

Abstract
N-type calcium channels contribute to the release of glutamate from primary afferent terminals synapsing onto nocisponsive neurons in the dorsal horn of the spinal cord, but little is known of functional adaptations to these channels in persistent pain states. Subtype-selective conotoxins and other drugs were used to determine the role of different calcium channel types in a rat model of inflammatory pain. Electrically evoked primary afferent synapses onto lumber dorsal horn neurons were examined three days after induction of inflammation with intraplantar complete Freund's adjuvant. The maximal inhibitory effect of the N-type calcium channel blockers, ω-conotoxins CVID and MVIIA, were attenuated in NK1 receptor-positive lamina I neurons after inflammation, but the potency of CVID was unchanged. This was associated with reduced inhibition of the frequency of asynchronous-evoked synaptic events by CVID studied in the presence of extracellular strontium, suggesting reduced N-type channel contribution to primary afferent synapses after inflammation. After application of CVID, the relative contributions of P/Q and L channels to primary afferent transmission and the residual current were unchanged by inflammation, suggesting the adaptation was specific to N-type channels. Blocking T-type channels did not affect synaptic amplitude under control or inflamed conditions. Reduction of N-type channel contribution to primary afferent transmission was selective for NK1 receptor-positive neurons identified by post hoc immunohistochemistry and did not occur at synapses in laminae IIo or IIi, or inhibitory synapses. These results suggest that inflammation selectively downregulates N-type channels in the terminals of primary afferents synapsing onto (presumed) nociceptive lamina I NK1 receptor-positive neurons.
Laminae I and II of the dorsal horn are primary targets for synaptic inputs from the small-diameter unmyelinated C-fibres and myelinated Aδ primary afferent fibres that relay nociceptive information from the periphery to the spinal cord (Christensen & Perl, 1970; Light & Perl, 1979; Sugiura et al. 1986; Bester et al. 2000; Craig et al. 2001). Incoming excitatory signals are subject to modulation by intrinsic inhibitory and excitatory interneurons within the spinal cord before projecting to deeper laminae and ascending pathways to higher centres in the brain (Dickenson et al. 1997). Synapses between primary afferents and instrinsic dorsal horn neurons are thought to be glutamatergic (Jahr & Jessell, 1985; Jessell et al. 1986), and a subpopulation of the small-diameter afferent fibres contain substance P (Hökfelt et al. 1975; Lawson et al. 1997), a neuropeptide shown to be released during noxious peripheral stimulation (Kuraishi et al. 1985; Go & Yaksh, 1987; Duggan & Hendry, 1986, Duggan et al. 1988).
Lamina I exhibits dense substance P and neurokinin 1 (NK1) receptor immunoreactivity (Liu et al. 1994; Brown et al. 1995; Littlewood et al. 1995), and contains projection neurons that transmit nociceptive information to supraspinal sites involved in pain (Ding et al. 1995; Craig, 1995; Marshall et al. 1996; Todd et al. 2000, 2002; Spike et al. 2003; Yu et al. 1999, 2005). Behavioural studies have shown that selective ablation of NK1 receptor-expressing lamina I neurons with a neurotoxic substance P–saporin conjugate produces reduced nociceptive responses during inflammation and after nerve ligation, implicating these neurons in chronic pain states (Mantyh et al. 1997; Nichols et al. 1999; Suzuki et al. 2002). In addition, large lamina I NK1 receptor-positive projection neurons exhibit long-term potentiation (LTP) after high-frequency primary afferent stimulation (Ikeda et al. 2003) and perhaps more interestingly at a physiologically relevant low intensity (Ikeda et al. 2006), mechanisms which may contribute to spinal amplification of pain signals and central sensitization in chronic inflammatory and neuropathic pain (Woolf & Salter, 2000; Ji et al. 2003).
Influx of calcium through presynaptic voltage-gated calcium channels (VGCC) at synapses on primary afferents' terminals controls neurotransmitter release. There are several types of VGCC classified as L (Cav1.X), N (Cav2.2), P/Q (Cav2.1), T (Cav3.X) and R (Cav2.3) according to their molecular, pharmacological and electrophysiological properties (Catterall et al. 2003). All subtypes are found in the dorsal horn but evidence suggests that the N- and P/Q-types are predominantly presynaptic (Westenbroek et al. 1992, 1995) and are present on the central terminations of primary afferents (Westenbroek et al. 1998). The N-type calcium channel is the most abundant in lamina I of the dorsal horn and colocalizes with substance P in primary afferents (Westenbroek et al. 1998). ω-Conotoxins specific for the N-type calcium channel (Lewis et al. 2000; Adams et al. 2003) can block substance P release in the spinal cord (Smith et al. 2002) and have been shown to be antinociceptive in various animals models of chronic pain (Chaplan et al. 1994; Bowersox et al. 1996; Wang et al. 2000; Scott et al. 2002; Smith et al. 2002) and in the clinic (Atanassoff et al. 2000; Staats et al. 2004).
Although others have used specific pharmacological tools to functionally identify contributions of different calcium channel types to normal primary afferent synaptic transmission (Bao et al. 1998; Heinke et al. 2004), little is known about adaptive changes in calcium channel function during chronic pain states. With the use of highly specific ω-conotoxins, this study has investigated physiological changes in the contribution of these calcium channel subtypes, with particular emphasis on the N-type, to excitatory and inhibitory synaptic transmission in the dorsal horn during chronic inflammation. We focused on lamina I cells expressing the NK1 receptor and found a downregulation of presynaptic N-type calcium channel function using the N-type calcium channel ω-conotoxin inhibitors, CVID, and in some experiments MVIIA, during synaptic transmission at this synapse. Understanding adaptive changes in the spinal cord physiology during inflammation may prove useful for the development of more effective analgesics.
Methods
Inflammatory pain model
Persistent inflammation was induced by intraplantar injection of 100–150 μl of complete Freund's adjuvant (CFA; Sigma, Australia) into the left hindpaw of male and female Sprague–Dawley rats (18–24 days old) under isoflurane anaesthesia (Aerrane; Baxter, Puerto Rico, USA) The animals were allowed to recover and were kept for 72 h before being used for electrophysiological experiments which is the peak for nociceptive responses associated with this pain model (Iadarola et al. 1988). Saline-injected rats show no significant changes in mechanical threshold or paw-withdrawal latencies (Iadarola et al. 1988; Gu & Huang, 2001), so naïve rats were used as controls. The total number of rats used in this study was 69, 34 of which were injected with CFA to induce inflammation. Development of erythema, oedema and soreness of the paw, which was restricted to the hindpaw, was monitored during this time and always confirmed prior to experiments. All experimental protocols were approved by the University of Sydney Animal Ethics Committee.
Preparation of spinal cord slices and electrophysiology
Seventy-two hours after injection of CFA, rats were anaesthetized with isoflurane, decapitated, the spinal cord removed and transverse slices from the lumbar region (350 μm) were cut using a vibratome in ice-cold sucrose-based artificial CSF (sACSF) of the following composition (mm): 100 sucrose, 63 NaCl, 2.5 KCl, 1.2 NaH2PO4, 1.2 MgCl2, 2.4 CaCl2, 25 glucose, and 2.5 NaHCO3. Slices were maintained at 34°C in a submerged chamber containing artificial CSF (ACSF) of the following composition (mm): 125 NaCl, 2.5 KCl, 1.2 NaH2PO4, 1.2 MgCl2, 2.4 CaCl2, 25 glucose, and 2.5 NaHCO3, and equilibrated with 95% O2 and 5% CO2. The slices were then transferred to a recording chamber and superfused continuously (2 ml min−1) with ACSF. Lamina I and II dorsal horn neurons were visualized on an upright microscope (BX50WI; Olympus Optical, Tokyo, Japan) using infrared Nomarski optics. Large lamina I cells with a capacitance ≥20 pF and dendrites in the lateral-medial orientation (Han et al. 1998; Cheunsuang & Morris, 2000) were selected to increase the likelihood of selecting cells expressing the NK1 receptor. Preliminary studies were performed to identify lamina I NK1 receptor-positive cells in living slices with a substance P–rhodamine fluorescent conjugate. The presence of the NK1 receptor was confirmed post hoc by filling neurons with biocytin and colabelling for NK1 receptor immunoreactivity (see below for details). Lamina II was identified as the translucent area adjacent to lamina I; dorsal cells in this region were considered in lamina IIo and those ventral considered lamina IIi. The position of biocytin-filled cells in lamina II was confirmed relative to IB4 immunolabelling post hoc. As IB4 labels small-diameter non-peptidergic afferent fibres in lamina II, neurons in lamina IIo were classified as within the dorsal region of the IB4-labelled band and cells ventral of the IB4 band considered lamina IIi neurons. Whole-cell voltage-clamp recordings of synaptic currents were made using an Axopatch 2D (Axon Instruments, Foster City, California, USA). A Cs+-based internal solution was used to record electrically evoked EPSCs (eEPSCs) containing (mm): 113 CsGluconate, 10 EGTA, 10 Hepes, 8 NaCl2, 3 QX-314, 0.2% biocytin, 2 MgATP and 0.3 NaGTP, pH 7.2 (osmolarity, 280–290 mosmol l−1). Patch electrodes were of 2–4 MΩ resistance and series resistance (≤15 MΩ) was compensated by 80%, and continuously monitored during experiments. Liquid junction potentials of –14 mV were not corrected. Cell capacitance was estimated from the axopatch capacitance compensation setting. eEPSCs were elicited by two consecutive stimuli (interstimulus interval 50 ms) of identical strength via bipolar tungsten stimulating electrodes (FHC, Bowdoin, ME, USA) placed in the dorsal root entry zone (rate, 0.01 Hz; stimuli, 2–30 V, 100 μs). Paired-pulse ratio (PPR) was calculated by dividing the second pulse by the first (PSC2/PSC1). All EPSCs were recorded in the presence of picrotoxin (100 μm) and strychnine (0.5 μm) and for eIPSCs CNQX (10 μm), AP5 (100 μm) and strychnine (0.5 μm). Neurons were voltage clamped at –60 mV eEPSCs and eIPSCs were quantified by averaging 20 consecutive responses for each condition (Axograph 4.6). Desynchronization of miniature currents underlying the eEPSC were obtained in the presence of 8 mm SrCl2 replacing CaCl2 in the ACSF solution. These miniature asynchronous evoked postsynaptic currents (aeEPSC) were filtered (2 kHz low-pass filter) and sampled at 10 kHz for online and later offline analysis (Axograph 4.6; Axon Instruments, Union City, CA, USA). Desynchronized miniature events above a preset threshold (3.5 s.d. above baseline noise) were automatically detected by a sliding-template algorithm for a window 100–500 ms post-stimulation to ensure the capture of asynchronous events related to stimulation, and then manually checked offline. Events were counted in 400 ms epochs every 10 s to construct time plots of the aeEPSC rate versus time, and ranked by amplitude to construct cumulative probability functions. Experiments were conducted in the presence of the ω-conotoxins, CVID (50 nm – 5 μm) and MVIIA (1 μm), provided by RJ Lewis, Institute for Molecular Bioscience, The University of Queensland to block N-type calcium channels.
Immunohistochemistry
At the end of electrophysiological experiments, slices were fixed in 4% paraformaldehyde, 12.5% picric acid in 0.1 m PBS, pH 7.1 for 1 h and then washed in 0.1 m Tris-buffered saline (TBS). For immunohistochemical detection of NK1 receptors, slices were washed in 0.1 m PBS, pretreated for 1 h in 5% normal horse serum, 1% bovine serum albumin and 0.3% Triton X-100 in 0.1 m PBS, followed by incubation with rabbit anti-NK1 receptor (Chemicon, Australia) 1: 1000–1: 2000 for 48–72 h at 4°C. For visualization of the primary antibody and biocytin-filled cells, slices were incubated with Alexa 488 donkey antirabbit IgG 1: 200 and Alexa 647 Streptavidin conjugate 1: 500 (both from Invitrogen, Australia) for 2 h at room temperature. All antibodies were diluted in 1% BSA and 0.3% Triton-X100 in 0.1 m PBS. For identification of cells recorded in lamina II, slices were incubated with FITC-conjugated Isolectin B4 (Sigma) and Alexa 647 Streptavidin conjugate for 2 h at room temperature. Slices were then washed in PBS, mounted onto slides, air dried for 2 h and then coverslipped with Fluoromount-G (ProSciTech, Thuringowa, Australia). Images of immunolabelled neurons were obtained using a confocal microscope (Olympus FV-300, Olympus, Australia). Optical sections were collected by sequential scanning with the relevant lasers and NK1 receptor immunopositivity and laminar localization of the recorded neurons were analysed from maximum-intensity projections or single optical sections.
Drugs, reagents, and solutions
ω-Agatoxin IVA was from Peptide Institute (Japan), QX-314 chloride from Alomone Laboratories (Jerusalem, Israel), AP5, mibefradil, nimodipine, picrotoxin, strychnine hydrochloride and substance P–rhodamine fluorescent conjugate from Sigma (Sydney, Australia), and CNQX from Tocris Cookson (Bristol, UK). Stock solutions of all drugs were made in distilled water, diluted to working concentrations in the extracellular solution immediately before use and applied by superfusion.
Data analysis
All pooled values are expressed as mean ± s.e.m., drug effect as a percentage of baseline (% baseline) or percentage inhibition ((before drug – after drug)/before drug) × 100). Statistical tests between treatment groups were made using a two-tailed unpaired t test and assuming unequal variance. Within-group comparisons were made using a two-tailed paired t test. When multiple contrasts were tested, ANOVA with Bonferroni post hoc test to correct for multiple comparisons was used. Concentration–response relationships were constructed using cumulative addition of 2–3 concentrations of CVID to each cell. Normalized concentration–response data were pooled and fitted with a logistic function (GraphPad Prism Software) using the following equation:
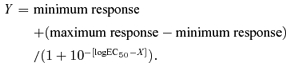 |
Significance was accepted at *P < 0.05, **P < 0.01 and ***P < 0.001 levels
Results
N-type calcium channels on primary afferent inputs to lamina I cells are downregulated after chronic inflammation
Lamina I neurons were identified and selected on the basis of large membrane capacitance and organization of dendrites to increase the likelihood of recording from a neuron expressing the NK1 receptor (see Methods). Estimated capacitances of 32 ± 2 pF (n = 16) in control and 29 ± 2 pF (n = 24) in inflamed neurons did not differ significantly.
To isolate excitatory evoked currents paired-pulse recordings were performed in the presence of picrotoxin (100 μm) and strychnine (10 μm), and were abolished in the presence of CNQX (10 μm). Stimulus intensity was adjusted to produce eEPSCs of similar amplitude in both groups, which did not differ significantly (977 ± 115 pA, n = 23 in control and 1061 ± 167 pA, n = 32 in inflamed). eEPSC amplitude and PPR was measured to determine the effect of the N-type calcium channel blocker, CVID, on excitatory synaptic transmission and release probability (Fig. 1A, C, E and F). In the majority of cases (11 cells of 16 for control and 20 cells of 24 in the inflamed group) paired pulse recordings exhibited a depression, with a PPR of less than 1, suggesting that the synapses between the primary afferents and most lamina I neurons have high-probability release sites. Twenty successive baseline recordings of eEPSCs were taken before CVID was applied. In the presence of CVID (1 μm), eEPSC amplitude was significantly decreased to 46 ± 3% of baseline in control (P < 0.001, n = 16) after 10–15 min, while the PPR significantly increased from 0.65 ± 0.09 to 0.9 ± 0.09 (P < 0.01, n = 16), indicating that blocking N-type calcium channels inhibits this synapse and changes release probability from a high to a low state.
Figure 1. N-type calcium channel blocker CVID inhibits eEPSC amplitude recorded from lamina I dorsal horn neurons.

Paired eEPSCs from a control lamina I cell (A) and from an inflamed animal (B) voltage clamped at –60 mV, before and after application of 1 μm CVID. C and D, time plots of the eEPSC peak amplitude from the same cells during application of 1 μm CVID, points sampled every 10 s for 15 min. E, bar chart displaying the mean effect of CVID (1 μm) and MVIIA (1 μm) on initial eEPSC amplitude for control (open bars) (n = 16, n = 8, respectively) and the inflamed group (filled bars) (n = 24, n = 8, respectively). Mean data ± s.e.m. are expressed as percentage of baseline before drug application. F, bar chart illustrates the baseline (open bars) PPR ± s.e.m. and after application of 1 μm CVID (hatched bars) for the control (n = 16) and inflamed group (n = 24). Asterisks denote significant difference from control: *P < 0.05; **P < 0.01; ***P < 0.001.
In the inflamed group, eEPSC amplitude was inhibited to a significantly lesser extent by CVID in comparison to control, exhibiting a reduction to only 60 ± 3% of baseline (P < 0.001, n = 24), 14% less than that observed in the control group (Fig. 1B, D, E and F). PPR was significantly increased by CVID (1 μm) from 0.66 ± 0.07 to 1.01 ± 0.06 (P < 0.001, n = 24). However when comparing PPR between control and inflamed groups, no significant differences were observed. Inhibition of eEPSC amplitude was also significantly reduced in the inflamed group when compared to control (P < 0.05) with another N-type selective ω-conotoxin, MVIIA (Fig. 1E). In control slices, MVIIA (1 μm) reduced eEPSC amplitude to 62 ± 5% (n = 8) of baseline and for the inflamed group to 80 ± 6% (n = 8).
Inhibition of the frequency of asynchronous-evoked-miniature events by the N-type calcium channel antagonist CVID is abolished after inflammation
To further elucidate the change in N-type calcium channel function at the synapse between the primary afferents and lamina I neurons, the frequency and amplitude of quantal events underlying eEPSCs were examined using strontium (8 mm) to replace calcium (2.4 mm) in the external recording solution. Strontium causes desychronization of the quantal events underlying the evoked synaptic current, so postsynaptic responses to individual asynchronous-evoked EPSCs (aeEPSCs) can be resolved (Goda & Stevens, 1994). Cells were discarded if the spontaneous EPSC rate was high in normal ACSF. The first 100 ms after each stimulation in the presence of strontium were excluded to reduce the likelihood of including synchronous events, and only currents measured up to 500 ms after stimulation were considered to be aeEPSCs. eEPSCs were evoked in normal ACSF and then exchanged for a solution in which strontium (8 mm) replaced calcium; eEPSC amplitude diminished as the occurrence of aeEPSCs increased. CVID (1 μm) was applied after the increase in aeEPSC frequency stablized (10–15 min). Examples of eEPSCs in the presence of normal ACSF, strontium and CVID (1 μm) are presented in Fig. 2A and B for control and inflamed, respectively. For the same neurons cumulative probability plots for amplitude (Fig. 2C and D) and interevent interval (Fig. 2E and F) are displayed. In the absence of CVID, there were no differences between groups in aeEPSC mean amplitude (23 ± 2 pA and 22 ± 2 pA) or interevent interval (24 ± 2 ms and 22 ± 2 ms) for control (n = 15) and the inflamed group (n = 12), respectively. However, these parameters may have been biased by sampling only cells with a low basal spontaneous EPSC rate. From Fig. 2G it is evident that CVID (1 μm) caused a significant reduction in aeEPSC frequency to 65 ± 5% of baseline (P ≤ 0.001) in control, whereas CVID had almost no effect (95 ± 5% of baseline, P > 0.05) in the inflamed group. The effect of CVID on aeEPSC frequency was significantly less in the inflamed group than the control group (P < 0.001). There was also a small but significant reduction in mean aeEPSC amplitude to 87 ± 3% of baseline (P < 0.01) in control and to 94 ± 3% in the inflamed group (P ≤ 0.05) in the presence of CVID (1 μm). However, these measurements were not significantly different when compared between groups (P > 0.05). Furthermore aeEPSC rise time remained unchanged in both groups, being 105 ± 4% and 103 ± 3% of baseline for control and inflamed groups respectively. These results suggest that the predominant effect of CVID is to inhibit release from primary afferents on to lamina I neurons, and this effect is reduced during inflammation due to a reduction of presynaptic N-type calcium function on primary afferent terminals.
Figure 2. N-type calcium channel blocker CVID reduces the frequency and amplitude of asynchronous miniature currents in the control but not the inflamed group.

eEPSCs were evoked in lamina I cells from control (A) and an inflamed animal (B) in normal calcium (upper traces) and in the same cells after calcium was replaced by 8 mm strontium to produce aeEPSCs before (middle trace) and after (lower trace) application of 1 μm CVID (Vh = –60 mV). Cumulative distribution plots of aeEPSC amplitude (C and D) and interevent interval (E and F) before (black line) and after application of 1 μm CVID (grey line) from control and an inflamed animal, respectively, in the same cells shown in A and B. G, bar chart displaying mean data ± s.e.m. expressed as percentage of baseline, for aeEPSC frequency (Freq), amplitude (Amp) and rise time (RT) for control (open bars) (n = 15) and inflamed (filled bars) (n = 12). ***Significant difference from control (P < 0.001).
A reduced maximal effect is responsible for the loss of inhibition of CVID after inflammation
The affinities of ω-conotoxins, including CVID, for N-type calcium channels are influenced by expression of auxiliary subunits (Mould et al. 2004). Concentration–response curves for inhibition of eEPSCs by CVID were therefore examined to determine whether loss of inhibition after inflammation was due to a reduction in affinity or maximal effect (Fig. 3). The maximum inhibition of eEPSC amplitude by CVID (5 μm) in the inflamed group was 41 ± 7% (n = 8) compared with 68 ± 7% (n = 5) in control (P < 0.05). Maximum inhibition was significantly reduced according to a two-way ANOVA with Bonferroni post hoc test to correct for multiple comparisons. The EC50 for CVID was not different between the control (EC50 = 105 nm, 95% confidence interval (CI) of estimate = 55 nm to 198 nm) and inflamed group (EC50 = 55 nm, 95% CI of estimate = 23 nm to 134 nm). Thus a downregulation of N-type calcium channel function occurs at the synapse between primary afferent inputs and lamina I neurons during inflammation, rather than a reduction in affinity of ω-conotoxins.
Figure 3. Maximal inhibition of eEPSC amplitude by CVID is significantly reduced after inflammation.

The maximal inhibition of eEPSC amplitude was reduced in the inflamed group (•) when compared to control (○), but the EC50 was unchanged (see text). Inhibition of eEPSC amplitude by CVID is concentration dependent; each point shows the mean ± s.e.m. of responses of several different neurons. Mean data ± s.e.m. is expressed as percentage inhibition. Lines represent the fitted logistic curves.
Downregulation of N-type calcium channel function during chronic inflammation is not paralleled by a change in the function of other calcium channel subtypes
To determine whether plasticity in calcium channel function was evident for other subtypes at this synapse, cumulative doses of CVID for N-type, ω-agatoxin IVA for P/Q-type and nimodipine for L-type channels were applied (Fig. 4A and B with time-plots Fig. 4C and D); all synaptic responses were abolished upon application of cadmium (200 μm) a non-specific calcium channel blocker. In slices from control animals, CVID (1 μm) inhibited eEPSC amplitude by 52 ± 5%, ω-agatoxin IVA (100 nm) by 14 ± 5% and nimodipine (3 μm) by 7 ± 2% (n = 9). For the inflamed group this was 33 ± 4%, 19 ± 9% and 8 ± 2% (n = 7), respectively. Mibefradil (5 μm) a non-selective antagonist for the T-type calcium channel was also applied at the end of each experiment but had no effect (–0.3 ± 2% inhibition in control and –2.7 ± 4% in the inflamed group). The residual current of each cell therefore represented the R-current (although this was not tested pharmacologically). In control this accounted for 27 ± 4% of the eEPSC amplitude and 39 ± 8% in the inflamed group. In both the control and inflamed group CVID, nimodipine and ω-agatoxin IVA reduced eEPSC amplitude. CVID was again found to inhibit eEPSC amplitude to significantly less in the inflamed group when compared to control (P ≤ 0.01). Considering that contributions of all calcium channel components to the eEPSC necessarily sum to 100%, we determined the relative contributions of the P/Q, L and R components of calcium channel currents to inhibition of eEPSC amplitude after N-type channels were blocked. As shown in Fig. 4E, the contributions of P/Q, L and R components to the residual eEPSC were unchanged after inflammation.
Figure 4. Inhibition of lamina I neuron eEPSC amplitude by N-, P/Q- and L-type calcium channels antagonists.

eEPSC traces recorded at a holding potential of –60 mV from control (A) and inflamed group (B) before and after cumulative application of CVID (1 μm), ω-agatoxin IVA (100 nm, Aga) and nimodipine (3 μm, Nim). C and D, time plots of the eEPSC peak amplitude from the same cells, points taken every 10 s. E, bars represent the percentage residual contribution of P/Q-type (black bars), L-type (grey bars) and R-type currents (open bars) to the peak eEPSC amplitude control group (n = 9) and inflamed group (n = 7) after application of CVID. Mean data ± s.e.m. are expressed as percentage inhibition.
Downregulation of N-type calcium channel function occurs on inputs to NK1 receptor-positive neurons and is exclusive to excitatory primary afferent inputs onto lamina I neurons
Spinal cord slices were processed post hoc to determine whether NK1 receptor immunoreactivity (Fig. 5A) was colocalized with biocytin labelling of the recorded neuron (Fig. 5B). NK1 receptor-positive neurons exhibited capacitances of 33 ± 4 pF (n = 7) and 34 ± 6 pF (n = 6) in the control and inflamed groups, respectively, which were not significantly different. In addition these capacitances did not differ significantly from the general population of large lamina I neurons selected for study (see above). To determine whether the differential effect of CVID after inflammation was exclusive to primary afferent–lamina I NK1 receptor-positive neuron synapses, we compared the effects of CVID (1 μm) on NK1 receptor-positive neurons with the whole group and also with those cells that were not identified as expressing the NK1 receptor (Fig. 5C). Neurons with confirmed NK1 receptor expression exhibited a significant difference in the inhibition of eEPSC amplitude in the control and inflamed groups by CVID (1 μm) (P < 0.05). For this population, control group eEPSC amplitude was reduced to 42 ± 4% of baseline (P < 0.001, n = 7) and for the inflamed group to 62 ± 6% of baseline (P ≤ 0.01, n = 6), 20% less than control. The remaining cells for which NK1 receptor expression could not be identified did not exhibit a significant difference in the effect of CVID between control and inflamed groups, with CVID (1 μm) inhibiting eEPSC amplitude by 50 ± 5% (n = 9) and 60 ± 4% (n = 18), respectively. This suggests that the difference in the effect of CVID between groups was enriched in the NK1 receptor-expressing population. However it should be noted that it is likely that the immunohistochemical method failed to detect NK1 receptor expression in an unknown proportion those non-identified neurons.
Figure 5. Effect of the N-type calcium channel blocker CVID on excitatory and inhibitory inputs onto NK1 receptor-expressing lamina I cells and lamina II neurons.

Fluorescent imagine of a lamina I neuron exhibiting colocalization of NK1 receptor immunoreactivity (A) with biocytin (B) that was present in the internal pipette solution for post hoc identification of the recorded neuron. Scale bar = 20 μm. C, reduced inhibition of eEPSC amplitude during inflammation is predominant in all cells (n = 16 for control, n = 24 for inflamed group) and those identified as expressing the NK1 receptor (NK1+) (n = 7 for control, n = 6 for inflamed group) in comparison with cells that were stained but could not be identified (Not NK1+) (n = 9 for control, n = 18 for inflamed group). D, eEPSCs recorded from cells in control and inflamed animals from lamina IIo (n = 13 and 8, respectively) and lamina IIi (n = 11 and 14, respectively) and eIPSCs recorded from NK1 receptor-positive lamina I neurons (n = 6 and 8, respectively) were reduced by CVID (1 μm) to the same extent in inflamed (filled bars and hatched bars) and control (open bars) animals, in contrast to eEPSCs recorded from NK1 receptor-positive lamina I neurons. Mean data ± s.e.m. are expressed as percentage of baseline. Asterisk denotes significant difference from control (*P < 0.05; ***P < 0.001).
To determine whether the differential effect of CVID after inflammation was exclusive to the primary afferent–lamina I cell synapse, we also investigated the effects of CVID (1 μm) on lamina II neurons (Fig. 5D). This region also receives inputs from nociceptive-specific C- and Aδ-fibres, and can be divided into two basic subdivisions outer (IIo) and inner (IIi). Lamina IIo cells are predominately nociceptive and thermoreceptive, whereas lamina IIi cells respond to more innocuous mechanical stimulation (Light et al. 1979). Neurons in laminae IIo and IIi were identified as lying dorsal or ventral to IB4 immunolabelling as described in the methods. Lamina IIo cells were smaller in size than lamina I neurons, with a mean capacitance of 13 ± 1 pF (n = 13) for control and 12 ± 1 pF (n = 8) for the inflamed group. Lamina IIi cells were slightly larger, with a mean capacitance of 15 ± 1 pF (n = 11) for control and 14 ± 1 (n = 10) for the inflamed group. eEPSCs from lamina II cells were pharmacologically isolated and elicited by stimulating the dorsal root entry zone. In control experiments, CVID (1 μm) reduced eEPSC amplitude of cells in laminae IIo (41 ± 7% of baseline) and IIi (38 ± 6% of baseline) to the same extent as that observed in lamina I. In contrast to lamina I, no significant difference regarding the extent of eEPSC amplitude inhibition by CVID (1 μm) in either lamina IIo (42 ± 5% of baseline) or IIi (42 ± 6% of baseline) was observed in the inflamed group.
It was also of interest to determine whether the downregulation of N-type calcium channel function during chronic inflammation also occurred at inhibitory inputs onto lamina I neurons. eIPSCs were recorded by stimulating inhibitory interneuron inputs from lamina II. To isolate inhibitory currents, paired eIPSCs were recorded in the presence of CNQX (10 μm), AP5 (100 μm) and strychnine (10 μm), and were abolished in the presence of picrotoxin (100 μm). For all cells described, NK1 receptor expression was confirmed. In contrast to the effect of CVID on excitatory inputs to lamina I cells, CVID (1 μm) inhibited eIPSC amplitude to a similar extent in the inflamed group: 46 ± 7% of baseline (n = 8) when compared to control 46 ± 9% (n = 6) (Fig. 5D). These results suggest that adaptation of N-type calcium channel function is indeed exclusive to excitatory primary afferent synaptic inputs onto lamina I neurons.
Discussion
We have shown that N-type calcium channel function changes in the rat dorsal horn during inflammation. A selective reduction in the contribution of this calcium channel to excitatory synaptic transmission recorded from NK1 receptor-positive neurons was observed in lamina I but not laminae IIo or IIi using the ω-conotoxin, CVID. The failure of the N-type calcium channel blocker CVID to significantly inhibit aeEPSC frequency during inflammation further suggests that a downregulation of presynaptic N-type calcium channel function occurs on primary afferent terminals synapsing onto lamina I dorsal horn neurons. No evidence was found for functional adaptations to other calcium channel types, because the relative proportions of eEPSCs attributable to P/Q, L and R channels were unaffected during inflammation, while T-type channels did not appear to contribute at all in either control or inflamed dorsal horn.
The observed reduction of eEPSC amplitude in lamina I by various calcium channel antagonists was similar to that found in previous reports (Bao et al. 1998; Heinke et al. 2004) with conotoxins selective for the N-type calcium channel having the greatest effect, followed by ω-agatoxin IVA for P/Q-types and finally nimodipine or verapamil for the L-type and a negligible effect of mibefradil or nickel for the T-type. The proportion of EPSC amplitudes inhibited by each of these VGCC antagonists is also consistent with their distribution within the dorsal horn and cellular location. Immunohistochemical data show the prevalence of N-type calcium channels over the other subtypes in lamina I with the P/Q-type channel being more concentrated in lamina II and deeper (Westenbroek et al. 1998; Murakami et al. 2004). In addition, N-type and P/Q-type calcium channels are predominately located presynaptically in nerve terminals and dendrites, suggesting they are more ideally located to inhibit eEPSC amplitude than the L-type channel, which is more often located on cell bodies and dendrites (Westenbroek et al. 1998). Immunohistochemical data for the T-type channel in the dorsal horn is lacking; pharmacologically it has been shown to be present in both the dorsal horn and DRG (Borgland et al. 2001; Heinke et al. 2004) although past and present studies show it does not contribute to the lamina I synapse. However in the present experiments the residual current proportion of the eEPSC recorded from lamina I neurons was quite substantial. Although cumulative concentrations of CVID, ω-agatoxin IVA, nimodipine and mibefradil were intended to maximally block the N-, P/Q-, L- and T-type components, respectively, without blocking this current pharmacologically the residual component cannot be concluded to be completely R-type mediated. However, considering that the residual current is the second largest component of the eEPSC, which in part may be mediated by the R-type current, we speculate that this calcium channel subtype plays a role at this synapse. Indeed, the work of Westenbroek et al. (1998) suggests that a proportion of R-type channels may be present presynaptically in the dorsal horn.
The increase in the PPR of eEPSCs and reduction in frequency of aeEPSCs observed in control slices indicate a presynaptic site of action of CVID. Paired-pulse depression (PPD), defined by a PPR of less than one, was usually observed at this synapse. This is indicative of a high-release-probability synapse which assumes a low release probability on the second pulse after depletion of neurotransmitter ready for release on the first pulse (Betz, 1970). A movement towards a PPR of one in the presence of CVID suggests a reduction in this high release probability, presumably due to a reduction of calcium entry through N-type calcium channels into the presynaptic terminal. Replacing external calcium with strontium gives rise to miniature currents evoked by the afferent barrage because the individual synaptic quantal events underlying the EPSC become asynchronous. In these experiments, analysis of aeEPSCs was important because they provide information on release probability and quantal amplitude of primary afferent synapses. This is particularly pertinent in the spinal cord as it minimizes the contribution of quantal events from excitatory synapses arising from intrinsic dorsal horn synapses, as occurs during mEPSC recordings. Indeed, differences have been reported in the contribution of the various calcium channels to mEPSCs and eEPSCs in laminae I and II of the dorsal horn (Bao et al. 1998). In control experiments a pronounced inhibition of aeEPSC frequency by the N-type calcium channel blocker CVID was observed, confirming a presynaptic site of action. After inflammation, CVID was almost completely ineffective, confirming a presynaptic loss of functional N-type calcium channels. The observation that CVID was less effective at reducing aeEPSC frequency than eEPSC amplitude could be due to strontium being less efficient than calcium at triggering neurotransmitter release (Xu-Friedman & Regehr, 2000). Reduced efficacy of conotoxins can be excluded because calcium channel blockers and toxins are equally potent at blocking strontium and calcium influx (Tokunaga et al. 2004).
A slight but significant decrease in aeEPSC amplitude was also observed in the presence of CVID in the control and inflamed group. However, considering that aeEPSC amplitude was bigger than the background spontaneous miniature current amplitude it is possible that this observation is due to contamination. Although precautions were taken to ensure recordings were made only from neurons with a low background rate of spontaneous EPSCs, most cells had some level of background activity. Considering that many of these miniature currents arise from a different population of terminals and are less affected by N-type calcium channel antagonists (Bao et al. 1998), these smaller currents would have been more likely to have remained in the presence of CVID and thus to have produced an artefactual reduction in the measured aeEPSC amplitude.
Loss of effectiveness of CVID to inhibit presynaptic N-type channel function on primary afferents synapsing onto lamina I neurons could be due to a change in channel subunit composition, kinetics, a reduction in channel number or a combination of these. In heterologous expression systems the potency of CVID is profoundly reduced by coexpression of calcium channel α2δ auxillary subunits (Mould et al. 2004), and expression of these subunits is upregulated in primary afferents in some models of persistent pain (Li et al. 2004). However, binding of CVID to the N-type channel was unchanged because the EC50 values did not differ between the control and inflamed group.
Only one study to date has investigated the expression levels of N-type calcium channels in spinal cord during inflammation. Seven days after carrageenan inflammation of the rat hind paw, an increase in N-type calcium channel protein expression in dorsal root ganglia (DRG) was detected (Yokoyama et al. 2003), but whether this occurred in nociceptive primary afferents was not determined. Nor is it known whether such changes produced an increase in N-type channel protein expression in central nerve terminals of DRG neurons. Comparisons with the present study are further limited by different time scales and models of inflammation used in that study.
A decrease in the presynaptic N-type calcium channel population at the lamina I synapse could result from increased endocytosis of the channel after inflammation. Beedle et al. (2004) described a direct interaction of N-type calcium channels with the opioid receptor-like receptor 1 (ORL-1, NOP). Through this direct interaction, internalization of ORL-1 during prolonged exposure to nociceptin, its endogenous ligand, was shown to internalize N-type calcium channels in small-diameter DRG neurons, which were then trafficked to the lysosomal degradation pathway (Altier et al. 2006). Nociceptin levels are increased in the spinal cord after CFA inflammation of the hindpaw (Jia et al. 1998), which could favour internalization of the ORL-1 receptor/N-type calcium channels complex under these conditions and thus account for our long-term loss of N-type calcium channel function.
The proximal C-terminal region of the N-type calcium channel also contains an alternatively spliced exon which is believed to affect channel density and kinetics (Castiglioni et al. 2006). Expression of exon 37a over the more ubiquitously encoded exon 37b is only present in sensory neurons, and particularly nociceptive neurons (Bell et al. 2004). Exon 37a confers a larger N-type current density than exon 37b, due to insertion of a greater number of functional channels and an increase in channel open time. Therefore it is possible that a preferential expression of the exon 37b in small-diameter nociceptive-specific neurons during inflammation could account for the reduced N-type calcium channel component and thus reduced effects of CVID found in this study. In addition to further investigating whether different N-type splice variants are upregulated in inflammation, it would be interesting to determine whether their presence or absence rendered the channel insensitive to ω-conotoxins.
It is intriguing that plasticity in N-type calcium channel function induced by inflammation was observed selectively at synapses onto lamina I neurons and was not observed at afferent eEPSCs recorded from laminae IIo and IIi. Considering that lamina IIo receives a large proportion of C- and Aδ-fibre inputs that are predominately nociceptive and thermoreceptive (Light et al. 1979) similar to lamina I, a change might also have been expected in this lamina. Plasticity might be less likely to occur in lamina IIi because it receives predominantly innocuous mechanoreceptive inputs (Light et al. 1979). Downregulation of N-type calcium channel function was also enriched in the lamina I NK1 receptor-expressing neuronal population, whereas inhibitory inputs onto the same cells from surrounding lamina II interneurons also failed to exhibit N-type calcium channel plasticity. Perhaps the basis of this highly selective adaptation at excitatory synapses during inflammation serves to change the balance between inhibitory and excitatory control of these NK1-positive neurons. Downregulation of N-type calcium channel function could exist as an auto-regulatory feedback mechanism which decreases excitation relative to inhibition to reinstate previous homeostasis in lieu of exacerbated peripheral inputs from the site of inflammation. It is of interest in this regard that the eEPSC PPR was unaffected by inflammation, suggesting that overall release probability of primary afferents synapsing onto NK1-positive lamina I neurons was maintained despite the loss of N-type calcium channel function.
An overwhelming body of evidence indicates crucial involvement NK1 receptor-expressing lamina I projection neurons in the nociceptive sensory pathways and development of persisitent pain (Mantyh et al. 1997; Nichols et al. 1999; Suzuki et al. 2002). Lamina I projection neurons are also of interest because LTP can be induced in this population which may underlie a mechanism of central sensitization during pain (Ikeda et al. 2003, 2006). The results of this and previous studies suggest that conotoxins targeting the N-type channel are effective antinociceptive agents (Cox, 2000; McGivern, 2006). However we have found that they are less effective at inhibiting excitatory inputs into the spinal cord during inflammation. Furthermore the imbalance of conotoxin inhibition of excitatory inputs and intrinsic inhibitory circuits in the superficial dorsal horn during inflammation may underlie some of their side-effect profiles. Overall, by understanding these limitations created by synaptic changes during pain states it is hoped that more effective therapies can be developed in the future.
Acknowledgments
ω-Conotoxin CVID was provided by R. J. Lewis, Institute for Molecular Bioscience, The University of Queensland. The work was supported by the National Health and Medical Research Council of Australia (Program Grant 351446, Fellowship 253799 to MJC) and NSW Spinal Cord and Other Neurological Conditions Program Grant R1PG5 (Chief Investigator Prof Janet Keast). KSV was also supported by the Australian and New Zealand College of Anaesthetists (05/009).
References
- Adams DJ, Smith AB, Schroeder CI, Yasuda T, Lewis RJ. Omega-conotoxin CVID inhibits a pharmacologically distinct voltage-sensitive calcium channel associated with transmitter release from preganglionic nerve terminals. J Biol Chem. 2003;278:4057–4062. doi: 10.1074/jbc.M209969200. [DOI] [PubMed] [Google Scholar]
- Altier C, Khosravani H, Evans RM, Hameed S, Peloquin JB, Vartian BA, Chen L, Beedle AM, Ferguson SS, Mezghrani A, Dubel SJ, Bourinet E, McRory JE, Zamponi GW. ORL1 receptor-mediated internalization of N-type calcium channels. Nat Neurosci. 2006;9:31–40. doi: 10.1038/nn1605. [DOI] [PubMed] [Google Scholar]
- Atanassoff PG, Hartmannsgruber MW, Thrasher J, Wermeling D, Longton W, Gaeta R, Singh T, Mayo M, McGuire D, Luther RR. Ziconotide, a new N-type calcium channel blocker, administered intrathecally for acute postoperative pain. Reg Anesth Pain Med. 2000;25:274–278. doi: 10.1016/s1098-7339(00)90010-5. [DOI] [PubMed] [Google Scholar]
- Bao J, Li JJ, Perl ER. Differences in Ca2+ channels governing generation of miniature and evoked excitatory synaptic currents in spinal laminae I and II. J Neurosci. 1998;18:8740–8750. doi: 10.1523/JNEUROSCI.18-21-08740.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beedle AM, McRory JE, Poirot O, Doering CJ, Altier C, Barrere C, Hamid J, Nargeot J, Bourinet E, Zamponi GW. Agonist-independent modulation of N-type calcium channels by ORL1 receptors. Nat Neurosci. 2004;7:118–125. doi: 10.1038/nn1180. [DOI] [PubMed] [Google Scholar]
- Bell TJ, Thaler C, Castiglioni AJ, Helton TD, Lipscombe D. Cell-specific alternative splicing increases calcium channel current density in the pain pathway. Neuron. 2004;41:127–138. doi: 10.1016/s0896-6273(03)00801-8. [DOI] [PubMed] [Google Scholar]
- Bester H, Chapman V, Besson J-M, Bernard J-F. Physiological properties of the lamina I spinoparabrachial neurons in the rat. J Neurophysiol. 2000;83:2239–2259. doi: 10.1152/jn.2000.83.4.2239. [DOI] [PubMed] [Google Scholar]
- Betz WJ. Depression of transmitter release at the neuromuscular junction of the frog. J Physiol. 1970;206:629–644. doi: 10.1113/jphysiol.1970.sp009034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borgland SL, Connor M, Christie MJ. Nociceptin inhibits calcium channel currents in a subpopulation of small nociceptive trigeminal ganglion neurons in mouse. J Physiol. 2001;536:35–47. doi: 10.1111/j.1469-7793.2001.t01-1-00035.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowersox SS, Gadbois T, Singh T, Pettus M, Wang YX, Luther RR. Selective N-type neuronal voltage-sensitive calcium channel blocker, SNX-111, produces spinal antinociception in rat models of acute, persistent and neuropathic pain. J Pharmacol Exp Ther. 1996;279:123–1249. [PubMed] [Google Scholar]
- Brown JL, Liu H, Maggio JE, Vigna SR, Mantyh PW, Basbaum AI. Morphological characterization of substance P receptor-immunoreactive neurons in the rat spinal cord and trigeminal nucleus caudalis. J Comp Neurol. 1995;356:327–344. doi: 10.1002/cne.903560302. [DOI] [PubMed] [Google Scholar]
- Castiglioni AJ, Raingo J, Lipscombe D. Alternative splicing in the C-terminus of CaV2.2 controls expression and gating of N-type calcium channels. J Physiol. 2006;576:119–134. doi: 10.1113/jphysiol.2006.115030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catterall WA, Striessnig J, Snutch TP, Perez-Reyes E. International Union of Pharmacology. XL. Compendium of voltage-gated ion channels: calcium channels. Pharmacol Rev. 2003;55:579–581. doi: 10.1124/pr.55.4.8. [DOI] [PubMed] [Google Scholar]
- Chaplan SR, Pogrel JW, Yaksh TL. Role of voltage-dependent calcium channel subtypes in experimental tactile allodynia. J Pharmacol Exp Ther. 1994;269:1117–1123. [PubMed] [Google Scholar]
- Cheunsuang O, Morris R. Spinal lamina I neurons that express neurokinin 1 receptors: morphological analysis. Neuroscience. 2000;97:335–345. doi: 10.1016/s0306-4522(00)00035-x. [DOI] [PubMed] [Google Scholar]
- Christensen BN, Perl ER. Spinal neurons specifically excited by noxious or thermal stimuli: marginal zone of the dorsal horn. J Neurophysiol. 1970;33:293–307. doi: 10.1152/jn.1970.33.2.293. [DOI] [PubMed] [Google Scholar]
- Cox B. Calcium channel blockers and pain therapy. Curr Rev Pain. 2000;4:488–498. doi: 10.1007/s11916-000-0073-8. [DOI] [PubMed] [Google Scholar]
- Craig AD. Distribution of brainstem projections from spinal lamina I neurons in the cat and the monkey. J Comp Neurol. 1995;361:225–248. doi: 10.1002/cne.903610204. [DOI] [PubMed] [Google Scholar]
- Craig AD, Krout K, Andrew D. Quantitative response characteristics of thermoreceptive and nociceptive lamina I spinothalamic neurons in the cat. J Neurophysiol. 2001;86:1459–1480. doi: 10.1152/jn.2001.86.3.1459. [DOI] [PubMed] [Google Scholar]
- Dickenson AH, Chapman V, Green GM. The pharmacology of excitatory and inhibitory amino acid-mediated events in the transmission and modulation of pain in the spinal cord. Gen Pharmacol. 1997;28:633–638. doi: 10.1016/s0306-3623(96)00359-x. [DOI] [PubMed] [Google Scholar]
- Ding YQ, Takada M, Shigernoto R, Mizurno N. Spinoparabrachial tract neurons showing substance P receptor-like immunoreactivity in the lumbar spinal cord of the rat. Brain Res. 1995;674:336–340. doi: 10.1016/0006-8993(95)00022-i. [DOI] [PubMed] [Google Scholar]
- Duggan AW, Hendry IA. Laminar localization of the sites of release of immunoreactive substance P in the dorsal horn with antibody-coated microelectrodes. Neurosci Lett. 1986;68:134–140. doi: 10.1016/0304-3940(86)90243-0. [DOI] [PubMed] [Google Scholar]
- Duggan AW, Hendry IA, Morton CR, Hutchison WD, Zhao ZQ. Cutaneous stimuli releasing immunoreactive substance P in the dorsal horn of the cat. Brain Res. 1988;451:261–273. doi: 10.1016/0006-8993(88)90771-8. [DOI] [PubMed] [Google Scholar]
- Go VL, Yaksh TL. Release of substance P from the cat spinal cord. J Physiol. 1987;391:141–167. doi: 10.1113/jphysiol.1987.sp016731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goda Y, Stevens CF. Two components of transmitter release at a central synapse. Proc Natl Acad Sci U S A. 1994;91:12942–12946. doi: 10.1073/pnas.91.26.12942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu Y, Huang LY. Gabapentin actions on N-methyl-d-aspartate receptor channels are protein kinase C-dependent. Pain. 2001;93:85–92. doi: 10.1016/S0304-3959(01)00297-4. [DOI] [PubMed] [Google Scholar]
- Han Z-S, Zhang E-T, Craig AD. Nociceptive and thermoreceptive lamina I neurons are anatomically distinct. Nat Neurosci. 1998;1:218–225. doi: 10.1038/665. [DOI] [PubMed] [Google Scholar]
- Heinke B, Balzer E, Sandkuhler J. Pre- and postsynaptic contributions of voltage-dependent Ca2+ channels to nociceptive transmission in rat spinal lamina I neurons. Eur J Neurosci. 2004;19:103–111. doi: 10.1046/j.1460-9568.2003.03083.x. [DOI] [PubMed] [Google Scholar]
- Hökfelt T, Kellerth JO, Nilsson G, Pernow B. Substance P: localization in the central nervous system and in some primary sensory neurons. Science. 1975;190:889–890. doi: 10.1126/science.242075. [DOI] [PubMed] [Google Scholar]
- Iadarola MJ, Douglass J, Civelli O, Naranjo JR. Differential activation of spinal cord dynorphin and enkephalin neurons during hyperalgesia: evidence using cDNA hybridization. Brain Res. 1988;455:205–212. doi: 10.1016/0006-8993(88)90078-9. [DOI] [PubMed] [Google Scholar]
- Ikeda H, Heinke B, Ruscheweyh R, Sandkuhler J. Synaptic plasticity in spinal lamina I projection neurons that mediate hyperalgesia. Science. 2003;299:1237–1240. doi: 10.1126/science.1080659. [DOI] [PubMed] [Google Scholar]
- Ikeda H, Stark J, Fischer H, Wagner M, Drdla R, Jager T, Sandkuhler J. Synaptic amplifier of inflammatory pain in the spinal dorsal horn. Science. 2006;312:1659–1662. doi: 10.1126/science.1127233. [DOI] [PubMed] [Google Scholar]
- Jahr CE, Jessell TM. Synaptic transmission between dorsal root ganglion and dorsal horn neurons in culture: antagonism of monosynaptic excitatory postsynaptic potentials and glutamate excitation by kynurenate. J Neurosci. 1985;5:2281–2289. doi: 10.1523/JNEUROSCI.05-08-02281.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jessell TM, Yoshioka K, Jahr CE. Amino acid receptor-mediated transmission at primary afferent synapses in rat spinal cord. J Exp Biol. 1986;124:239–258. doi: 10.1242/jeb.124.1.239. [DOI] [PubMed] [Google Scholar]
- Ji RR, Kohno T, Moore KA, Woolf CJ. Central sensitization and LTP: do pain and memory share similar mechanisms? Trends Neurosci. 2003;26:696–705. doi: 10.1016/j.tins.2003.09.017. [DOI] [PubMed] [Google Scholar]
- Jia Y, Linden DR, Serie JR, Seybold VS. Nociceptin/orphanin FQ binding increases in superficial laminae of the rat spinal cord during persistent peripheral inflammation. Neurosci Lett. 1998;250:21–24. doi: 10.1016/s0304-3940(98)00430-3. [DOI] [PubMed] [Google Scholar]
- Kuraishi Y, Hirota N, Sato Y, Hino Y, Satoh M, Takagi H. Evidence that substance P and somatostatin transmit separate information related to pain in the spinal dorsal horn. Brain Res. 1985;325:294–298. doi: 10.1016/0006-8993(85)90326-9. [DOI] [PubMed] [Google Scholar]
- Lawson SN, Crepps BA, Perl ER. Relationship of substance P to afferent characteristics of dorsal root ganglion neurones in guinea-pig. J Physiol. 1997;505:177–191. doi: 10.1111/j.1469-7793.1997.00177.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis RJ, Nielsen KJ, Craik DJ, Loughnan ML, Adams DA, Sharpe IA, Luchian T, Adams DJ, Bond T, Thomas L, Jones A, Matheson JL, Drinkwater R, Andrews PR, Alewood PF. Novel ω-conotoxins from Conus catus discriminate among neuronal calcium channel subtypes. J Biol Chem. 2000;275:35335–35344. doi: 10.1074/jbc.M002252200. [DOI] [PubMed] [Google Scholar]
- Li CY, Song YH, Higuera ES, Luo ZD. Spinal dorsal horn calcium channel alpha2delta-1 subunit upregulation contributes to peripheral nerve injury-induced tactile allodynia. J Neurosci. 2004;24:8494–8499. doi: 10.1523/JNEUROSCI.2982-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Light AR, Perl ER. Spinal termination of functionally identified primary afferent neurons with slowly conducting myelinated fibers. J Comp Neurol. 1979;186:133–150. doi: 10.1002/cne.901860203. [DOI] [PubMed] [Google Scholar]
- Littlewood NK, Todd AJ, Spike RC, Watt C, Shehab SA. The types of neuron in spinal dorsal horn which possess neurokinin-1 receptors. Neuroscience. 1995;66:597–608. doi: 10.1016/0306-4522(95)00039-l. [DOI] [PubMed] [Google Scholar]
- Liu H, Brown JL, Jasmin L, Maggio JE, Vigna SR, Mantyh PW, Basbaum AI. Synaptic relationship between substance P and the substance P receptor: light and electron microscopic characterization of the mismatch between neuropeptides and their receptors. Proc Natl Acad Sci U S A. 1994;91:1009–1013. doi: 10.1073/pnas.91.3.1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantyh PW, Rogers SD, Honore P, Allen BJ, Ghilardi JR, Li J, Daughters RS, Lappi DA, Wiley RG, Simone DA. Inhibition of hyperalgesia by ablation of lamina I spinal neurons expressing the substance P receptor. Science. 1997;278:275–279. doi: 10.1126/science.278.5336.275. [DOI] [PubMed] [Google Scholar]
- Marshall GE, Shehab SA, Spike RC, Todd AJ. Neurokinin-1 receptors on lumbar spinothalamic neurons in the rat. Neuroscience. 1996;72:255–263. doi: 10.1016/0306-4522(95)00558-7. [DOI] [PubMed] [Google Scholar]
- McGivern JG. Targeting N-type and T-type calcium channels for the treatment of pain. Drug Discov Today. 2006;11:245–253. doi: 10.1016/S1359-6446(05)03662-7. [DOI] [PubMed] [Google Scholar]
- Mould J, Yasuda T, Schroeder CI, Beedle AM, Doering CJ, Zamponi GW, Adams DJ, Lewis RJ. The α2δ auxiliary subunit reduces affinity of _ω-conotoxins for recombinant N-type (Cav2.2) Calcium Channels. J Biol Chem. 2004;279:34705–34714. doi: 10.1074/jbc.M310848200. [DOI] [PubMed] [Google Scholar]
- Murakami M, Nakagawasai O, Suzuki T, Mobarakeh II, Sakurada Y, Murata A, Yamadera F, Miyoshi I, Yanai K, Tan-No K, Sasano H, Tadano T, Iijima T. Antinociceptive effect of different types of calcium channel inhibitors and the distribution of various calcium channel alpha 1 subunits in the dorsal horn of spinal cord in mice. Brain Res. 2004;1024:122–129. doi: 10.1016/j.brainres.2004.07.066. [DOI] [PubMed] [Google Scholar]
- Nichols ML, Allen BJ, Rogers SD, Ghilardi JR, Honore P, Luger NM, Finke MP, Li J, Lappi DA, Simone DA, Mantyh PW. Transmission of chronic nociception by spinal neurons expressing the substance P receptor. Science. 1999;286:1558–1561. doi: 10.1126/science.286.5444.1558. [DOI] [PubMed] [Google Scholar]
- Scott DA, Wright CE, Angus JA. Actions of intrathecal omega-conotoxins CVID, GVIA, MVIIA, and morphine in acute and neuropathic pain in the rat. Eur J Pharmacol. 2002;451:279–286. doi: 10.1016/s0014-2999(02)02247-1. [DOI] [PubMed] [Google Scholar]
- Smith MT, Cabot PJ, Ross FB, Robertson AD, Lewis RJ. The novel N-type calcium channel blocker, AM336, produces potent dose-dependent antinociception after intrathecal dosing in rats and inhibits substance P release in rat spinal cord slices. Pain. 2002;96:119–127. doi: 10.1016/s0304-3959(01)00436-5. [DOI] [PubMed] [Google Scholar]
- Spike RC, Puskár Z, Andrew D, Todd AJ. A quantitative and morphological study of projection neurons in lamina I of the rat lumbar spinal cord. Eur J Neurosci. 2003;18:2433–2448. doi: 10.1046/j.1460-9568.2003.02981.x. [DOI] [PubMed] [Google Scholar]
- Staats PS, Yearwood T, Charapata SG, Presley RW, Wallace MS, Byas-Smith M, Fisher R, Bryce DA, Mangieri EA, Luther RR, Mayo M, McGuire D, Ellis D. Intrathecal ziconotide in the treatment of refractory pain in patients with cancer or AIDS: a randomized controlled trial. JAMA. 2004;291:63–70. doi: 10.1001/jama.291.1.63. [DOI] [PubMed] [Google Scholar]
- Sugiura Y, Lee CL, Perl ER. Central projections of identified unmyelinated (C) afferent fibres innervating mammalian skin. Science. 1986;234:358–361. doi: 10.1126/science.3764416. [DOI] [PubMed] [Google Scholar]
- Suzuki R, Morcuende S, Webber M, Hunt SP, Dickenson AH. Superficial NK1-expressing neurons control spinal excitability through activation of descending pathways. Nat Neurosci. 2002;5:1319–1326. doi: 10.1038/nn966. [DOI] [PubMed] [Google Scholar]
- Todd AJ, McGill MM, Shehab SA. Neurokinin 1 receptor expression by neurons in laminae I, III and IV of the rat spinal dorsal horn that project to the brainstem. Eur J Neurosci. 2000;12:689–700. doi: 10.1046/j.1460-9568.2000.00950.x. [DOI] [PubMed] [Google Scholar]
- Todd AJ, Puskar Z, Spike RC, Hughes C, Watt C, Forrest L. Projection neurons in lamina I of rat spinal cord with the neurokinin 1 receptor are selectively innervated by substance P-containing afferents and respond to noxious stimulation. J Neurosci. 2002;22:4103–4113. doi: 10.1523/JNEUROSCI.22-10-04103.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tokunaga T, Miyazaki K, Koseki M, Mobarakeh JI, Ishizuka T, Yawo H. Pharmacological dissection of calcium channel subtype-related components of strontium inflow in large mossy fibre boutons of mouse hippocampus. Hippocampus. 2004;14:570–585. doi: 10.1002/hipo.10202. [DOI] [PubMed] [Google Scholar]
- Wang YX, Pettus M, Gao D, Phillips C, Scott Bowersox S. Effects of intrathecal administration of ziconotide, a selective neuronal N-type calcium channel blocker, on mechanical allodynia and heat hyperalgesia in a rat model of postoperative pain. Pain. 2000;84:151–158. doi: 10.1016/s0304-3959(99)00197-9. [DOI] [PubMed] [Google Scholar]
- Westenbroek RE, Hell JW, Warner C, Dubel SJ, Snutch TP, Catterall WA. Biochemical properties and subcellular distribution of an N-type calcium channel alpha 1 subunit. Neuron. 1992;9:1099–1115. doi: 10.1016/0896-6273(92)90069-p. [DOI] [PubMed] [Google Scholar]
- Westenbroek RE, Hoskins L, Catterall WA. Localization of Ca2+ channel subtypes on rat spinal motor neurons, interneurons, and nerve terminals. J Neurosci. 1998;18:6319–6330. doi: 10.1523/JNEUROSCI.18-16-06319.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westenbroek RE, Sakurai T, Elliott EM, Hell JW, Starr TV, Snutch TP, Catterall WA. Immunochemical identification and subcellular distribution of the alpha 1A subunits of brain calcium channels. J Neurosci. 1995;15:6403–6418. doi: 10.1523/JNEUROSCI.15-10-06403.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolf CJ, Salter MW. Neuronal plasticity: increasing the gain in pain. Science. 2000;288:1765–1769. doi: 10.1126/science.288.5472.1765. [DOI] [PubMed] [Google Scholar]
- Xu-Friedman MA, Regehr WG. Probing fundamental aspects of synaptic transmission with strontium. J Neurosci. 2000;20:4414–4422. doi: 10.1523/JNEUROSCI.20-12-04414.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokoyama K, Kurihara T, Makita K, Tanabe T. Plastic change of N-type Ca channel expression after preconditioning is responsible for prostaglandin E2-induced long-lasting allodynia. Anesthesiol. 2003;99:1364–1370. doi: 10.1097/00000542-200312000-00019. [DOI] [PubMed] [Google Scholar]
- Yu XH, Ribeiro-da-Silva A, De Koninck Y. Morphology and neurokinin 1 receptor expression of spinothalamic lamina I neurons in the rat spinal cord. J Comp Neurol. 2005;491:56–68. doi: 10.1002/cne.20675. [DOI] [PubMed] [Google Scholar]
- Yu XH, Zhang ET, Craig AD, Shigemoto R, Ribeiro-da-Silva A, De Koninck Y. NK-1 receptor immunoreactivity in distinct morphological types of lamina I neurons of the primate spinal cord. J Neurosci. 1999;19:3545–3555. doi: 10.1523/JNEUROSCI.19-09-03545.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]