

Abstract
We hypothesize that remodelling of action potential and intracellular calcium (Cai) dynamics in the peri-infarct zone contributes to ventricular arrhythmogenesis in the postmyocardial infarction setting. To test this hypothesis, we performed simultaneous optical mapping of Cai and membrane potential (Vm) in the left ventricle in 15 rabbit hearts with myocardial infarction for 1 week. Ventricular premature beats frequently originated from the peri-infarct zone, and 37% showed elevation of Cai prior to Vm depolarization, suggesting reverse excitation–contraction coupling as their aetiology. During electrically induced ventricular fibrillation, the highest dominant frequency was in the peri-infarct zone in 61 of 70 episodes. The site of highest dominant frequency had steeper action potential duration restitution and was more susceptible to pacing-induced Cai alternans than sites remote from infarct. Wavebreaks during ventricular fibrillation tended to occur at sites of persistently elevated Cai. Infusion of propranolol flattened action potential duration restitution, reduced wavebreaks and converted ventricular fibrillation to ventricular tachycardia. We conclude that in the subacute phase of myocardial infarction, the peri-infarct zone exhibits regions with steep action potential duration restitution slope and unstable Cai dynamics. These changes may promote ventricular extrasystoles and increase the incidence of wavebreaks during ventricular fibrillation. Whereas increased tissue heterogeneity after subacute myocardial infarction creates a highly arrhythmogenic substrate, dynamic action potential and Cai cycling remodelling also contribute to the initiation and maintenance of ventricular fibrillation in this setting.
The incidence of sudden cardiac death is highest in the first month after myocardial infarction (Solomon et al. 2005). Myocardial infarction induces electrophysiological and structural remodelling of the peri-infarct zone, leading to fixed tissue heterogeneity that enhances the incidence of arrhythmogenesis after myocardial infarction (Gough et al. 1985; Bolick et al. 1986; Dillon et al. 1988; Wit et al. 1990; Zaitsev et al. 2003). However, recently we have shown that in addition to fixed tissue heterogeneity, dynamic factors are also important in the ventricular arrhythmogenesis (Weiss et al. 2005, 2006). Dynamic factors refer to cellular properties of the cardiac action potential and intracellular calcium (Cai) cycling that destabilize wave propagation in tissue. Dynamic action potential factors include action potential duration (APD) restitution, conduction velocity restitution, short-term cardiac memory, and electrotonic currents. Dynamic Cai cycling factors include primary Cai alternans and spontaneous (non-voltage gated) Cai release events. These factors act synergistically with fixed tissue heterogeneity to promote wavebreak and ventricular fibrillation. Most studies investigating the role of dynamic factors in ventricular arrhythmogenesis have been conducted in normal ventricles (Chudin et al. 1999; Choi et al. 2002; Omichi et al. 2004). The importance of dynamic factors in ventricular arrhythmogenesis in subacute myocardial infarction has not been fully investigated. In this study, we have investigated the importance of APD restitution and Cai dynamics in arrhythmogenesis in the setting of subacute myocardial infarction. We hypothesized that subacute myocardial infarction causes electrical remodelling that alters APD restitution and Cai dynamics in the peri-infarct zone. These factors, in addition to fixed tissue heterogeneity, contribute to the arrhythmogenesis after subacute myocardial infarction. We performed simultaneous voltage and Cai mapping in Langendorff-perfused rabbit hearts with subacute (1 week) myocardial infarction to test this hypothesis.
Methods
The research protocol was approved by the Animal Care and Use Committee of each institution and conformed to the American Heart Association Guidelines. Fifteen adult New Zealand white rabbits (3.5–5.9 kg) were used for mapping studies.
Coronary artery ligation
Myocardial infarction was created using a modification of a published protocol (Ng et al. 1998). Rabbits were premedicated with intravenous injection of ketamine (4 mg) and xylazine (8 mg), intubated and anaesthetized with isoflurane. When the rabbits were fully anaesthetized and unresponsive to physical stimulus, the chests were opened via a left thoracotomy. The anaesthetic agents were adjusted during the experiments to insure that the rabbits were fully anaesthetized and not responsive to surgical procedures. The obtuse marginal branch of the left circumflex artery was identified and ligated halfway between the atrioventricular groove and the cardiac apex. The limb lead electrocardiogram was continuously monitored. Myocardial infarction was documented by significant ST segment elevation on electrocardiogram, purple-red discolouration and decreased wall motion. The wound was closed and the rabbits were treated with subcutaneous injection of buprenorphine (0.05–0.1 mg kg−1) for 2 days. No anti-arrhythmic drugs were used any time during the procedure.
Optical mapping
The rabbits underwent a second surgery 1 week after myocardial infarction under intravenous injection of ketamine (10 mg) and xylazine (20 mg) anaesthesia. The heart was rapidly removed, and the rabbits were killed by exsanguination during general anaesthesia. The ascending aorta was cannulated and secured for retrograde perfusion with warm (36.5 ± 0.5°C) oxygenated Tyrode solution equilibrated with 95% O2–5% CO2 to maintain a pH of 7.4 ± 0.05 at a rate of 35–45 ml min−1. The Tyrode solution contained (mm): NaCl 125, KCl 4.5, NaH2PO4 1.8, NaHCO3 24, CaCl2 2.7, MgCl2 0.5 and glucose 5.5, and 50 mg l−1 albumin in deionized water. The coronary perfusion pressure was regulated and maintained at 70–80 mmH2O.
Fifteen hearts were used for simultaneous dual optical mapping of membrane potential (Vm) and Cai. A calcium-sensitive dye (0.5 mg acetoxymethly ester (AM) form of rhod-2, Kd, 0.57; Molecular Probes) was dissolved in 1 ml dimethyl sulfoxide containing Pluronic F-127 (20% wt/vol). A previous study showed that rhod-2 loading into subcellular compartments, determined by manganese quenching, was minimal (< 5%) (Del Nido et al. 1998). This solution was further diluted in 300 ml Tyrode solution to achieve a final rhod-2 concentration of 1.5 μm and was infused into the heart over a 10 min period. Afterwards, the hearts were perfused with dye-free Tyrode solution for 15 min for de-esterification of rhod-2/AM followed by direct injection of a voltage-sensitive dye (RH 237, Molecular Probes) into the perfusion system with 10–20 μl of the 1 mg ml−1 solution dissolved in dimethyl sulfoxide (Choi & Salama, 2000). The hearts were illuminated with a solid-state, frequency-doubled laser (Verdi, Coherent Inc.) at a wavelength of 532 nm. The emitted fluorescence was filtered through a 585 ± 20 nm bandpass filter for the Cai signal recording and a 710 nm long-pass filter for Vm recording. The fluorescence was then acquired simultaneously with two charge-coupled device cameras (CA-D1-0128T, Dalsa Inc.) at 4 ms frame−1. The digital images (128 × 128 pixels) were gathered from the epicardium of the left ventricle (25 mm × 25 mm area), resulting in a spatial resolution of 0.2 mm × 0.2 mm per pixel. We inserted four pins into the corners of the mapped surface for reference. Using these reference points, the mapped fields of the cameras were spatially matched. Motion artifacts were suppressed by 5 μm cytochalasin D (Hayashi et al. 2003). We have previously demonstrated no overlap between voltage and Cai signals (Omichi et al. 2004).
Experiment protocol
APD restitution and Cai transient restitution curves were determined by dynamic pacing and standard S1 (baseline pacing) S2 (premature stimulation) pacing protocols, respectively (Koller et al. 1998). A quadripolar catheter was inserted into the right ventricle for pacing and sensing. Electrical stimuli of two times diastolic threshold were delivered through the distal two electrodes. In dynamic pacing protocol, the ventricles initially were paced at a constant pacing cycle length of 300 ms. After 15–20 stimuli had been delivered at this pacing cycle length, 1000 frames of optical images (4 s) were acquired. The pacing cycle length was shortened with 20 ms decrement for pacing cycle length of 200 ms and with 10 ms decrement for pacing cycle length < 200 ms until loss of 1: 1 ventricular capture, or when ventricular fibrillation was induced. In S1S2 pacing protocol, after eight baseline (S1) beats at the pacing cycle length of 400 ms, the S1S2 interval was set from 300 ms and shortened successively in steps of 20 ms for S1S2 of 200 ms and in steps of 10 ms for S1S2 < 200 ms until loss of ventricular capture or induction of ventricular fibrillation. At the completion of the pacing protocols, ventricular fibrillation was induced by burst pacing. Three to five episodes of ventricular fibrillation were mapped consecutively within the first 2 min after induction. These episodes, each containing 1000 frames of optical images, were used for analyses of dominant frequency.
We studied the effects of propranolol on APD restitution and ventricular fibrillation in 6 out of 15 hearts. After completing the protocols as described above, we attempted to perform defibrillation with 300 V, 6 ms biphasic shocks delivered via epicardial patch electrodes. If defibrillation was unsuccessful, we allowed ventricular fibrillation to continue. Propranolol (1 μm) was then infused for 15 min to determine whether it resulted in chemical conversion of ventricular fibrillation. The pacing protocols were again performed after the resumption of sinus rhythm.
Infarct size measurement
The hearts (n = 8) were cut horizontally into six sections (Fig. 1) and immersed in triphenyl tetrazolium chloride solution (Fishbein et al. 1981) for 1 h. The surviving myocardium was stained brick red. Scion Image (Scion Cooperation) was used to calculate the areas of infarction (white) and normal zones (brick red) of five cross-sections. The percentage of myocardial infarction was the ratio between the white (infarcted) area and the total area of left ventricle.
Figure 1. Two types of subacute myocardial infarction.

A and C, show the results of triphenyl tetrazolium chloride staining and the methods used to determine infarct size. The sections were taken at sites shown by the horizontal lines in B and D. E, shows low power (4 ×) view of trichrome-stained peri-infarct zone showing transmural myocardial infarction and adjacent peri-infarct zone. Residual necrotic, ‘mummified’ myocardium in the centre of the infarct is seen in the left upper corner of the field (arrow). F, shows a higher power (20 ×) view of the field within the black square in E, demonstrating surviving myocytes (red) surrounded by fibrous tissue (blue). OM, obtuse marginal branch of left circumflex artery; LAD, left anterior descending artery; MI, myocardial infarction.
Data analysis
We divided the mapped field outside the infarct area equally into three zones: the peri-infarct zone, remote zone 1 (R1) and remote zone 2 (R2). Restitution curves for the interval between the phase 0 and the time of 80% repolarization (APD80) were constructed at selected evenly spaced sites in the remote zones and peri-infarct zone. We first determined the membrane potential associated with 80% repolarization. Each restitution curve was constructed by plotting APD80 against the preceding diastolic interval, defined by the interval between 80% repolarization and the onset of the next action potential. If an action potential occurred before the preceding action potential reached 80% repolarization, the data were not included in the analyses. The slopes of APD restitution were calculated by first-order exponential fitting with ORIGIN software (Microcal). After curve fitting, the slope of APD restitution curve was calculated for the shortest diastolic interval that elicited an action potential. By definition, this is the maximum slope for a rising mono-exponential curve. The Cai alternans ratio is defined as 1−(B/A), where B is the amplitude of the smaller transient and A is the amplitude of the larger transient. Cai transient restitution curves were constructed at selected evenly spaced points in the remote zones and peri-infarct zone by plotting B/A against the pacing cycle length, and estimated by first-order exponential fitting with ORIGIN. After curve fitting, the maximum slope of Cai transient restitution curve was calculated.
Construction and interpretation of two dimensional maps
The basic physiological mechanism we were examining is the coupling between electrical activation and calcium release during ventricular fibrillation in rabbit hearts with subacute myocardial infarction. To better understand the relationship between electrical activation (Vm changes) and the Cai dynamics, we developed methods to simultaneously map the Vm and Cai, and used two dimensional colour maps to illustrate their changes during paced rhythm and during ventricular arrhythmias, including ventricular fibrillation. To create colour maps, the average fluorescence level (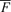 ) over the entire data window was first calculated for each pixel. At each pixel, the change in fluorescence level (the difference between fluorescence level and the
) over the entire data window was first calculated for each pixel. At each pixel, the change in fluorescence level (the difference between fluorescence level and the 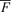 ) at each time point was colour coded to generate the colour maps. Individual Vm maps show the depolarized areas in shades of red and repolarized areas in shades of blue. The red colour (leading edge) of sequential maps indicates the direction of wavefront propagation. Similarly, the shades of red on individual Cai maps show the areas with above-average Cai while shades of blue show the areas with below-average Cai. By examining sequential Cai maps, one can determine the temporal sequence of Cai elevation at different sites. Focal discharge was defined by an activation arising from within the mapped region and propagating away from that site in all directions (Chou et al. 2005).
) at each time point was colour coded to generate the colour maps. Individual Vm maps show the depolarized areas in shades of red and repolarized areas in shades of blue. The red colour (leading edge) of sequential maps indicates the direction of wavefront propagation. Similarly, the shades of red on individual Cai maps show the areas with above-average Cai while shades of blue show the areas with below-average Cai. By examining sequential Cai maps, one can determine the temporal sequence of Cai elevation at different sites. Focal discharge was defined by an activation arising from within the mapped region and propagating away from that site in all directions (Chou et al. 2005).
The phase maps were constructed using a time-delay embedding method (Bray et al. 2001). Different phases were colour coded and plotted on the mapped region. Phase singularity, which correlates well with the site of wavebreak (Liu et al. 2003), is an area with ambiguous phase where neighbouring elements exhibit a continuous progression of phase from −π to +π. A cumulative phase singularity map was constructed by plotting phase singularities of 100 consecutive frames (400 ms) in the same map to determine the spatial dispersion of the phase singularities (Chou et al. 2005).
Fast Fourier transform was used to determine the frequency at each pixel during 4 s ventricular fibrillation. Dominant frequency was defined as the largest spectral peak for each pixel. The dominant-frequency maps were constructed by plotting the dominant frequency of activation at different mapped regions. These maps are also colour coded to demonstrate the distribution of dominant frequency during fibrillation.
Statistical methods
Continuous variables were expressed as the mean ± s.d. unpaired student's t tests were used to compare the means of two groups. ANOVA and Neuman–Keuls post hoc test were used to compare the differences of dominant frequency at the peri-infarct, R1 and R2 zones. A P value of ≤ 0.05 was considered statistically significant.
Results
Two different types of myocardial infarction were created in the rabbit ventricles. The first type (n = 12) involved both the anterolateral wall and the apex (Fig. 1A and B) and the second type (n = 3) spared the apex (Fig. 1C and D). Figure 1B and D shows the epicardial view of the transmural infarct (white). The estimated infarct size (yellow in the schematic diagrams below the photographs in Fig. 1A and C) averaged 28.4 ± 9% of the left ventricle. Figure 1E and F shows a representative cross-section of trichrome-stained left ventricular myocardium at the peri-infarct zone. In the peri-infarct zone, there were surviving epicardial myocardial cells isolated and surrounded by a much larger amount of connective tissue and inflammatory cells than in the non-infarcted myocardium.
Focal discharges from the peri-infarct zone
Reverse excitation–contraction coupling (ter Keurs et al. 1998; Boyden & ter Keurs, 2001) can lead to focal electrical discharges, and is detected by a rise in Cai prior to the rise in Vm at the same site (Cai prefluorescence) (Choi et al. 2002; Chou et al. 2005; Lakkireddy et al. 2006). We documented 27 episodes of ventricular premature beats arising from within the peri-infarct zone during ventricular pacing in six rabbit hearts (excluding two ventricular premature beats arising at the border of the mapped field in one rabbit heart), of which 10 (37%) showed Cai prefluorescence. Figure 2 shows an example of two consecutive ventricular premature beats. Figure 2A shows simultaneous Cai and Vm mapping of the first ventricular premature beat (beat 2, Fig. 2B). The rise of Cai occurred prior to the onset of Vm (Cai prefluorescence). The number below each frame is the time (ms) with the onset of data acquisition as time zero. The onset of rise in Cai was recorded at time 2608 ms (yellow arrow, upper row), preceding the earliest Vm signal (yellow arrow, frame 2616 ms, lower row) by 8 ms. The Cai prefluorescence at the focal site suggests that the Vm-independent calcium release from the sarcoplasmic reticulum (SR) was the cause for this focal discharge. However, once depolarization occurs at the focal site, the subsequent propagation occurs through the activation of the Na+ channel. Therefore, the sites distant from the focal site were activated by the usual Na+ channel-based propagation. The normal electromechanical coupling accounts for the delay between Vm and Cai maps in frames 2624 ms and 2636 ms. White arrows in frames 2624 ms and 2636 ms indicate the wavefront that propagates away from the focal discharge site. Figure 2B shows simultaneous Cai (orange) and Vm (black) tracings during rapid ventricular pacing (pacing cycle length, 180 ms) at the site corresponding to the asterisks in Fig. 2A. The blue upward arrow indicates Cai prefluorescence leading to a ventricular premature beat (beat 2). The following ventricular premature beat (beat 3) did not show Cai rise preceding Vm rise. Figure 2C shows the isochronal activation maps of beats labelled in Fig. 2B. Each colour represents a different range of activation time according to the colour bar at the right end of the panel. White arrows indicate the direction of wavefront propagation. Beat 1 was a paced beat coming from the right ventricle. Beats 2 and 3 (ventricular premature beats) originated from the centre of the mapped region, in the peri-infarct zone.
Figure 2. Cai prefluorescence and ventricular premature beat.

A, upper and lower rows show the maps of Cai and Vm, respectively, of the ventricular premature beat as marked by an upward blue arrow in B. The number below each panel in A is the time with the beginning of data acquisition as time zero. A wave front originated from the peri-infarct zone (asterisk) and propagated centrifugally in all directions. The leading edge of the wavefront is marked by white upward arrows. B, shows the optical recordings of Vm (black) and Cai transients (orange). Cai prefluorescence (upward blue arrow) preceded and triggered an action potential (beat 2). C, shows the isochronal maps of the beats marked in B. These isochronal maps were drawn based on optical recordings of Vm. Consecutive arrows are used to indicate the direction of wavefront propagation.
Induction of ventricular fibrillation from the peri-infarct zone
Figure 3 shows an example in which rapid ventricular pacing at pacing cycle length of 180 ms induced repetitive focal discharges from the peri-infarct zone, followed by ventricular fibrillation. Figure 3A shows the mapped field (marked by a black square). Figure 3B is the dominant-frequency map of ventricular fibrillation. The dominant frequency is used to indicate how fast the site was activated. The highest dominant frequency (white region) was located in the peri-infarct zone. Figure 3C shows the Vm tracings at sites labelled a (peri-infarct) and b in Fig. 3B. The dominant frequency at site a (21.9 Hz) was higher than at site b (15.7 Hz). Figure 3D shows simultaneous Cai (orange) and Vm tracings (black) recorded from site a during initiation of ventricular fibrillation at the period labelled in Fig. 3C (red bar). Beat 4 came from the right border of the mapped field. Beats 5–7 were consecutive focal discharges from site a. Figure 3E shows isochronal maps illustrating the origin (white arrows) and activation patterns of beats 2–7 and beat 10. In beat 5, the colour change indicates that the earliest site of activation was in the peri-infarct region (blue, as shown by white arrow), followed by the areas surrounding that earliest site. The site of origin of focal discharge subsequently became the site of highest dominant frequency during ventricular fibrillation (Fig. 3B). An interesting finding of this episode is that there was Cai prefluorescence at beats 5, 6, 7 and 10, suggesting reverse excitation–contraction coupling as their aetiology. Figure 3F shows the Cai and Vm frame shots of beats 5–7. The red colour indicates elevated Cai on the Cai maps and depolarized tissues on the Vm maps. The blue colour shows reduced Cai or repolarized tissues, respectively, on the Cai and the Vm maps. The white arrows on Cai frames 580 ms, 676 ms and 792 ms indicate the first occurrence of Cai fluorescence for these beats, and the earliest Vm activation came 8–12 ms later (white arrows, Vm frames 588 ms, 688 ms and 800 ms). In addition to Cai prefluorescence, we also noted that a region of low Cai surrounded by high Cai (Cai sinkhole) was present prior to the focal discharge on the Cai maps (white arrow, frames 660 ms and 756 ms). The Cai sinkholes had both lower Cai and shorter APD, and occurred prior to the subsequent focal discharges from the same site. The same findings (Cai sinkholes preceding consecutive focal discharges) were found in one more episode of ventricular fibrillation induction in a different heart. The remaining episodes of ventricular fibrillation were induced by multiple wavebreaks during rapid pacing. The presence of a Cai sinkhole preceding the spontaneous (voltage-independent) Cai release is consistent with reverse excitation–contraction coupling due to a highly calcium-loaded SR in this region (ter Keurs et al. 1998; Boyden & ter Keurs, 2001).
Figure 3. Consecutive focal discharges with Cai prefluorescence at the onset of ventricular fibrillation.

A, shows the mapped region. B, shows dominant frequency map during ventricular fibrillation. C, shows an example of optical recording. D, shows the rise of Cai (orange) preceding the elevation of Vm in beats 5, 6, 7 and 10 (downward blue arrows) at site labelled a during the onset of ventricular fibrillation. E, shows isochronal maps of beats 2–7 and beat 10, which included the paced beat before ventricular fibrillation (beat 2) and beats during the development of ventricular fibrillation. These isochronal maps were drawn based on optical recordings of Vm. White arrows indicate the direction of wavefront propagation (beats 2–4) or focal discharges (beats 5–7 and 10) from the peri-infarct zone. F, shows Cai and Vm maps that demonstrated Cai prefluorescence in beats 2–4 at onset of ventricular fibrillation. DF, dominant frequency; OM, obtuse marginal branch of left circumflex artery; LAD, left anterior descending artery.
Cai and wavebreaks
Figure 4 shows an example of wavebreak during ventricular fibrillation. Figure 4A shows Vm and Cai maps during ventricular fibrillation. There were depolarized tissues (red coloured areas) at different parts of the mapped region. Green arrows point to a region of transient Cai elevation. The membrane potential at that location and time (frame 368 ms) was already repolarized. The Vm map shows that a focal discharge from the peri-infarct zone (yellow arrow, frame 368 ms) was not able to conduct into that region (frame 388 ms), resulting in conduction block (white dotted line) and re-entrant wavefronts (frame 396 ms). Similarly, the wavefronts in the surrounding area (red coloured region) were also not able to conduct into the region with high Cai, resulting in conduction block and wavebreaks. The yellow arrows in frame 396 ms indicate the direction of wave propagation after conduction block. Note that the wavefront had to circle around the site of block to eventually depolarize the tissues on the other side of the dotted line. Figure 4B shows the isochronal map. The site of focal discharge is marked by a white arrow. Note that the arrow points to a region with blue colour, which according to the colour bar is the site of earliest activation. Figure 4C shows the phase map, with phase singularities marked by white arrows. The formation of phase singularities indicates wavebreak as a result of conduction block at those sites. These findings suggest that persistently elevated Cai prolongs local refractoriness to facilitate wavebreak. The incidence of diastolic Cai elevation causing wavebreaks in ventricular fibrillation was 2.2 ± 0.8 s−1 in all rabbit hearts studied.
Figure 4. Wavebreak induced by elevated Cai.

A, maps of Vm and Cai during ventricular fibrillation. B, isochronal maps of propagation. C, phase map showing multiple phase singularities (arrows) around the site of elevated Cai.
Spatial distribution of dominant frequency during ventricular fibrillation
Ventricular fibrillation could be induced in all 15 rabbit hearts. In 61 of 70 pacing-induced ventricular fibrillation episodes (87%), the dominant-frequency maps showed that the highest dominant frequency was located in the peri-infarct zone (see Figs 3B, 5B and 6A and 7B). For these 61 ventricular fibrillation episodes, the spatial distribution of dominant frequency remained largely unchanged in each heart throughout the episodes of ventricular fibrillation. The mean maximum dominant frequency within the peri-infarct zone was 19.5 ± 4.8 Hz (n = 15 rabbit hearts) during ventricular fibrillation. The mean dominant frequency of these ventricular fibrillation episodes was also determined at six selected evenly spaced points at the midline of the peri-infarct, R1 and R2 zones, and were 18.6 ± 2.6, 14.8 ± 1.9 and 13.0 ± 1.5 Hz, respectively (P < 0.001). This finding (high dominant frequency in the peri-infarct zone) is similar to that reported by Zaitsev et al. (2003) in an acute regional ischaemic pig model.
Figure 5. Cai and Vm alternans.

Data from the same rabbit as in Fig. 4A, a and b indicate sites with the earliest Cai transient alternans at the peri-infarct zone and at left ventricle base, respectively. B, dominant-frequency map during ventricular fibrillation. The highest dominant frequency was located at the peri-infarct zone (yellow arrow), corresponding to site a in A. C, shows Cai (orange) and Vm (black) recordings at sites a and b when the ventricles were paced at pacing cycle lengths of 300 ms, 180 ms and 150 ms. The numbers below each Vm tracings are APD80 values. S, small amplitude; L, large amplitude; 1−(B/A), alternans ratio; R1 and R2, remote zones 1 and 2, respectively.
Figure 6. Heterogeneous action potential duration restitution (A) and Cai restitution (B) distribution.

CaiR, Cai transient restitution.
Figure 7. Propranolol converts baseline ventricular fibrillation (Ba) to type 2 ventricular fibrillation (Bb) then ventricular tachycardia (Bc).

A, the mapped field. B, the dominant-frequency maps. C, Vm tracings at the asterisks in B. D, cumulative phase singularity maps of baseline ventricular fibrillation (a), type 2 ventricular fibrillation (b) and ventricular tachycardia (c).
Cai and APD alternans in the peri-infarct zone
Spatially heterogeneous Cai alternans was frequently observed during pacing. In 12 out of 15 hearts studied, the peri-infarct zones exhibiting focal discharges and the highest dominant frequency were also the sites where Cai alternans first occurred. We progressively shortened the pacing interval until Cai alternans occurred. While the other parts of the ventricles showed no alternans, Cai alternans occurred prior to APD alternans in the peri-infarct zone at a mean pacing cycle length of 200 ± 51 ms in seven of 12 hearts. Figure 5 shows an example from the same heart as that shown in Fig. 4. Optical signals at sites labelled a and b in Fig. 5A indicate the site of earliest Cai alternans and a site from the left ventricle base, respectively. Among them, site a (white colour on Fig. 5B) was the site with the highest dominant frequency during ventricular fibrillation. Figure 5C shows the Cai (orange) and Vm (black) recordings at sites a and b. At a pacing cycle length of 300 ms, neither Cai nor APD alternans occurred at site a. The Cai alternans first appeared at site a when pacing cycle length was shortened to 180 ms. Upon further shortening of the pacing cycle length, both APD and Cai alternans occurred at sites a and b. These findings show that Cai alternans was induced more easily in the peri-infarct zone than in areas remote from the site of myocardial infarction. The alternans ratio (1−B/A) is shown on the right-hand side. We compared the degree of Cai alternans at these sites. At pacing cycle length of 150 ms, the alternans ratio at site b (0.38) was much larger than that at site a (0.16). Whereas site a was the site of the earliest alternans, the alternans ratio did not grow as much as at the base of the heart when pacing cycle length was progressively shortened.
APD restitution in the peri-infarct zone
The mean APD80 values in the peri-infarct zone were 145 ± 18 (pacing cycle length 300 ms, n = 15 rabbit hearts) and 92 ± 14 ms (pacing cycle length 150 ms, n = 13 rabbit hearts), which were significantly shorter than those at remote zones (pacing cycle length 300 ms, 154 ± 16 ms; pacing cycle length 150 ms, 100 ± 14 ms, P < 0.001 for both comparisons). The steepest APD restitution occurred in the peri-infarct zone in 11 of 15 hearts studied, with the mean steepest APD restitution slope of 1.80 ± 0.15. In these 11 rabbit hearts, 51 of 52 episodes (98%) of ventricular fibrillation showed the highest dominant frequency within the peri-infarct zone. Figure 6A shows a typical example of APD restitution curve distribution. The dominant frequency map of ventricular fibrillation (inset) shows that the highest dominant frequency during ventricular fibrillation occurred within the peri-infarct zone, colocalizing with the site of steepest APD restitution slope. The Cai transient restitution curves at various sites are also shown in Fig. 6B. The slope of Cai restitution was generally flatter in the peri-infarct zone than at remote sites.
Effects of propranolol infusion
Propranolol infusion reduced the maximal APD restitution slope from 1.09 ± 0.05 to 0.75 ± 0.19 (P = 0.02, n = 6 rabbit hearts), whereas repeated 300 V shocks failed to convert ventricular fibrillation in 4 of 6 rabbit hearts at baseline; propranolol facilitated successful electrical conversion in all four resistant rabbit hearts and converted ventricular fibrillation to ventricular tachycardia in one heart. A typical example is shown in Fig. 7. the mapped region is shown in Fig. 7A, while Fig. 7B shows the dominant-frequency maps during baseline (a), after 10 min (b) and 14 min (c) of propranolol infusion. The highest dominant frequency was located within the peri-infarct zone (asterisks in Fig. 7Ba and Bb). Figure 7C shows the Vm tracings at asterisks in Fig. 7B. After 10 min propranolol infusion, the baseline ventricular fibrillation was converted to type 2 ventricular fibrillation (Wu et al. 2002) and the highest dominant frequency of ventricular fibrillation decreased from 18.6 to 9.5 Hz. After 14 min of propranolol infusion, type 2 ventricular fibrillation was converted to ventricular tachycardia and the dominant frequency of ventricular tachycardia further decreased to 7.6 Hz. Figure 7D shows the respective cumulative phase singularity maps at baseline ventricular fibrillation (Fig. 7Da), type 2 ventricular fibrillation (Fig. 7Db) and ventricular tachycardia (Fig. 7Dc). At baseline (Fig. 7Da), phase singularities (white dots) clustered at the peri-infarct zone, interventricular sulcus and left ventricle base. In all six rabbit hearts the mean maximum dominant frequency of ventricular fibrillation was significantly suppressed by propranolol (21.4 ± 6.3 versus 11.0 ± 1.8 Hz, P = 0.004), and in four rabbit hearts type 2 ventricular fibrillation occurred during propranolol infusion. The phase singularity maps (Fig. 7Db) showed reduced number of phase singularities as compared with Da. Finally, when ventricular tachycardia occurred (Fig. 7Dc), even fewer phase singularities were detected. No ventricular premature beat was induced by dynamic pacing protocol during propranolol infusion. These data show that propranolol flattened APD restitution slope and suppressed ventricular premature beat, resulting in significant antifibrillatory effects in rabbit hearts with subacute myocardial infarction.
Discussion
The major finding of this study is that dynamic properties of the action potential and Cai cycling are heterogeneously altered in the peri-infarct zone during the subacute phase of myocardial infarction. Combined with increased fixed tissue heterogeneity, these factors enhance the initiation and maintenance of ventricular arrhythmias in this setting. Extrasystoles typically originated from the peri-infarct zone, and in 37% of extrasystoles, a rise in Cai preceded membrane depolarization, suggesting that they were triggered by reverse excitation–contraction coupling. The peri-infarct zone, although heterogeneous, contained the region with the steepest APD restitution slope, and APD and Cai alternans, known factors that promote wavebreak (Weiss et al. 2006), were more easily induced at these sites. During ventricular fibrillation, transient regional Cai elevations often preceded wavebreak in the peri-infarct zone. These findings indicate that in addition to structural remodelling, dynamic action potential and Cai cycling remodelling also contribute to the initiation and maintenance of ventricular fibrillation in subacute myocardial infarction.
Mechanisms of abnormal Cai dynamics after subacute myocardial infarction
In ventricles after subacute myocardial infarction, many investigators have found significant remodelling of calcium handling protein expression and ion current activities, including reduction of sarco(endo)plasmic reticulum Ca2+-ATPase (SERCA) and up-regulation of phospholamban mRNA (Sun et al. 2005). On the other hand, the Na+–Ca2+ exchanger activity has been reported to increase, decrease or remain unchanged (Sipido et al. 2002). The remodelling can be heterogeneous, resulting in different expression levels in the epicardium and in the endocardium (Quinn et al. 2003). In addition to changes of SERCA and Na+–Ca2+ exchange activities, ischaemia also modulates the open probability of the ryanodine receptor (Eisner et al. 2000). Ischaemia leads to a reduction in ATP, which impairs both calcium uptake by SR and ryanodine receptor function. The net result might be either maintained or increased SR calcium content (Eisner et al. 2000). These complex interactions among different calcium handling mechanisms can together result in instability of the Cai cycling dynamics and contribute to the predisposition of the peri-infarct zone to Cai alternans. Consistent with this prediction, both theoretical (Shiferaw et al. 2003; Weiss et al. 2006) and experimental (Pruvot et al. 2004) studies have shown that a reduced ability of the SR to sequester and release calcium promotes Cai alternans. Moreover, depending on the mode of APD–Cai coupling (Shiferaw et al. 2003; Weiss et al. 2006), Cai cycling instabilities and steep APD restitution slope may interact synergistically to further potentiate alternans and enhance the likelihood of wavebreak and re-entry. This is consistent with the finding that propranolol, which has both adrenergic β-blocking and ion channel inhibition effects (Varro et al. 1990), decreased APD restitution slope and suppressed ventricular fibrillation.
Wavebreaks during ventricular fibrillation
During ventricular fibrillation, the peri-infarct zone contained the site with the highest dominant frequency. This peri-infarct region also had the steepest APD restitution slope, and exhibited earlier onset of Cai alternans during pacing. The high dominant frequency in the peri-infarct zone can be attributed to both the shorter baseline APD in this region, and the steeper APD restitution slope, so that the APD shortened even more dramatically as the activation rate increased during ventricular fibrillation. Moreover, unstable Cai dynamics in the peri-infarct zone may also have contributed to enhanced APD dispersion, as APD is influenced by the Cai transient amplitude via Ca2+-sensitive currents. This factor may have also led to increased wavebreak in the peri-infarct zone, which also increases dominant frequency. It is interesting that wavebreak during ventricular fibrillation tended to occur in regions with persistently high Cai. This is consistent with positive coupling between APD and Cai in the rabbit heart (Shiferaw et al. 2003), such that high Cai prolongs refractoriness by stimulating Na+–Ca2+ exchanger current and thereby prolonging APD. Persistently elevated Cai may have other effects promoting wavebreak (e.g. through affecting gap junction resistance), but the precise mechanisms and relative importance are unclear and deserve further study.
Study limitation
We only mapped the epicardium of the anterior aspect of hearts. It is possible that ventricular premature beats originating from subepicardial sites caused an underestimation of the percentage of ventricular premature beats with Cai prefluorescence. Although Cai prefluorescence prior to a ventricular premature beat suggests reverse excitation-contraction coupling, it does not prove it as a mechanism. Finally, we used cytochalasin D as an excitation–contraction uncoupler, and cannot rule out non-selective effects on electrophysiological and Cai cycling parameters that may have affected the results of the study.
Acknowledgments
This study was supported by a National Science Council (NSC) grant 94-2314-B-182 A-193 and a fellowship grant from the Chang Gung Memorial Hospital, Taipei, Taiwan to C-C.C., National Institutes of Health/National Heart, Lung, and Blood Institute (NIH/NHLBI) grants P01 HL078931, P50 HL52319, R01 HL66389, R01 HL71140 and R01 HL58533, an American Heart Association (AHA) Scientist Development Grant 0335308 N and a Pauline and Harold Price Endowment. We thank Avile McCullen, Lei Lin, Sarah Whiting and Elaine Lebowitz for their assistance.
References
- Bolick DR, Hackel DB, Reimer KA, Ideker RE. Quantitative analysis of myocardial infarct structure in patients with ventricular tachycardia. Circulation. 1986;74:1266–1279. doi: 10.1161/01.cir.74.6.1266. [DOI] [PubMed] [Google Scholar]
- Boyden PA, ter Keurs HE. Reverse excitation-contraction coupling: Ca2+ ions as initiators of arrhythmias. J Cardiovasc Electrophysiol. 2001;12:382–385. doi: 10.1046/j.1540-8167.2001.00382.x. [DOI] [PubMed] [Google Scholar]
- Bray MA, Lin S-F, Aliev RR, Roth BJ, Wikswo JP., Jr Experimental and theoretical analysis of phase singularity dynamics in cardiac tissue. J Cardiovasc Electrophysiol. 2001;12:716–722. doi: 10.1046/j.1540-8167.2001.00716.x. [DOI] [PubMed] [Google Scholar]
- Choi BR, Burton F, Salama G. Cytosolic Ca2+ triggers early afterdepolarizations and Torsade de Pointes in rabbit hearts with type 2 long QT syndrome. J Physiol. 2002;543:615–631. doi: 10.1113/jphysiol.2002.024570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi BR, Salama G. Simultaneous maps of optical action potentials and calcium transients in guinea-pig hearts: mechanisms underlying concordant alternans. J Physiol. 2000;529:171–188. doi: 10.1111/j.1469-7793.2000.00171.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou C-C, Nihei M, Zhou S, Tan AY, Kawase A, Macias ES, Fishbein MC, Lin S-F, Chen P-S. Intracellular calcium dynamics and anisotropic reentry in isolated canine pulmonary veins and left atrium. Circulation. 2005;111:2889–2297. doi: 10.1161/CIRCULATIONAHA.104.498758. [DOI] [PubMed] [Google Scholar]
- Chudin E, Goldhaber J, Garfinkel A, Weiss J, Kogan B. Intracellular Ca2+ dynamics and the stability of ventricular tachycardia. Biophys J. 1999;77:2930–2941. doi: 10.1016/S0006-3495(99)77126-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Nido PJ, Glynn P, Buenaventura P, Salama G, Koretsky AP. Fluorescence measurement of calcium transients in perfused rabbit heart using rhod 2. Am J Physiol Heart Circ Physiol. 1998;274:H728–H741. doi: 10.1152/ajpheart.1998.274.2.H728. [DOI] [PubMed] [Google Scholar]
- Dillon SM, Allessie MA, Ursell PC, Wit AL. Influences of anisotropic tissue structure on reentrant circuits in the epicardial border zone of subacute canine infarcts. Circ Res. 1988;63:182–206. doi: 10.1161/01.res.63.1.182. [DOI] [PubMed] [Google Scholar]
- Eisner DA, Choi HS, Diaz ME, O'Neill SC, Trafford AW. Integrative analysis of calcium cycling in cardiac muscle. Circ Res. 2000;87:1087–1094. doi: 10.1161/01.res.87.12.1087. [DOI] [PubMed] [Google Scholar]
- Fishbein MC, Meerbaum S, Rit J, Lando U, Kanmatsuse K, Mercier JC, Corday E, Ganz W. Early phase acute myocardial infarct size quantification: validation of the triphenyl tetrazolium chloride tissue enzyme staining technique. Am Heart J. 1981;101:593–600. doi: 10.1016/0002-8703(81)90226-x. [DOI] [PubMed] [Google Scholar]
- Gough WB, Megra R, Restivo M, Zeiler RH, El-Sherif N. Reentrant ventricular arrhythmias in the late myocardial infarction period in the dog. 13. Correlation of activation and refractory maps. Circ Res. 1985;57:432–445. doi: 10.1161/01.res.57.3.432. [DOI] [PubMed] [Google Scholar]
- Hayashi H, Miyauchi Y, Chou CC, Karagueuzian HS, Chen PS, Lin SF. Effects of cytochalasin D on electrical restitution and the dynamics of ventricular fibrillation in isolated rabbit heart. J Cardiovasc Electrophysiol. 2003;14:1077–1084. doi: 10.1046/j.1540-8167.2003.03234.x. [DOI] [PubMed] [Google Scholar]
- Koller ML, Riccio ML, Gilmour RF., Jr Dynamic restitution of action potential duration during electrical alternans and ventricular fibrillation. Am J Physiol Heart Circ Physiol. 1998;275:H1635–H1642. doi: 10.1152/ajpheart.1998.275.5.H1635. [DOI] [PubMed] [Google Scholar]
- Lakkireddy V, Bub G, Baweja P, Syed A, Boutjdir M, El Sherif N. The kinetics of spontaneous calcium oscillations and arrhythmogenesis in the in vivo heart during ischemia/reperfusion. Heart Rhythm. 2006;3:58–66. doi: 10.1016/j.hrthm.2005.09.018. [DOI] [PubMed] [Google Scholar]
- Liu YB, Peter A, Lamp ST, Weiss JN, Chen PS, Lin SF. Spatiotemporal correlation between phase singularities and wavebreaks during ventricular fibrillation. J Cardiovasc Electrophysiol. 2003;14:1103–1109. doi: 10.1046/j.1540-8167.2003.03218.x. [DOI] [PubMed] [Google Scholar]
- Ng GA, Cobbe SM, Smith GL. Non-uniform prolongation of intracellular Ca2+ transients recorded from the epicardial surface of isolated hearts from rabbits with heart failure. Cardiovasc Res. 1998;37:489–502. doi: 10.1016/s0008-6363(97)00255-1. [DOI] [PubMed] [Google Scholar]
- Omichi C, Lamp ST, Lin SF, Yang J, Baher A, Zhou S, et al. Intracellular Ca dynamics in ventricular fibrillation. Am J Physiol Heart Circ Physiol. 2004;286:H1836–H1844. doi: 10.1152/ajpheart.00123.2003. [DOI] [PubMed] [Google Scholar]
- Pruvot EJ, Katra RP, Rosenbaum DS, Laurita KR. Role of calcium cycling versus restitution in the mechanism of repolarization alternans. Circ Res. 2004;94:1083–1090. doi: 10.1161/01.RES.0000125629.72053.95. [DOI] [PubMed] [Google Scholar]
- Quinn FR, Currie S, Duncan AM, Miller S, Sayeed R, Cobbe SM, Smith GL. Myocardial infarction causes increased expression but decreased activity of the myocardial Na+–Ca2+ exchanger in the rabbit. J Physiol. 2003;553:229–242. doi: 10.1113/jphysiol.2003.050716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiferaw Y, Watanabe MA, Garfinkel A, Weiss JN, Karma A. Model of intracellular calcium cycling in ventricular myocytes. Biophys J. 2003;85:3666–3686. doi: 10.1016/S0006-3495(03)74784-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sipido KR, Volders PG, Vos MA, Verdonck F. Altered Na/Ca exchange activity in cardiac hypertrophy and heart failure: a new target for therapy? Cardiovasc Res. 2002;53:782–805. doi: 10.1016/s0008-6363(01)00470-9. [DOI] [PubMed] [Google Scholar]
- Solomon SD, Zelenkofske S, McMurray JJ, Finn PV, Velazquez E, Ertl G, et al. Sudden death in patients with myocardial infarction and left ventricular dysfunction, heart failure, or both. N Engl J Med. 2005;352:2581–2588. doi: 10.1056/NEJMoa043938. [DOI] [PubMed] [Google Scholar]
- Sun YL, Hu SJ, Wang LH, Hu Y, Zhou JY. Effect of beta-blockers on cardiac function and calcium handling protein in postinfarction heart failure rats. Chest. 2005;128:1812–1821. doi: 10.1378/chest.128.3.1812. [DOI] [PubMed] [Google Scholar]
- ter Keurs HE, Zhang YM, Miura M. Damage-induced arrhythmias: reversal of excitation-contraction coupling. Cardiovasc Res. 1998;40:444–455. doi: 10.1016/s0008-6363(98)00263-6. [DOI] [PubMed] [Google Scholar]
- Varro A, Lathrop DA, Surawicz B. Effects of propranolol on premature action potentials in canine Purkinje and ventricular muscle. J Cardiovasc Pharmacol. 1990;16:757–763. doi: 10.1097/00005344-199011000-00010. [DOI] [PubMed] [Google Scholar]
- Weiss JN, Karma A, Shiferaw Y, Chen PS, Garfinkel A, Qu Z. From pulsus to pulseless: the saga of cardiac alternans. Circ Res. 2006;98:1244–1253. doi: 10.1161/01.RES.0000224540.97431.f0. [DOI] [PubMed] [Google Scholar]
- Weiss JN, Qu Z, Chen PS, Lin SF, Karagueuzian HS, Hayashi H, Garfinkel A, Karma A. The dynamics of cardiac fibrillation. Circulation. 2005;112:1232–1240. doi: 10.1161/CIRCULATIONAHA.104.529545. [DOI] [PubMed] [Google Scholar]
- Wit AL, Dillon SM, Coromilas J, Saltman AE, Waldacker B. Anisotropic reentry in epicardial border zone in myocardial infarcts. Ann N Y Acad Sci. 1990;591:86–108. doi: 10.1111/j.1749-6632.1990.tb15083.x. [DOI] [PubMed] [Google Scholar]
- Wu T-J, Lin S-F, Weiss JN, Ting C-T, Chen P-S. Two types of ventricular fibrillation in isolated rabbit hearts: importance of excitability and action potential duration restitution. Circulation. 2002;106:1859–1866. doi: 10.1161/01.cir.0000031334.49170.fb. [DOI] [PubMed] [Google Scholar]
- Zaitsev AV, Guha PK, Sarmast F, Kolli A, Berenfeld O, Pertsov AM, De Groot JR, Coronel R, Jalife J. Wavebreak formation during ventricular fibrillation in the isolated, regionally ischemic pig heart. Circ Res. 2003;92:546–553. doi: 10.1161/01.RES.0000061917.23107.F7. [DOI] [PubMed] [Google Scholar]