the proximal artery (Kelly et al. 2006a). It has been shown that a cytochrome-related endothelial-derived hyperpolarizing factor is involved in shear stress-mediated vasodilation in the human radial artery (Bellien et al. 2006) However, it seems from the experiments in which l-NAME was administered that if this mechanism has a role in shear stress mediated dilatation in the iliac artery, it would be a very small role. This reflects the decrease in the importance of EDHF as arteries get bigger.
Iliac preparation
In previous experiments (Snow et al. 1994, 2001; Snow, 2002; Kelly et al. 2006b) describing the effects of flow-induced vasodilatation in the iliac artery of the dog and pig, the mean and amplitude of pulsatile flow were increased by reducing peripheral resistance by either an intra-arterial infusion of acetylcholine (distal to the site of diameter measurement) or reactive hyperaemia and stimulation of the sympathetic nerves to the heart. These techniques for increasing the mean and amplitude of flow have major disadvantages: it is not possible to obtain precise predetermined increases, nor can any increases be maintained steady for a given period of time. Also, uncontrolled changes in the arterial pressure, which may alter the diameter of the artery, often occur and any re-circulating acetylcholine would have a direct effect on arterial diameter at the site of measurement. Thus, the above techniques only allow at best a semi-quantitative description of the effects of flow on arterial diameter. The shunt preparation used in the present series of experiments, overcomes these disadvantages: acetylcholine is not used and peripheral resistance is controlled by the screw clamp, allowing precise control of the magnitude and duration of mean flow. The amplitude of the pulsatile component is controlled independently of the mean by varying the amount of air in the ‘windkessel’ capacitor. Falls in arterial pressure (∼10 mmHg) do occur when the shunt is abruptly fully opened (Figs 3A and 4) causing a relatively small decrease in arterial diameter. When the artery dilates, causing a significant decrease in wall shear stress, no observable change in blood flow takes place demonstrating that the decrease in resistance in the iliac artery is not significant. Importantly, changes in the amplitude of flow are obtained with no change in the amplitude of the arterial pressure pulse. These features of the shunt preparation allow a quantitative analysis of the effects of wall shear stress on arterial diameter in vivo. Also, using the shunt preparation, the artery is perfused with blood and the flow pulses are generated by the heart. Perfusion with blood is essential if quantitative results applicable to the intact animal are to be obtained, since haemoglobin rapidly binds NO, restricting its activity to that of an apocrine hormone without long-lasting systemic effects.
Threshold stimulus
Basal blood flow in the intact iliac artery of the anaesthetized pig is in the region of 100 ml min−1 (Kelly et al. 2006b). In the present series of experiments, the average flow at the threshold for an increase in diameter was 132 ml min−1 and the corresponding wall shear stress 4.49 N m−2. It may be concluded that at basal blood flow, NO is being released at a rate sufficient to have a detectable effect on arterial diameter. In previous experiments in vitro in the absence of Hb (Hecker et al. 1993; Mochizuki et al. 2003), shear stresses lower than the threshold observed in the present experiments caused production of nitric oxide or vasodilatation. Taken together these results would suggest that nitric oxide could be produced at shear stresses lower than the threshold observed in vivo, but that the amounts produced are too low to cause vasodilatation in the presence of haemoglobin.
Response time of the artery
The time course of the response of an increase in arterial diameter is slow, an average of 47 s to achieve 50% of the maximum increase in diameter. Whether this delay occurs in the mechano-transduction of the signal at the luminal surface and membrane of the endothelial cell e.g. at the level of the glycocalyx (Mochizuki et al. 2003; Weinbaum et al. 2003; van den Berg et al. 2006a) or cytoskeleton (Sun et al. 2001), or in the production, diffusion or elimination of NO or in the mechano-transduction in the smooth muscle cell is uncertain. The glycocalyx is a highly negatively charged structure, rich in anionic sites mostly represented by the sialic acid moieties of glycoproteins and the sulphate and carboxyl groups of heparan-sulphate proteoglycans. Squire et al. (2001) found that the glycocalyx had a thickness of 100–200 nm. The glycocalyx may be a significant structure in mechano-transduction in several ways: (1) transmembrane glycoproteins extending into the extracellular space occupied by the glycocalyx may be physically displaced by the frictional haemodynamic shear stress to provoke an intracellular response, (2) the fluid layer associated with the glycocalyx represents a microenvironment for agonist receptor interactions (Davies, 1995). Weinbaum et al. (2003) suggest that the glycocalyx transduces the response via its proteoglycans which interact via their trans-membrane core protein with the cell cytoskeleton. This is consistent with the findings of Thi et al. (2004) who showed that, in the presence of an intact glycocalyx, shear stress resulted in a dramatic redistribution of actin filaments in cultured endothelial cells which was not seen in endothelial cells in which the glycocalyx was disrupted.
The results obtained in the stimulus–duration experiments (Fig. 6) where the predicted minimum duration of a large stimulus is 0.8 s indicate that overall the system is capable of responding to a stimulus of relatively short duration. Therefore shorter transient increases in wall shear stress (e.g. similar to those associated with peak flow during each cardiac cycle, Fig. 7), which are well above the threshold, should not be able to cause dilatation and by implication activate nitric oxide synthase, a conclusion corroborated by the lack of effect of increases in the amplitude of wall shear stress. These results indicate that the response time of the overall mechano-transduction system in the glycocalyx and endothelium leading to the production of NO cannot fully account for the overall delay. The distance for NO to diffuse from the endothelium to the outermost smooth muscle of the artery is at the most 0.4 mm and even if the diffusion time was as great as 10 s it is unlikely that NO would remain unbound to any substance containing a haem group for such a period of time. Also Sorop et al. (2003) showed that superoxide dismutase shortened the response time to shear stress in pig arterioles, presumably by allowing the concentration of NO to rise to threshold more quickly. It is therefore speculated that the long delay (47 s) in the response is most likely to be in the balance between the relative kinetics and capacities of the processes of production and elimination of NO and therefore the time required to reach a concentration sufficient to activate the guanyl cyclase in smooth muscle.
Decay
When the stimulus is removed the response decays with a half-life of ∼7 min. Thus, a wall shear stress stimulus lasting for 2 min has significant effects for a further ∼28 min. In previous experiments (Snow et al. 1994) it was shown that NO released upstream in the iliac artery did not cause dilatation at a downstream site from which the endothelium had been removed and it was concluded that NO in amounts released by the endothelial constitutive enzyme nitric oxide synthase into the bloodstream was rapidly deactivated. Therefore the long-lasting effects are related to either the continued production of NO or long-lasting effects of NO on the guanyl cyclase–cyclic GMP system and smooth muscle relaxation. Kharitonov et al. (1997) found that the half-life of the NO–guanyl cyclase complex was approx 5 s in the presence of the substrate for the reaction, GTP, and a study by Russwurm & Koesling (2002) found a half-life of 4 s for the deactivation of NO-sensitive guanyl cyclase. The long half-life could be mediated by events downstream of guanyl cyclase in the arterial smooth muscle, such as elevated levels of cGMP-dependent kinase, decreased levels of calcium or increased myosin light chain phosphatase. Etter et al. (2001) showed that cGMP induced a large transient increase in myosin light chain phosphatase which would accelerate myosin light chain dephosphorylation attributable to the decline in myosin light chain kinase and enhance the drop in force. They also showed that after myosin light chain phosphatase activity returned to basal levels there was a sustained low force. This finding is in agreement with the findings of Rembold et al. (2000) who found that sustained exposure of smooth muscle to NO caused relaxation, without proportional myosin light chain dephosphorylation, by causing phosphorylation of heat shock protein 20. They suggest that heat shock protein 20 does this by interfering with or inhibiting the normal function of the actin-containing thin filaments despite activation of myosin by myosin light chain phosphorylation. This sustained low force may account for the long decay of the response of the artery seen in our study. The short duration (2 min) of the shear stress stimulus in the present experiments make it unlikely that any genomic effects on protein expression are involved. Such effects may well be involved in the long-term adaptation to repeated stimuli (Cheng et al. 2005) as occurs during physical training. In our opinion the continuing production of NO is the most likely reason for the long decay. NO is known to prevent cell adhesion and platelet aggregation and continued production after a period of increased blood flow could be of importance in explaining the beneficial effects of increased blood flow during exercise on atherosclerosis and ischaemic heart disease.
Effects of amplitude of shear stress on reponse to mean
In a previous series of experiments on the iliac artery of the anaesthetized dog (Snow et al. 2001; Markos et al. 2002) a multiple regression analysis of the effects of the mean and amplitude of wall shear stress on arterial diameter indicated that the amplitude component was negatively correlated with arterial diameter and that this correlation was abolished by the endothelin antagonist tezosentan. However, it was not possible to conclude that the amplitude component of wall shear stress was solely responsible for this effect since increases in amplitude were always accompanied by increases in both the mean and amplitude of the arterial pulse pressure. Thus, the endothelium was subjected to changes in both shear and circumferential stresses which may account for the involvement of endothelin. In the present series of experiments in the pig, using the shunt preparation, the amplitude of the pulsatile component of wall shear stress was increased without causing changes in the mean and amplitude of the arterial pulse pressure, allowing a more precisely defined effect to be obtained on arterial diameter of the amplitude component of wall shear stress at constant mean, e.g. Fig. 7. Under these conditions no effect was observed. A possible explanation for this lack of an effect is the inability of the endothelial cell to respond to transient increases in shear stresses shorter than 1 s (Fig. 6). However in a separate series of experiments where mean wall shear stress was increased at either constant low or high amplitude of shear stress, the sensitivity of the response to mean wall shear stress was reduced at high amplitude. How to reconcile these apparent conflicting results is unclear, but it is probable that under some circumstances the amplitude component of wall shear stress reduces either the production of NO or its ability to cause arterial dilatation. It is possible that a vasodilator is inhibited or that a vasoconstrictor is produced. In experiments carried out by Qiu & Tarbell (2000)in vitro, it was shown that steady shear stress stimulates the highest production of vasodilators whereas oscillatory shear stress stimulates the highest vasoconstrictor response. Also in experiments carried out by Ziegler et al. (1998) where bovine aortic endothelial cells were subjected to a non-reversing pulsatile shear stress and a highly reversing shear stress with a low mean value, it was found that the reversing shear stress caused an approximately 6-fold increase in endothelin-1 receptor expression whereas the non-reversing shear stress only caused a 3.7-fold increase in endothelin-1 receptor expression. Parent & Lavallee (2000) reported that agents triggering NO formation or increasing cGMP formation impaired endothelin-dependent responses and prevented stimulated endothelin release in vitro. Therefore, they hypothesized that nitric oxide may normally suppress an endothelin-dependent component triggered by elevated shear stress levels in vivo.
Cheng et al. (2005) found that low mean shear stress and oscillatory shear stress down-regulated eNOS gene expression while high mean shear stress increased gene expression. An increase in phosphor eNOS was observed in high shear stress regions compared with low shear stress (6-fold) and oscillatory shear stress (2-fold) regions.
Feedback mechanism
The ability of large-conduit arteries to dilate in response to an increase in wall shear stress, caused by an increase in blood flow and thereby reducing the initial increase in wall shear stress without having a major effect on flow, suggests the existence of a negative feedback control mechanism (Fig. 3A). The endothelium senses wall shear stress through a mechanism involving the glycocalyx (Weinbaum et al. 2003; Kelly et al. 2006b; van den Berg et al. 2006a) and minimizes increases in wall shear stress by producing NO, causing relaxation of smooth muscle, dilatation of the artery and consequent reduction in wall shear stress. As discussed above, high shear stresses are known to activate platelet and neutrophil adhesion, processes which are also inhibited by NO. Thus, the endothelial cell has a double protection system against these processes. First, by minimizing the increases in shear stress associated with increasing blood flow and second, by inhibiting the process of cell adhesion to the wall of the artery. Such a negative feedback system is able to operate in large-conduit arteries because changes in diameter within the physiological range (∼30%) have no significant effect on the overall resistance to blood flow, as demonstrated in Fig. 3, where an increase in diameter of the iliac artery has no measurable effect on blood flow. The contribution of the iliac artery to peripheral resistance can be estimated using Poiseuille's equation. For example, in the current experiment (Table 1) the overall peripheral resistance in the control period was 42 mmHg ml−1 s of which the calculated resistance of the iliac artery was 2 mmHg ml−1 s, 5% of the total. When the screw clip was opened, the overall resistance was reduced to 7.9 mmHg ml−1 s of which the iliac artery component was 25%. Flow increased and the iliac artery dilated reducing resistance to 1 mmHg ml−1 s−1, 12.5% of the overall resistance.
In the intact animal, flow is determined by the small-resistance vessels. Flow-mediated dilation in small-resistance arteries in situ is technically difficult to demonstrate since their diameter is predominantly affected by the physical and metabolic activity of the surrounding tissue, as for example, in the heart, and not by shear stress alone as demonstrated by Bakker et al. (2003). Nevertheless, Sorop et al. (2003), by isolating small-resistance arteries from the pig heart, clearly demonstrated the existence of flow-mediated dilatation and showed that oscillating flow without a mean forward component did not cause dilatation except in the presence of superoxide dismutase. They concluded that ‘During steady flow, the bioavailabilty of NO is sufficient to cause vasodilation. During oscillating flow, NO is quenched by the O2−, suppressing vasodilation’. In the present experiments, such an effect of superoxide could also explain the effect of the pulsatile component of wall shear stress on the initial response to mean wall shear stress when the production of NO would be relatively low. More relevant to the present experiments are those of Kartamyshev et al. (2007) who showed that the constrictor effect of noradrenaline on the femoral arterial bed of the cat was greater under constant flow than under constant pressure perfusion. They concluded that the difference in constrictor responses at constant flow and pressure perfusions depends mainly on the smooth muscle relaxation caused by increased wall shear stress. These results demonstrate that shear stress-mediated vasodilatation occurs in resistance vessels. The resistance vessels at a constant pressure gradient flow (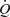 ) increases as D4 and wall shear stress is proportional to
) increases as D4 and wall shear stress is proportional to 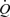 /D3. Therefore, if wall shear stress were the only determinant of vessel diameter, a positive rather than a negative feedback would operate and the vessel would exist in two stable states of fully open or fully closed. Such is clearly not the case, but this analysis illustrates the difficulties encountered in interpreting experiments designed to determine the role of flow-mediated dilatation and NO in the metabolic regulation of blood flow (Poucher, 1995).
/D3. Therefore, if wall shear stress were the only determinant of vessel diameter, a positive rather than a negative feedback would operate and the vessel would exist in two stable states of fully open or fully closed. Such is clearly not the case, but this analysis illustrates the difficulties encountered in interpreting experiments designed to determine the role of flow-mediated dilatation and NO in the metabolic regulation of blood flow (Poucher, 1995).
Pathological significance of the mean and amplitude of shear stress in the arterial system
It has been known for some time (Caro et al. 1971; Glagov et al. 1988; van den Berg, 2006a, b) that areas of the arterial system with a low mean wall shear stress are predisposed to the development of atheroma, the ‘low mean shear stress hypothesis’. In general, mean wall shear stress is low in the large central conduit arteries and increases towards the periphery as arterial diameter decreases (Caro et al. 1971), therefore atheroma tends to be confined to the conduit arteries and localized to areas of low mean wall shear stress, e.g. the inner wall of bends and outer wall of bifurcations. Many of these areas also have relatively high amplitudes of wall shear stress, e.g. the coronary arteries and in the abdominal aorta at rest. Moore et al. (1994) demonstrated a significant correlation between an index of oscillatory shear stress, which is effectively a ratio between the amplitude and mean of wall shear stress, and early atherosclerotic disease in man, suggesting that it is the combination of low mean and high amplitude which predisposes to atheroma. Transient high shear stresses stimulate mononuclear leucocyte adhesion to cultured human endothelial cells (Chappell et al. 1998) and platelets can be caused to aggregate in vivo by exposure to transient high stresses (144 ± 15 N m−2) such as occur in the Folts model of coronary arterial thrombosis (Folts et al. 1976). These acute effects on a large-conduit artery have also been shown to occur in small-resistance vessels (e.g. Sorop et al. 2003) and their repeated application, causing remodelling of blood vessels, may explain the beneficial effects of regular exercise on hypertension and atherosclerosis (Pries et al. 2005).
Conclusion
We have provided a quantitative description of the short-term effects of flow-mediated dilatation in the iliac artery of the pig and demonstrated the existence of a negative feedback system, which, by the production of NO in response to the stimulus of mean wall shear stress, reduces both the mean and amplitude of shear stress by dilatation of the artery and thereby the stimulus to cell adhesion. Furthermore, NO has a direct inhibitory effect on platelet aggregation. Thus, overall, this mechanism serves to protect the endothelium from cell adhesion and the consequent development of atheroma and offers an explanation for their uneven distribution throughout the arterial system.
References
- Bakker EN, Versluis JP, Sipkema P, Van Teeffelen JW, Rolf TM, Spaan JA, VanBavel E. Differential structural adaptation to haemodynamics along single rat cremaster arterioles. J Physiol. 2003;548:549–555. doi: 10.1113/jphysiol.2002.035907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellien J, Iacob M, Gutierrez L, Isabelle M, Lahary A, Thuillez C, Joannides R. Crucial role of NO and endothelium-derived hyperpolarizing factor in human sustained conduit artery flow-mediated dilatation. Hypertension. 2006;48:1088–1094. doi: 10.1161/01.HYP.0000246672.72188.bd. [DOI] [PubMed] [Google Scholar]
- Caro CG, Fitz-Gerald JM, Schroter RC. Atheroma and arterial wall shear. Observation, correlation and proposal of a shear dependent mass transfer mechanism for atherogenesis. Proc R Soc Lond B Biol Sci. 1971;177:109–159. doi: 10.1098/rspb.1971.0019. [DOI] [PubMed] [Google Scholar]
- Chappell DC, Varner SE, Nerem RM, Medford RM, Alexander RW. Oscillatory shear stress stimulates adhesion molecule expression in cultured human endothelium. Circ Res. 1998;82:532–539. doi: 10.1161/01.res.82.5.532. [DOI] [PubMed] [Google Scholar]
- Cheng C, van Haperen R, de Waard M, van Damme LC, Tempel D, Hanemaaijer L, van Cappellen GW, Bos J, Slager CJ, Duncker DJ, van der Steen AF, de Crom R, Krams R. Shear stress affects the intracellular distribution of eNOS: direct demonstration by a novel in vivo technique. Blood. 2005;106:3691–3698. doi: 10.1182/blood-2005-06-2326. [DOI] [PubMed] [Google Scholar]
- Davies PF. Flow mediated endothelial transduction. Physiol Rev. 1995;75:519–559. doi: 10.1152/physrev.1995.75.3.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Etter EF, Eto M, Wardle RL, Brautigan DL, Murphy RA. Activation of myosin light chain phosphatase in intact arterial smooth muscle during nitric oxide-induced relaxation. J Biol Chem. 2001;276:34681–34685. doi: 10.1074/jbc.M104737200. [DOI] [PubMed] [Google Scholar]
- Folts JD, Crowell EB, Jr, Rowe GG. Platelet aggregation in partially obstructed vessels and its elimination with aspirin. Circulation. 1976;54:365–370. doi: 10.1161/01.cir.54.3.365. [DOI] [PubMed] [Google Scholar]
- Glagov S, Zarins C, Giddens DP, Ku DN. Hemodynamics and atherosclerosis. Insights and perspectives gained from studies of human arteries. Arch Pathol Laboratory Med. 1988;112:1018–1031. [PubMed] [Google Scholar]
- Griffith TM, Edwards DH, Lewis MJ, Newby AC, Henderson AH. The nature of endothelium-derived vascular relaxant factor. Nature. 1984;308:645–647. doi: 10.1038/308645a0. [DOI] [PubMed] [Google Scholar]
- Hecker M, Mulsch A, Bassenge E, Busse R. Vasoconstriction and increased flow: two principal mechanisms of shear stress-dependent endothelial autacoid release. Am J Physiol. 1993;265:H828–H833. doi: 10.1152/ajpheart.1993.265.3.H828. [DOI] [PubMed] [Google Scholar]
- Hunter A. Applied Mathematics. Cork: University College Cork; 2003. Analysis of blood flow in large arteries using Fourier techniques; p. 152. [Google Scholar]
- Kartamyshev SP, Balashov SA, Melkumyants AM. Role of endothelium sensitivity to shear stress in noradrenaline-induced constriction of feline femoral arterial bed under constant flow and constant pressure perfusions. J Vasc Res. 2007;44:1–10. doi: 10.1159/000097750. [DOI] [PubMed] [Google Scholar]
- Kelly R, O'Hora T, Noble MIM, Snow HM. Res Cardiovasc Sci. Epub; 2006a. Antidromic signalling does not contribute to the vasodilatory response of the iliac artery to acetylcholine in the anaesthetised pig. [Google Scholar]
- Kelly R, Ruane-O'Hora T, Noble MI, Drake-Holland AJ, Snow HM. Differential inhibition by hyperglycaemia of shear stress- but not acetylcholine-mediated dilatation in the iliac artery of the anaesthetized pig. J Physiol. 2006b;573:133–145. doi: 10.1113/jphysiol.2006.106500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly R, Snow H. Effects of mean and pulsatile wall shear stress on the diameter of the iliac artery in the anaesthetised pig. pA2. 2005;3 [Google Scholar]
- Kharitonov VG, Russwurm M, Magde D, Sharma VS, Koesling D. Dissociation of nitric oxide from soluble guanylate cyclase. Biochem Biophys Res Commun. 1997;239:284–286. doi: 10.1006/bbrc.1997.7470. [DOI] [PubMed] [Google Scholar]
- Markos F, Hennessy BA, Fitzpatrick M, O'Sullivan J, Snow HM. The effect of tezosentan, a non-selective endothelin receptor antagonist, on shear stress-induced changes in arterial diameter of the anaesthetized dog. J Physiol. 2002;544:913–918. doi: 10.1113/jphysiol.2002.030478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melkumyants AM, Balashov SA, Khayutin VM. Endothelium dependent control of arterial diameter by blood viscosity. Cardiovasc Res. 1989;23:741–747. doi: 10.1093/cvr/23.9.741. [DOI] [PubMed] [Google Scholar]
- Mochizuki S, Vink H, Hiramatsu O, Kajita T, Shigeto F, Spaan JA, Kajiya F. Role of hyaluronic acid glycosaminoglycans in shear-induced endothelium-derived nitric oxide release. Am J Physiol Heart Circ Physiol. 2003;285:H722–H726. doi: 10.1152/ajpheart.00691.2002. [DOI] [PubMed] [Google Scholar]
- Moore JE, Jr, Xu C, Glagov S, Zarins CK, Ku DN. Fluid wall shear stress measurements in a model of the human abdominal aorta: oscillatory behavior and relationship to atherosclerosis. Atherosclerosis. 1994;110:225–240. doi: 10.1016/0021-9150(94)90207-0. [DOI] [PubMed] [Google Scholar]
- Parent R, Lavallee M. Endothelin-dependent effects limit flow-induced dilation of conductance coronary vessels after blockade of nitric oxide formation in conscious dogs. Cardiovasc Res. 2000;45:470–477. doi: 10.1016/s0008-6363(99)00366-1. [DOI] [PubMed] [Google Scholar]
- Pedley T. The Fluid Mechanics of Large Blood Vessels. Cambridge: Cambridge University Press; 1980. [Google Scholar]
- Poucher SM. The effect of NG-nitro-l-arginine methyl ester upon hindlimb blood flow responses to muscle contraction in the anaesthetized cat. Exp Physiol. 1995;80:237–247. doi: 10.1113/expphysiol.1995.sp003843. [DOI] [PubMed] [Google Scholar]
- Pries AR, Reglin B, Secomb TW. Remodeling of blood vessels: responses of diameter and wall thickness to hemodynamic and metabolic stimuli. Hypertension. 2005;46:725–731. doi: 10.1161/01.HYP.0000184428.16429.be. [DOI] [PubMed] [Google Scholar]
- Pyke KE, Tschakovsky ME. The relationship between shear stress and flow-mediated dilatation: implications for the assessment of endothelial function. J Physiol. 2005;568:357–369. doi: 10.1113/jphysiol.2005.089755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu Y, Tarbell JM. Numerical simulation of pulsatile flow in a compliant curved tube model of a coronary artery. J Biomech Eng. 2000;122:77–85. doi: 10.1115/1.429629. [DOI] [PubMed] [Google Scholar]
- Rembold CM, Foster DB, Strauss JD, Wingard CJ, Eyk JE. cGMP-mediated phosphorylation of heat shock protein 20 may cause smooth muscle relaxation without myosin light chain dephosphorylation in swine carotid artery. J Physiol. 2000;524:865–878. doi: 10.1111/j.1469-7793.2000.00865.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russwurm M, Koesling D. Isoforms of NO-sensitive guanylyl cyclase. Mol Cell Biochem. 2002;230:159–164. [PubMed] [Google Scholar]
- Schretzenmayr A. Uber kreislaufregulatorische Vorgange an den grossen Arterien bei der Muskelarbeit. Pflugers Arch Ges Physiol. 1933;232:743–748. [Google Scholar]
- Snow HM. Conway Review Lecture. Atheroma and the mechanics of blood flow in arteries. Ir J Med Sci. 2002;171:165–169. doi: 10.1007/BF03170508. [DOI] [PubMed] [Google Scholar]
- Snow HM, McAuliffe SJ, Moors JA, Brownlie R. The relationship between blood flow and diameter in the iliac artery of the anaesthetized dog: the role of endothelium-derived relaxing factor and shear stress. Exp Physiol. 1994;79:635–645. doi: 10.1113/expphysiol.1994.sp003796. [DOI] [PubMed] [Google Scholar]
- Snow HM, Markos F, O'Regan D, Pollock K. Characteristics of arterial wall shear stress which cause endothelium-dependent vasodilatation in the anaesthetized dog. J Physiol. 2001;531:843–848. doi: 10.1111/j.1469-7793.2001.0843h.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorop O, Spaan JA, Sweeney TE, VanBavel E. Effect of steady versus oscillating flow on porcine coronary arterioles: involvement of NO and superoxide anion. Circ Res. 2003;92:1344–1351. doi: 10.1161/01.RES.0000078604.47063.2B. [DOI] [PubMed] [Google Scholar]
- Squire JM, Chew M, Nneji G, Neal C, Barry J, Michel C. Quasi-periodic substructure in the microvessel endothelial glycocalyx: a possible explanation for molecular filtering? J Struct Biol. 2001;136:239–255. doi: 10.1006/jsbi.2002.4441. [DOI] [PubMed] [Google Scholar]
- Sun D, Huang A, Sharma S, Koller A, Kaley G. Endothelial microtubule disruption blocks flow-dependent dilation of arterioles. Am J Physiol Heart Circ Physiol. 2001;280:H2087–H2093. doi: 10.1152/ajpheart.2001.280.5.H2087. [DOI] [PubMed] [Google Scholar]
- Thi MM, Tarbell JM, Weinbaum S, Spray DC. The role of the glycocalyx in reorganization of the actin cytoskeleton under fluid shear stress: a ‘bumper-car’ model. Proc Natl Acad Sci U S A. 2004;101:16483–16488. doi: 10.1073/pnas.0407474101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Berg BM, Nieuwdorp M, Stroes ES, Vink H. Glycocalyx and endothelial (dys) function: from mice to men. Pharmacol Rep. 2006b;58(Suppl.):75–80. [PubMed] [Google Scholar]
- van den Berg BM, Spaan JA, Rolf TM, Vink H. Atherogenic region and diet diminish gycocalyx dimension and increase intima-to-media ratios at murione carotid artey bifurcation. Am J Physiol Heart Circ Physiol. 2006a;290:H915–H920. doi: 10.1152/ajpheart.00051.2005. [DOI] [PubMed] [Google Scholar]
- Weinbaum S, Zhang X, Han Y, Vink H, Cowin SC. Mechanotransduction and flow across the endothelial glycocalyx. Proc Natl Acad Sci U S A. 2003;100:7988–7995. doi: 10.1073/pnas.1332808100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziegler T, Bouzourene K, Harrison VJ, Brunner HR, Hayoz D. Influence of oscillatory and unidirectional flow environments on the expression of endothelin and nitric oxide synthase in cultured endothelial cells. Arterioscler Thromb Vasc Biol. 1998;18:686–692. doi: 10.1161/01.atv.18.5.686. [DOI] [PubMed] [Google Scholar]