

Abstract
Animal and tissue studies have indicated that the carotid bodies are sensitive to glucose concentrations within the physiological range. This glucose sensitivity may modulate the ventilatory response to hypoxia, with hyperglycaemia suppressing the hypoxic response and hypoglycaemia stimulating it. This study was designed to determine whether hypo- and hyperglycaemia modulate the hypoxic ventilatory response in humans. In 11 normal research participants, glucose levels were clamped at 2.8 and 11.2 mmol l−1 for 30 min. At the start and end of each clamp, blood was drawn for hormone measurement and the isocapnic hypoxic ventilatory response was measured. Because generation of reactive oxygen species may be a common pathway for the interaction between glucose and oxygen levels, the experiments were repeated with and without pretreatment for 1 week with vitamins C and E. Hypoglycaemia caused an increase in the counter-regulatory hormones, a 54% increase in isocapnic ventilation, and a 108% increase in the hypoxic ventilatory response. By contrast, hyperglycaemia resulted in small but significant increases in both ventilation and the hypoxic ventilatory response. Antioxidant vitamin pretreatment altered neither response. In conclusion, the stimulant effect of hypoglycaemia on the hypoxic ventilatory response is consistent with a direct effect on the carotid body, but an indirect effect through the activation of the counter-regulatory response cannot be excluded. The mechanisms behind the mild stimulating effect of hyperglycaemia remain to be elucidated.
Oxygen and glucose are the two critical substrates for cellular metabolism that need to be delivered to the cells. When faced with a lack of external sources of either substrate (e.g. high altitude or starvation) or with increased demands (e.g. exercise), the autonomic nervous system is immediately activated and over a longer time adaptive changes take place. For glucose, considerable internal stores can be mobilized to maintain blood glucose levels; however for oxygen, the lack of internal stores means that mechanisms to transport oxygen from the environment must be quickly increased. Thus, although both hypoxia and hypoglycaemia activate the autonomic nervous system, differences in the patterns of activation are apparent. This commonality presents possibilities for interactions between glucose and oxygen regulation that may occur at multiple levels within the autonomic nervous system and within the cells utilizing the substrates.
The carotid bodies are multimodal arterial chemoreceptors responding to changes in oxygen and carbon dioxide tensions (Andronikou et al. 1988), potassium concentrations (Linton & Band, 1990) and osmolarity (Molnar et al. 2003); for a recent review see also Kumar & Bin-Jaliah (2007). The type 1 cells within the carotid bodies are thought to be involved in all these responses, which activate the autonomic nervous system and primarily increase ventilation. Recently, a response to changes in glucose level has also been observed in type 1 cells in a thin-slice carotid body preparation (Pardal & Lopez-Barneo, 2002; Lopez-Barneo, 2003) and in petrosal neurons co-cultured with type 1 cells (Nurse, 2005; Zhang et al. 2007).
While there are many glucose response elements involved in the regulatory response to changes in glucose level (Levin et al. 2004), the physiological role of the carotid bodies in glucose regulation has not been fully explored (Koyama et al. 2000). However, the pathway from oxygen sensing to an increase in ventilation has been intensively studied. Whereas there is still considerable controversy about the details of the oxygen signal transduction process leading to an increase in the carotid sinus nerve firing rate (and thus an increase in ventilation), there is an emerging consensus about many of the steps involved (Gonzalez et al. 1994; Prabhakar, 2001; Lopez-Barneo et al. 2001). Decreased tissue O2 tension causes membrane depolarization in carotid body type 1 cells through closure (decreasing the open probability) of oxygen-sensitive potassium channels (Peers, 1997; Buckler et al. 2000; Sanchez et al. 2002). The membrane depolarization causes an influx of Ca2+ through voltage-sensitive Ca2+ channels (Andronikou et al. 1988; Urena et al. 1994; Lopez-Barneo et al. 1999). This increase in intracellular Ca2+ concentration causes an exocytosis of neurotransmitters, currently thought to be the co-release of ATP and acetylcholine, that excite the postsynaptic receptors in the afferent endings of the carotid sinus nerve (Nurse, 2005).
The transduction mechanism whereby hypoxia closes the K+ channel has been an area of intense focus with respect to the carotid body. The generation of reactive oxygen species (ROS) has been viewed as a key link between O2 partial pressure and K+ channels (Kourie, 1998; Patel & Honoré, 2001). The term ROS refers to free radical as well as non-radical compounds that can cause oxidation, including singlet oxygen, superoxide radical, hydroxyl radicals and hydrogen peroxide. The superoxide radical can be generated in the mitochondria when oxygen is not completely reduced to water, or by various NAD(P)H oxidases (Droge, 2002), and superoxide dismutase converts superoxide to hydrogen peroxide enzymatically. While ROS generation is a key mechanism for the cellular toxicity of both high glucose and oxygen, it is also a key common intracellular messenger that may underlie the physiological signalling for both oxygen and glucose (Droge, 2002; Gonzalez et al. 2002). Most of the regulatory effects of ROS are not mediated by superoxide directly but rather through an ROS derivative.
Strong interactions between the stimuli that excite the carotid bodies have been found, with the most studied being the oxygen–carbon dioxide interaction (Lahiri & Delaney, 1975). However, the sites where the interactions take place within the carotid body signal transduction sequence are not well known. The data from thin-slice tissue cultures would also indicate a strong oxygen–glucose interaction (Pardal & Lopez-Barneo, 2002), but Lopez-Barneo (2003) has also postulated that the interaction takes place at the K+ channel and thus is additive. However, in a rat model, Bin-Jaliah et al. (2004, 2005) found that hypoglycaemia stimulated ventilation and the hypercapnic ventilatory response, although without an increase in the carotid body firing rate. In a recent study, Zhang et al. 2007) found that the carotid body receptors are direct ‘glucosensors’ within the physiological range of both glucose and oxygen.
This study was designed to determine the effect of hypoglycaemia and hyperglycaemia on the hypoxic ventilatory response (HVR) in humans. Because of the potential role of ROS in both glucose and oxygen sensing, the effect of pretreatment with antioxidant vitamins was also studied. In addition, it has been reported that antioxidant vitamin pretreatment blocked the depressant effect of halothane on the HVR (Teppema et al. 2002). Some of the results of these studies have been previously reported in abstract form (Ward, 2006a,b,c).
Methods
The study was completed at the University of Rochester General Clinical Research Center (GCRC) and approved by the local institutional review board. The studies conformed to the standards set by the Declaration of Helsinki and all subjects gave written, informed consent. All subjects were free of significant cardiopulmonary disease and took no medications.
The subjects initially came to the centre for screening tests (urinalysis, haemoglobin level, vitamin levels, ECG and pregnancy test if indicated) and for a practice hypoxic breathing test. They were instructed regarding a standard diet and asked not to make substantial changes in their diets during the study. Each subject was then scheduled for four experimental days. The four arms of the protocol were hypo- and hyperglycaemic clamps, each with antioxidant vitamin or placebo pretreatment. At least 2 weeks were scheduled between experimental days. The sequence of the four experiments was randomized, except that sequences which required adjacent experiments with antioxidant pretreatment were not used, in order to eliminate possible accumulation of the vitamins. No sequence was repeated. The subjects were admitted to the GCRC the evening prior to the experiment. At 07.00 h on the morning of the experiment, two intravenous lines were used, one for insulin and glucose infusion and the other for blood sampling. All infusions were made with a pump (Alaris Medley Pump, Module 8100 Series; Alaris Medley Medical Systems, San Diego, CA, USA). The catheter for blood sampling was placed in the dorsum of the hand in retrograde fashion and was flushed with a small amount of dilute heparin solution after a sample was drawn. The hand was then placed in a clear plastic box warmer thermostatically controlled at 46–47°C to arterialize the venous blood for sampling. The subjects remained in bed for the duration of the experiment (except for bathroom visits as needed) with the head elevated approximately 30–45 deg. In between breathing runs, subjects were allowed to sleep, read or watch television; during the breathing runs, subjects were told to keep their eyes open and most watched television. After the final experimental measurements, the subjects were given a meal, intravenous catheters were removed, and the subjects were discharged home. A follow-up phone call was made the next day.
Anti-oxidant treatment
Vitamins E and C were used as antioxidants. Each research participant was given a 7-day supply of either the vitamins or placebo, packaged in identical capsules. Vitamin C (250 mg; Goldline Laboratories, Ft Lauderdale, FL, USA) and vitamin E (200 mg; Major Pharmaceuticals, Apotex Corp., Livonia, MI, USA) were taken at 07.00 and 19.00 h. The final doses were given on the morning of the experiment.
Glucose clamp
The target glucose levels were 2.8 mmol l−1 for the hypoglycaemia clamps (Mitrakou et al. 1991) and 11.2 mmol l−1 for the hyperglycaemia clamps (DeFronzo et al. 1979). For the hypoglycaemia clamps, following baseline measurement of ventilation and the HVR and after blood was drawn for hormone measurement, an insulin infusion was begun at 1 mU kg −1min−1. An infusion of 25% dextrose was adjusted, as needed, to maintain a glucose level of 2.8 mmol l−1 at 45 min; this glucose level was held for another 15 min prior to the measurements. The glucose level was measured at the bedside every 5 min (Beckman Glucose Analyser 2; Beckman Coulter Inc., Brea, CA, USA).
The insulin infusion was increased only if the target hypoglycaemia was not reached even after the glucose infusion had been titrated to zero. After blood samples and ventilatory measurements were made 30 min apart, the insulin infusion was terminated and the glucose infusion was adjusted to achieve euglycaemia in approximately 1 h.
The hyperglycaemic protocol was similar, except that no insulin was infused and the 25% dextrose infusion was adjusted to maintain a blood glucose level of 11.2 mmol l−1. Some subjects had an initial bolus of 25% glucose (0.8 ml kg−1), but most only had the infusion started at an initial rate of 1.5 ml kg −1min−1 and then adjusted as needed.
Hormone measurements
Blood samples for hormone measurements were taken prior to the start of the clamp (Pre-Clamp), at the time the clamp was stabilized at approximately 1 h after the start of the insulin or glucose infusion (Start-Clamp), after clamping the glucose for 30 min (End-Clamp) and approximately 1 h after the termination of the clamp when the glucose level returned to normal (Post-Clamp). These blood samples were obtained prior to measuring the HVR. At the times these samples were drawn, the subjects rated their symptoms using a previously published rating scale (Mitrakou et al. 1991). The symptoms were divided into those related to the autonomic response (autonomic symptoms) and those related to central nervous system (CNS) glucose deprivation (neuroglycopenic symptoms).
Measurements of insulin, glucagon, cortisol, adrenaline and noradrenaline levels were made on each of these four samples. In addition, on the Start-Clamp sample, vitamins E and C levels were measured.
Vitamin C levels were determined by spectrophotometer (ZeptoMetrix Oxidative Stress Tests Services, Buffalo, NY, USA). Vitamin E levels were determined by high-performance liquid chromatography using a reverse-phase column and ultraviolet absorbance detection (University of Rochester Medical Center Clinical Laboratories, Rochester, NY, USA). The glucagon, insulin and cortisol determinations were made in the GCRC laboratory. Glucagon and insulin levels were measured using the appropriate double-antibody radioimmunoassay kit (Linco Research Inc., St Charles, MO, USA). Cortisol was measured with a coated-tube radioimmunoassay kit (Diagnostic Systems Laboratories Inc., Webster, TX, USA). Adrenaline and noradrenaline were determined by Kat-Combi radioimmunoassay kit (KMI Diagnostics, Minneapolis, MN, USA).
Ventilatory measurements
The techniques for ventilatory measurements have been described in detail previously (Cartwright et al. 1998). Briefly, subjects breathed from a gas-mixing chamber via a face mask (Vital Signs, Totawa, NJ, USA). Inhaled and exhaled volumes were measured with a bi-directional impeller flow meter (VMM 110; Interface Associates, Laguna Niguel, CA, USA). Airway gases were sampled continuously by a combined infrared (CO2) and paramagnetic (O2) analyser (Gemini Respiratory Monitor; CWE Inc., Ardmore, PA, USA) or a mass spectrometer (MGA 110; Perkin-Elmer, Pomona, CA, USA) in some experiments. Heart rate and saturation were obtained from a pulse oximeter (Model 3900; Datex-Ohmeda Inc., Madison, WI, USA). Subjects were continuously monitored with an ECG for safety. Computer-driven, high-flow (total flow of 60 l min−1), mass-flow controllers (Omega Engineering, Inc, Stamford, CT, USA) provided N2, CO2 and O2 to the gas mixing chamber at desired concentrations on a breath-by-breath basis. Ventilation (l min−1), tidal volume (l), breathing frequency (breaths min−1), end-tidal gas concentrations (end-tidal partial pressure of O2 (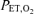 ) and CO2 (
) and CO2 (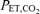 ), mmHg), haemoglobin oxygen saturation by pulse oximetry (%) and heart rate (beats min−1) were determined and data were collected using the TIDAL software package running under the LINUX operating system (Jenkins et al. 1989).
), mmHg), haemoglobin oxygen saturation by pulse oximetry (%) and heart rate (beats min−1) were determined and data were collected using the TIDAL software package running under the LINUX operating system (Jenkins et al. 1989).
To study the ventilatory response to isocapnic hypoxia (HVR), a computer-driven dynamic end-tidal forcing technique was utilized (Cartwright et al. 1998). With this technique, the 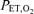 and
and 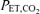 are dynamically forced to follow a prescribed pattern in time by manipulation of the inspired gas concentrations independent of the ventilatory response. In this study, step transitions in
are dynamically forced to follow a prescribed pattern in time by manipulation of the inspired gas concentrations independent of the ventilatory response. In this study, step transitions in 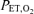 were used with a background of constant
were used with a background of constant 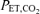 individually determined for each subject. The
individually determined for each subject. The 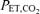 was determined by an initial 10 min breathing run while inhaling 1% CO2 for each subject on each experimental day. This permitted the setting of a control level of
was determined by an initial 10 min breathing run while inhaling 1% CO2 for each subject on each experimental day. This permitted the setting of a control level of  that would not cause too high a normoxic ventilation but yet could be determined by chemoreceptor drives. The
that would not cause too high a normoxic ventilation but yet could be determined by chemoreceptor drives. The  for all subsequent runs for that subject on that experimental day was set at 1–2 mmHg above the average
for all subsequent runs for that subject on that experimental day was set at 1–2 mmHg above the average  during this initial period. The HVR was determined by a 10 min breathing period which started with 250 s of normoxia (
during this initial period. The HVR was determined by a 10 min breathing period which started with 250 s of normoxia (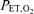 , 100 mmHg), followed by a step-wise decrease in
, 100 mmHg), followed by a step-wise decrease in  , with targets of 75.2, 64.0, 57.0, 52.0, 48.2 and 45 mmHg, to give equally spaced saturation values in 50 s intervals, except with the final
, with targets of 75.2, 64.0, 57.0, 52.0, 48.2 and 45 mmHg, to give equally spaced saturation values in 50 s intervals, except with the final 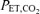 being held for 100 s using the protocol of Mou et al. (1995).
being held for 100 s using the protocol of Mou et al. (1995).
Data analysis and statistics
The HVR slope was determined by linear regression of the ventilation on the saturation calculated from the measured 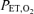 using the equations suggested by Severinghaus (1979). The breath-by-breath data were averaged over the last 20 s at each level of hypoxia, except for the first level (100 mmHg), where 120 s was used, and the last level (45 mmHg), where 40 s was used. The HVR was calculated by linear regression of the averaged ventilation on the averaged saturation for these seven intervals. The averaged ventilation, exhaled tidal volume, breathing frequency and heart rate for the initial isocapnic normoxic period were also analysed. Data are reported as means ±s.e.m. unless otherwise noted.
using the equations suggested by Severinghaus (1979). The breath-by-breath data were averaged over the last 20 s at each level of hypoxia, except for the first level (100 mmHg), where 120 s was used, and the last level (45 mmHg), where 40 s was used. The HVR was calculated by linear regression of the averaged ventilation on the averaged saturation for these seven intervals. The averaged ventilation, exhaled tidal volume, breathing frequency and heart rate for the initial isocapnic normoxic period were also analysed. Data are reported as means ±s.e.m. unless otherwise noted.
Statistical analysis was performed using STATA (Stata Corp., College Station, TX, USA). The measured vitamin levels were compared for control (measured during the initial screening visit), placebo and vitamin treatment using analysis of variance (ANOVA). Statistical comparisons were not made between the hyper- and hypoglycaemic clamps. Within each clamp, ANOVA was used with subject, drug (placebo or antioxidant), time (Pre-Clamp, Start-Clamp, End-Clamp and Post-Clamp) and drug–time interaction. If no drug effect was found, ANOVA was repeated with subject and time as the dependent variables and a possible time effect was determined by post hoc testing using the false discovery rate procedure with P < 0.05 used as the significant level (Curran-Everett, 2000). If the drug effect was found to be significant, subsequent testing for a time effect was performed on the placebo and antioxidant data separately.
Results
Eleven participants (five male and six female) enrolled and completed the protocol. Except for one subject who developed a superficial thrombosis in the vein that had received the 25% glucose bolus, there were no adverse reactions. That subject completed the protocol and for the other subjects the initial glucose bolus was deleted and the hyperglycaemic runs were initiated solely by beginning the 25% infusion. The average age and body mass index (±s.d.) were 27 ± 6 years and 25.6 ± 5.5 kg m2 (19.4–36.8), respectively.
No subject reported any problems with taking the vitamin capsules and all completed the treatments. The control (obtained at the screening visit) vitamin C level (57.0 ± 6.7 μmol l−1) was within the expected normal range (28.5–85.5 μmol l−1) and the vitamin E level (25.9 ± 3.3 μmol l−1) was slightly above the expected normal range (12–21.6 μmol l−1) in these healthy, non-smoking subjects. As expected, the placebo treatment did not cause any significant change in levels from control, whereas the administration of the active vitamins caused a significant increase in both vitamin levels (vitamin C, 82.1 ± 7.4 μmol l−1; vitamin E, 39.6 ± 3.0 μmol l−1; both values were significantly different from placebo, P < 0.05); however, the water-soluble vitamin C levels were still within the normal range.
The glucose clamps were successfully accomplished at the target values. Insulin levels increased via exogenous administration for the hypoglycaemic clamp and via endogenous response for the hyperglycaemic clamp (Fig. 1). Figure 2 shows a recording of the HVR and the regression analysis.
Figure 1. Glucose and insulin measurements for both the hypoglycaemic and the hyperglycaemic clamps.

No statistical difference was found between the placebo and vitamin treatments and the averages are given for both treatments combined. Statistical testing (see Methods) within the hypoglycaemic and hyperglycaemic clamps: *significantly different from Pre-Clamp value; +significantly different from Post-Clamp value; #significantly different from Start-Clamp value.
Figure 2. Example of hypoxic ventilatory response during hypoglycaemia.

The left panel shows the instantaneous measurement of airway gases and tidal volume. The end-tidal 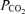 and
and 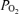 are controlled by manipulation of the inspired gas concentration. The right panel shows the ventilation versus saturation for each of the seven defined time intervals (see text) and the linear regression line.
are controlled by manipulation of the inspired gas concentration. The right panel shows the ventilation versus saturation for each of the seven defined time intervals (see text) and the linear regression line.
Overall the  was well controlled. There were no differences in either the normoxic (37.8 ± 2.9 mmHg) or hypoxic values (37.6 ± 2.8 mmHg) across the time periods or treatments. The somewhat large standard deviations resulted from the individualization of the
was well controlled. There were no differences in either the normoxic (37.8 ± 2.9 mmHg) or hypoxic values (37.6 ± 2.8 mmHg) across the time periods or treatments. The somewhat large standard deviations resulted from the individualization of the  used for each subject and not from any variation across multiple runs for an individual subject. Except as noted below, no drug effect was detected via ANOVA; thus the results are reported for both vitamin treatments combined.
used for each subject and not from any variation across multiple runs for an individual subject. Except as noted below, no drug effect was detected via ANOVA; thus the results are reported for both vitamin treatments combined.
The primary hypothesis of this study was that the glucose level would modulate the HVR. As predicted, the hypoglycaemic clamp greatly increased the HVR but unexpectedly, the HVR was also increased by the hyperglycaemic clamp (Fig. 3). For both the hyper- and hypoglycaemic clamps, the Post-Clamp measurement did not return completely to the initial Pre-Clamp value. For the hyperglycaemic clamp, the first measurement at the start of the clamp was not significantly greater than the Pre-Clamp value. For the hypoglycaemic clamp, both clamp measurements (Start-Clamp and End-Clamp) were increased above the Pre-Clamp measurement and were not different from each other.
Figure 3. Hypoxic ventilatory response for both the hypoglycaemic and hyperglycaemic clamps.

No statistical difference was found between the placebo and vitamin treatments and the averages are given for both treatments combined. Statistical testing (see Methods) within the hypoglycaemic and hyperglycaemic clamps: *significantly different from Pre-Clamp value; +significantly different from Post-Clamp value.
The normoxic ventilation was also increased by both clamps (Fig. 4), but the pattern of breathing was different. For the hypoglycaemic clamp, the increase in ventilation was caused primarily by an increase in breathing frequency, but for the hyperglycaemic clamp, there was primarily an increase in tidal volume. There was only a small increase in heart rate during both clamps; hypoglycaemic clamp: from 68.1 ± 7.2 beats min−1 Pre-Clamp to a maximum of 75.4 ± 9.9 beats min−1 (P < 0.05) in the End-Clamp period and returning to 66.1 ± 6.9 beats min−1 (n.s. from Pre-Clamp) in the Post-Clamp period; hyperglycaemic clamp: from 67.3 ± 6.6 beats min−1 Pre-Clamp to 71.4 ± 6.7 beats min−1 at the end of the clamp and then a non-significant further increase to 73.7 ± 7.3 beats min−1 in the Post-Clamp period.
Figure 4. Ventilation, tidal volume and breathing frequency averaged over the last 2 min of the normoxic period prior to the hypoxic periods for the four time periods in both the hypoglycaemic and hyperglycaemic clamps.

Ventilation showed a significant increase during the clamp periods for both hypoglycaemia and hyperglycaemia but the frequency was increased for hypoglycaemia and tidal volume was increased for hyperglycemia. Statistical testing (see Methods): *significantly different from Pre-Clamp value; +significantly different from Post-Clamp value.
The hypoglycaemic clamp elicited a brisk counter-regulatory response with increases in glucagon, adrenaline, noradrenaline and cortisol levels (Fig. 5). Glucagon and cortisol levels remained elevated in the Post-Clamp period. Both sets of reported symptoms (neuroglycopenic and autonomic) increased during the hypoglycaemic clamp (Fig. 6), whereas the neuroglycopenic symptoms remained elevated in the Post-Clamp period because of hunger (when hunger was removed from the neuroglycopenic symptoms, the Post-Clamp value was no longer significantly different from the Pre-Clamp value).
Figure 5. Hormonal response to hypoglycaemic and hyperglycaemic clamps.

Except for noradrenaline level during the hyperglycaemic clamp, there was no difference between the placebo and antioxidant treatments and the responses for the two treatments are shown averaged together. For noradrenaline, there was no effect of the clamp time period, but the values following vitamin treatment were significantly (P < 0.01) higher than for placebo (see Results). Statistical testing (see Methods): *significantly different from Pre-Clamp value; +significantly different from Post-Clamp value; #significantly different from Start-Clamp value.
Figure 6. Neuroglycopenic and autonomic symptoms scores Average of the symptom score.

shown as the symptoms related to an increase in autonomic activity and those related to neuroglycopenia (Mitrakou et al. 1991) for the hypoglycaemic clamp. Statistical testing (see Methods): *significantly different from Pre-Clamp value; +significantly different from Post-Clamp value.
The noradrenaline levels for the hyperglycaemic tests were significantly (P < 0.001) higher for vitamin pretreatment (vitamin pre-treatment, 867 ± 107 pmol l−1; placebo, 597 ± 75 pmol l−1), with no effects from the clamp period. The adrenaline levels were not changed by the hyperglycaemic clamp but the glucagon levels were suppressed (Fig. 5). Cortisol showed a monotonic decline from 414 ± 90 nmol l−1 in the Pre-Clamp period to 306 ± 54 nmol l−1 in the Post-Clamp period that did not reach statistical significance in the ANOVA for a clamp time period effect (P= 0.07). None of the symptoms showed a statistically significant increase.
Discussion
The potential for an interaction between glucose levels and ventilation has been studied in tissue cultures (Pardal & Lopez-Barneo, 2002; Nurse, 2005; Zhang et al. 2007) and animal preparations (Alvarez-Buylla & Alvarez-Buylla, 1988; Bin-Jaliah et al. 2004, 2005) but we have been unable to find any prior reports of the interaction between glucose and ventilation in humans. The study of interaction between glucose and oxygen has focused on the carotid bodies, because they are the primary stimulatory sensors for hypoxia. The observation that the carotid bodies are also sensitive to glucose has resulted in their designation as ‘metabolic sensors’ (Nurse, 2005). Recently Zhang et al. (2007) investigated specific mechanisms of glucose sensing in the rat carotid body in vitro. Their results indicate a sensitivity to glucose changes within the physiological range and an effect that was additive to hypoxic stimulation. It is interesting that in their experiments the carotid body type 1 cell membrane depolarization during hypoglycaemia was associated with a decrease in cell input resistance in contrast to the increase seen during hypoxia. However, this metabolic sensitivity in vitro may not translate to a physiological or clinical importance in vivo. In studying the effects of both hypo- and hyperglycaemia on the HVR in humans, clear interactions have been demonstrated which differ in certain respects from those predicted by the tissue and animal studies.
Hypoglycaemia
The hormonal and symptom responses to the induced hypoglycaemia found in this study are similar to those previously reported (Mitrakou et al. 1991; Porcellati et al. 2003). The brisk counter-regulatory response triggered by the hypoglycaemia is the result of autonomic nervous system activation and attempts to return the glucose levels to normal.
While hypoglycaemia clearly stimulated resting ventilation and greatly increased the HVR, this could be either a direct or an indirect stimulation. A direct stimulation would be consistent with the tissue culture experiments of Pardal & Lopez-Barneo (2002), Nurse (2005) and Zhang et al. (2007). Other experiments have shown that the carotid bodies in animals are responsive to hypoglycaemia and play a role in the development of the counter-regulatory response (Koyama et al. 2000; Montero et al. 2006). This would most probably imply an interaction between glucose and the hypoxic sensitivity at the carotid bodies because, although not extensively studied, no selectivity to carotid body activation has been found in the carotid sinus nerve fibres (Vidruk et al. 2001). It is important to note that older studies of the carotid bodies have emphasized the need for sufficient substrate (e.g. glucose) to be available for adequate chemoresponsiveness of the cells (Spergel et al. 1992), while other studies have found an increasing responsiveness with decreasing intracellular ATP levels (Obeso et al. 1986). Regardless, the in vivo levels of glucose obtained in these experiments would not imply a lack of metabolic substrate sufficient to alter cell function.
In rats, Bin-Jaliah et al. (2004, 2005) found that hypoglycaemia stimulated ventilation and increased the hyperoxic hypercapnic ventilatory response and that these effects were eliminated by carotid sinus section. However, when the carotid sinus nerve firing rate was measured in vitro, no increase in firing rate was seen when the glucose level in the superfusate was lowered (Bin-Jaliah et al. 2004). These authors concluded that while hypoglycaemia stimulated the carotid bodies, this effect was due to a metabolic factor other than glucose. Although not measured in these experiments, the autonomic response to hypoglycaemia could provide this indirect link.
Weissman et al. (1986) gave human subjects adrenaline, cortisol and glucagon by infusion according to the protocol developed by Shamoon et al. (1981). Although Weissman et al. (1986) did not measure hormone plasma levels, the levels measured by Shamoon et al. (1981) were comparable to the levels measured in this study. Resting ventilation was increased by approximately 30%, primarily via an increase in tidal volume and not in breathing frequency, and caused a mild hypocapnia. In the current study we found a 54% increase in ventilation, but with the increase due to a different pattern of breathing: increased frequency and not tidal volume. Additionally, in the current study, the increase in ventilation was measured isocapnically, which could account for some of the increased effect. It is also important to note that Shamoon et al. (1981) found that the infusion of all three hormones caused a marked hyperglycaemia (glucose of approximately 11 mmol l−1 after 2 h) and a mild hyperinsulinaemia (approximately 30 μU ml−1), in contrast to the hypoglycaemia and marked hyperinsulinaemia in this protocol. Although other explanations are also possible, the difference in the breathing pattern and the larger increase in ventilation may be caused by direct carotid body stimulation by hypoglycaemia. The HVR was not measured by Weissman et al. (1986), but they did find that the hyperoxic hypercapnic response was shifted to the left without an increase in slope. This is in contrast to the findings of Bin-Jaliah et al. (2005) that hypoglycaemia increased both the metabolic rate and the slope of the ventilatory response to carbon dioxide.
Other studies have investigated the effects of catecholamine infusions on the HVR. Heistad et al. (1972) observed that noradrenaline and isoprenaline (isoproterenol; a β-adrenergic agonist) each increased the hypoxic sensitivity by approximately 20% (estimated from their reported aggregate data). Interestingly, Linton et al. (1992) found that adrenaline increased the firing rate of the carotid sinus nerve in the anaesthetized cat, but only to a level that would have been predicted by the increase in arterial [K+] and metabolic rate. They concluded that adrenaline did not have a direct effect on the carotid chemoreceptors. In the present protocol, the arterial [K+] would be expected to decrease (Strakosch et al. 1976), resulting in a lessening of the carotid body sensitivity and thus not explaining the increase that was found.
Hyperglycaemia
Contrary to the predictions from in vitro studies (Pardal & Lopez-Barneo, 2002; Zhang et al. 2007), a decrease in HVR was not found during hyperglycaemia, but rather a mild stimulation was observed. The counter-regulatory response activated by the induced hyperglycaemia consisted of an increase in insulin (Fig. 1) and an increase in glucagon (Fig. 5). Cortisol also showed a slight, insignificant, slow decrease (Fig. 5) which could be the normal morning circadian decrease. Although not measured, somatostatin would be expected to increase and [K+] to decrease during the hyperglycaemic period, but both of these changes should reduce the HVR (Maxwell et al. 1986). It is possible that the glucose levels used in this protocol were not high enough to suppress the sensitivity of the carotid bodies to hypoxia.
Although based on in vivo studies, the interaction between hyperglycaemia and hypoxia was hypothesized to occur at the carotid bodies; this interaction could also occur within the central respiratory controller. There are glucose-excited neurons (neurons that increase their activity as ambient glucose levels increase) in many regions of the brain (Levin et al. 2002) including in the nucleus tractus solitarius (Mizuno & Oomura, 1984), which is known to be a primary site for the integration of respiratory stimuli (Paton et al. 1999).
Glucose sensing in CNS neurons is not fully understood (Levin et al. 2004) but ATP-dependent K+ (KATP) channels and glucokinase seem to be key elements in the transduction process (Levin et al. 2001; Dunn-Meynell et al. 2002). Although various K+ channels have been implicated in O2 sensitivity in the carotid bodies (Lopez-Barneo et al. 2004; Weir et al. 2005), KATP channels are not thought to be involved in the response to hypoxia because in the carotid bodies it occurs at an arterial  level still high enough to cause no reduction of intracellular ATP.
level still high enough to cause no reduction of intracellular ATP.
The slow increase in the HVR with hyperglycaemia and its persistence with the return to euglycaemia (Fig. 3) could imply a slow modification by glucose of the hypoxic sensing mechanisms. Although glucose and oxygen interact in the expression of several genes and in the stability of hypoxia-inducible factor 1α (Kietzmann et al. 2002; Catrina et al. 2004), these effects are likely to be too slow to explain our results. Hyperglycaemia has been postulated to induce an intracellular ‘pseudohypoxia’ with an increase in NADH/NAD+ even in normoxia (Williamson et al. 1993). Clarification of these hypotheses awaits mechanistic studies in reduced preparations.
Antioxidants
The interaction of inhalational anaesthetics with the HVR is thought to occur via ROS in the carotid body; this effect is blocked by antioxidant vitamins (Teppema et al. 2002). Although ROS have been shown to play significant roles in the mediating effects of both oxygen and glucose (Williamson et al. 1993), we found no effects of antioxidant vitamin pretreatment except for a significant increase in noradrenaline levels by the vitamin treatment in the hyperglycaemic clamp experiments. The hyperglycaemic periods did not affect the noradrenaline levels for either placebo or vitamin treatments (Fig. 5). As the Pre-Clamp periods for both the hypo- and hyperglycaemic clamps are identical, the effects of vitamin pretreatment on noradrenaline level were assessed on the combine data for the Pre-Clamp periods alone. This analysis did not show a significant difference in noradrenaline level (P= 0.24), so the difference found for the hyperglycaemic test alone is difficult to interpret.
Although oxygen and glucose are two vital metabolic substrates, apparently no prior studies have examined the effects of alterations in glucose levels on ventilation in humans. This study has demonstrated that hypoglycaemia is a potent ventilatory stimulant and increases the HVR. Although the carotid bodies are likely to be the site of action, the relative contribution of indirect stimulation via counter-regulatory hormones cannot be determined from this study. Hyperglycaemia did not suppress the HVR.
Acknowledgments
Assistance from Dr John Gerich, Linda Palmer and the nursing and laboratory staff of the GCRC is gratefully acknowledged. We wish to acknowledge the American Diabetes Association Clinical Research Grant (1-04-CR-37), University of Rochester Clinical Research Center (5 M01 RR00044 from the National Center for Research Resources, NIH) and the Department of Anaesthesiology for financial support.
References
- Alvarez-Buylla R, Alvarez-Buylla ER. Carotid sinus receptors participate in glucose homeostasis. Respir Physiol. 1988;72:347–359. doi: 10.1016/0034-5687(88)90093-x. [DOI] [PubMed] [Google Scholar]
- Andronikou S, Shirahata M, Mokashi A, Lahiri S. Carotid body chemoreceptor and ventilatory responses to sustained hypoxia and hypercapnia in the cat. Respir Physiol. 1988;72:361–374. doi: 10.1016/0034-5687(88)90094-1. [DOI] [PubMed] [Google Scholar]
- Bin-Jaliah I, Maskell PD, Kumar P. Indirect sensing of insulin-induced hypoglycaemia by the carotid body in the rat. J Physiol. 2004;556:255–266. doi: 10.1113/jphysiol.2003.058321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bin-Jaliah I, Maskell PD, Kumar P. Carbon dioxide sensitivity during hypoglycaemia-induced, elevated metabolism in the anaesthetized rat. J Physiol. 2005;563:883–893. doi: 10.1113/jphysiol.2004.080085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckler KJ, Williams BA, Honoré E. An oxygen-, acid- and anaesthetic-sensitive TASK-like background potassium channel in rat arterial chemorecptor cells. J Physiol. 2000;525:135–142. doi: 10.1111/j.1469-7793.2000.00135.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cartwright CR, Henson LC, Ward DS. Effects of alfentanil on the ventilatory response to sustained hypoxia. Anesthesiology. 1998;89:612–619. doi: 10.1097/00000542-199809000-00009. [DOI] [PubMed] [Google Scholar]
- Catrina SB, Okamoto K, Pereira T, Brismar K, Poellinger L. Hyperglycemia regulates hypoxia-inducible factor-1a protein stability and function. Diabetes. 2004;53:3226–3232. doi: 10.2337/diabetes.53.12.3226. [DOI] [PubMed] [Google Scholar]
- Curran-Everett D. Multiple comparisons: philosophies and illustrations. Am J Physiol Regul Integr Comp Physiol. 2000;279:R1–R8. doi: 10.1152/ajpregu.2000.279.1.R1. [DOI] [PubMed] [Google Scholar]
- DeFronzo RA, Tobin JD, Andres R. Glucose clamp technique: a method for quantifying insulin secretion and resistance. Am J Physiol Endocrinol Metab. 1979;237:E214–E223. doi: 10.1152/ajpendo.1979.237.3.E214. [DOI] [PubMed] [Google Scholar]
- Droge W. Free radicals in the physiological control of cell function. Physiol Rev. 2002;82:47–95. doi: 10.1152/physrev.00018.2001. [DOI] [PubMed] [Google Scholar]
- Dunn-Meynell AA, Routh VH, Kang L, Gaspers L, Levin BE. Glucokinase is the likely mediator of glucosensing in both glucose-excited and glucose-inhibited central neurons. Diabetes. 2002;51:2056–2065. doi: 10.2337/diabetes.51.7.2056. [DOI] [PubMed] [Google Scholar]
- Gonzalez C, Almaraz L, Obeso A, Rigual R. Carotid body chemoreceptors: from natural stimuli to sensory discharges. Physiol Rev. 1994;74:829–898. doi: 10.1152/physrev.1994.74.4.829. [DOI] [PubMed] [Google Scholar]
- Gonzalez C, Sanz-Alfayate G, Agapito MT, Gomez-Nino A, Rocher A, Obeso A. Significance of ROS in oxygen sensing in cell systems with sensitivity to physiological hypoxia. Respir Physiol Neurobiol. 2002;132:17–41. doi: 10.1016/s1569-9048(02)00047-2. [DOI] [PubMed] [Google Scholar]
- Heistad DD, Wheeler RC, Mark AL, Schmid PG, Abboud FM. Effects of adrenergic stimulation on ventilation in man. J Clin Invest. 1972;51:1469–1475. doi: 10.1172/JCI106943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenkins JS, Valcke CP, Ward DS. A programmable system for acquisition and reduction of respiratory physiology data. Ann Biomed Eng. 1989;17:93–108. doi: 10.1007/BF02364275. [DOI] [PubMed] [Google Scholar]
- Kietzmann T, Krones-Herzig A, Jungermann K. Signaling cross-talk between hypoxia and glucose via hypoxia-inducible factor 1 and glucose response elements. Biochem Pharmacol. 2002;64:903–911. doi: 10.1016/s0006-2952(02)01160-7. [DOI] [PubMed] [Google Scholar]
- Kourie JI. Interaction of reactive oxygen species with ion transport mechanisms. Am J Physiol Cell Physiol. 1998;275:C1–C24. doi: 10.1152/ajpcell.1998.275.1.C1. [DOI] [PubMed] [Google Scholar]
- Koyama Y, Coker RH, Stone EE, Lacy DB, Jabbour K, Williams PE, Wasserman DH. Evidence that carotid bodies play an important role in glucoregulation in vivo. Diabetes. 2000;49:1434–1442. doi: 10.2337/diabetes.49.9.1434. [DOI] [PubMed] [Google Scholar]
- Kumar P, Bin-Jaliah I. Adequate stimuli of the carotid body: more than an oxygen sensor? Respir Physiol Neurobiol. 2007 doi: 10.1016/j.resp.2007.01.007. in press. [DOI] [PubMed] [Google Scholar]
- Lahiri S, Delaney RG. Stimulus interaction in the responses of carotid body chemoreceptor single afferent fibers. Respir Physiol. 1975;24:249–266. doi: 10.1016/0034-5687(75)90017-1. [DOI] [PubMed] [Google Scholar]
- Levin BE, Dunn-Meynell AA, Routh VH. Brain glucosensing and the KATP channel. Nat Neurosci. 2001;4:459–460. doi: 10.1038/87405. [DOI] [PubMed] [Google Scholar]
- Levin BE, Dunn-Meynell AA, Routh VH. CNS sensing and regulation of peripheral glucose levels. Int Rev Neurobiol. 2002;51:219–258. doi: 10.1016/s0074-7742(02)51007-2. [DOI] [PubMed] [Google Scholar]
- Levin BE, Routh VH, Kang L, Sanders NM, Dunn-Meynell AA. Neuronal glucosensing: what do we know after 50 years? Diabetes. 2004;53:2521–2528. doi: 10.2337/diabetes.53.10.2521. [DOI] [PubMed] [Google Scholar]
- Linton RA, Band DM. Potassium and breathing. News Physiol Sci. 1990;5:104–107. [Google Scholar]
- Linton RA, Band DM, Wolff CB. Carotid chemoreceptor discharge during epinephrine infusion in anesthetized cats. J Appl Physiol. 1992;73:2420–2424. doi: 10.1152/jappl.1992.73.6.2420. [DOI] [PubMed] [Google Scholar]
- Lopez-Barneo J. Oxygen and glucose sensing by carotid body glomus cells. Curr Opin Neurobiol. 2003;13:493–499. doi: 10.1016/s0959-4388(03)00093-x. [DOI] [PubMed] [Google Scholar]
- Lopez-Barneo J, del Toro R, Levitsky KL, Chiara MD, Ortega-Saenz P. Regulation of oxygen sensing by ion channels. J Appl Physiol. 2004;96:1187–1195. doi: 10.1152/japplphysiol.00929.2003. [DOI] [PubMed] [Google Scholar]
- Lopez-Barneo J, Pardal R, Montoro RJ, Smani T, Garcia-Hirschfeld J, Urena J. K+ and Ca2+ channel activity and cytosolic [Ca2+] in oxygen-sensing tissues. Respir Physiol. 1999;115:215–227. doi: 10.1016/s0034-5687(99)00016-x. [DOI] [PubMed] [Google Scholar]
- Lopez-Barneo J, Pardal R, Ortega-Saenz P. Cellular mechanisms of oxygen sensing. Annu Rev Physiol. 2001;63:259–287. doi: 10.1146/annurev.physiol.63.1.259. [DOI] [PubMed] [Google Scholar]
- Maxwell DL, Chahal P, Nolop KB, Hughes JMB. Somatostatin inhibits the ventilatory response to hypoxia in humans. J Appl Physiol. 1986;60:997–1002. doi: 10.1152/jappl.1986.60.3.997. [DOI] [PubMed] [Google Scholar]
- Mitrakou A, Ryan C, Veneman T, Mokan M, Jenssen T, Kiss I, Durrant J, Cryer P, Gerich J. Hierarchy of glycemic thresholds for counterregulatory hormone secretion, symptoms, and cerebral dysfunction. Am J Physiol Endocrinol Metab. 1991;260:E67–E74. doi: 10.1152/ajpendo.1991.260.1.E67. [DOI] [PubMed] [Google Scholar]
- Mizuno Y, Oomura Y. Glucose responding neurons in the nucleus tractus solitarius of the rat: in vitro study. Brain Res. 1984;307:109–116. doi: 10.1016/0006-8993(84)90466-9. [DOI] [PubMed] [Google Scholar]
- Molnar Z, Petheo GL, Fulop C, Spat A. Effects of osmotic changes on the chemoreceptor cell of rat carotid body. J Physiol. 2003;546:471–481. doi: 10.1113/jphysiol.2002.024125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montero SA, Cadenas JL, Lemus M, Roces de Alvarez-Buylla E, Alvarez-Buylla R. Nitric oxide in brain glucose retention after carotid body receptors stimulation with cyanide in rats. Adv Exp Med Biol. 2006;580:293–300. doi: 10.1007/0-387-31311-7_45. [DOI] [PubMed] [Google Scholar]
- Mou XB, Howard LS, Robbins PA. A protocol for determining the shape of the ventilatory response to hypoxia in humans. Respir Physiol. 1995;101:139–143. doi: 10.1016/0034-5687(95)00027-b. [DOI] [PubMed] [Google Scholar]
- Nurse CA. Neurotransmission and neuromodulation in the chemosensory carotid body. Auton Neurosci. 2005;120:1–9. doi: 10.1016/j.autneu.2005.04.008. [DOI] [PubMed] [Google Scholar]
- Obeso A, Almaraz L, Gonzalez C. Effects of 2-deoxy-D-glucose on in vitro cat carotid body. Brain Res. 1986;371:25–36. doi: 10.1016/0006-8993(86)90806-1. [DOI] [PubMed] [Google Scholar]
- Pardal R, Lopez-Barneo J. Low glucose-sensing cells in the carotid body. Nat Neurosci. 2002;5:197–198. doi: 10.1038/nn812. [DOI] [PubMed] [Google Scholar]
- Patel AJ, Honoré E. Molecular physiology of oxygen-sensitive potassium channels. Eur Respir J. 2001;18:221–227. doi: 10.1183/09031936.01.00204001. [DOI] [PubMed] [Google Scholar]
- Paton JF, Li YW, Kasparov S. Reflex response and convergence of pharyngoesophageal and peripheral chemoreceptors in the nucleus of the solitary tract. Neuroscience. 1999;93:143–154. doi: 10.1016/s0306-4522(99)00098-6. [DOI] [PubMed] [Google Scholar]
- Peers C. Oxygen-sensitive ion channels. Trends Pharmacol Sci. 1997;18:405–408. doi: 10.1016/s0165-6147(97)01120-6. [DOI] [PubMed] [Google Scholar]
- Porcellati F, Pampanelli S, Rossetti P, Cordoni C, Marzotti S, Scionti L, Bolli GB, Fanelli CG. Counterregulatory hormone and symptom responses to insulin-induced hypoglycemia in the postprandial state in humans. Diabetes. 2003;52:2774–2783. doi: 10.2337/diabetes.52.11.2774. [DOI] [PubMed] [Google Scholar]
- Prabhakar NR. Oxygen sensing during intermittent hypoxia: cellular and molecular mechanisms. J Appl Physiol. 2001;90:1986–1994. doi: 10.1152/jappl.2001.90.5.1986. [DOI] [PubMed] [Google Scholar]
- Sanchez D, Lopez-Lopez JR, Perez-Garcia MT, Sanz-Alfayate G, Obeso A, Ganfornina MD, Gonzalez C. Molecular identification of Kv alpha subunits that contribute to the oxygen-sensitive K+ current of chemoreceptor cells of the rabbit carotid body. J Physiol. 2002;542:369–382. doi: 10.1113/jphysiol.2002.018382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Severinghaus JW. Simple, accurate equations for human blood O2 dissociation computations. J Appl Physiol. 1979;46:599–602. doi: 10.1152/jappl.1979.46.3.599. [DOI] [PubMed] [Google Scholar]
- Shamoon H, Hendler R, Sherwin RS. Synergistic interactions among antiinsulin hormones in the pathogenesis of stress hyperglycemia in humans. J Clin Endocrinol Metab. 1981;52:1235–1241. doi: 10.1210/jcem-52-6-1235. [DOI] [PubMed] [Google Scholar]
- Spergel D, Lahiri S, Wilson DF. Dependence of carotid chemosensory responses on metabolic substrates. Brain Res. 1992;596:80–88. doi: 10.1016/0006-8993(92)91535-m. [DOI] [PubMed] [Google Scholar]
- Strakosch CR, Stiel JN, Gyory AZ. Hypokalaemia occurring during insulin-induced hypoglycaemia. Aust N Z J Med. 1976;6:314–316. doi: 10.1111/imj.1976.6.4.314. [DOI] [PubMed] [Google Scholar]
- Teppema L, Nieuwenhuijs D, Sarton E, Romberg R, Olievier CN, Ward DS, Dahan A. Antioxidants prevent depression of the acute hypoxic ventilatory response by subanaesthetic halothane in men. J Physiol. 2002;544:931–938. doi: 10.1113/jphysiol.2002.025999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urena J, Fernandez-Chacon R, Benot AR, de Toledo GA, Lopez-Barneo J. Hypoxia induces voltage-dependent Ca2+ entry and quantal dopamine secretion in carotid body glomus cells. Proc Natl Acad Sci U S A. 1994;91:10208–10211. doi: 10.1073/pnas.91.21.10208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vidruk EH, Olson EB, Jr, Ling L, Mitchell GS. Responses of single-unit carotid body chemoreceptors in adult rats. J Physiol. 2001;531:165–170. doi: 10.1111/j.1469-7793.2001.0165j.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward DS. Modelling and the Control of Breathing. Calgary: Oxford Conference; 2006a. The effects of glucose on ventilation and the hypoxic ventilatory response (HVR) p. 166. [Google Scholar]
- Ward DS. Hyperglycemia stimulates ventilation and the hypoxic ventilatory response. Anesthesiology. 2006b;105:A1561. [Google Scholar]
- Ward DS. Hypoglycemia (HG) and the hypoxic ventilatory response (HVR) FASEB J. 2006c;20:A376–A377. [Google Scholar]
- Weir EK, Lopez-Barneo J, Buckler KJ, Archer SL. Acute oxygen-sensing mechanisms. N Engl J Med. 2005;353:2042–2055. doi: 10.1056/NEJMra050002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weissman C, Askanazi J, Forse RA, Hyman AI, Milic-Emili J, Kinney JM. The metabolic and ventilatory response to the infusion of stress hormones. Ann Surg. 1986;203:408–412. doi: 10.1097/00000658-198604000-00012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williamson JR, Chang K, Frangos M, Hasan KS, Ido Y, Kawamura T, van den Nyengaard JREM, Kilo C, Tilton RG. Hyperglycemic pseudohypoxia and diabetic complications. Diabetes. 1993;42:801–813. doi: 10.2337/diab.42.6.801. [DOI] [PubMed] [Google Scholar]
- Zhang M, Buttigieg J, Nurse CA. Neurotransmitter mechanisms mediating low-glucose signalling in cocultures and fresh tissue slices of rat carotid body. J Physiol. 2007;578:735–750. doi: 10.1113/jphysiol.2006.121871. [DOI] [PMC free article] [PubMed] [Google Scholar]