

Abstract
Autologous stem cell transplantation (ASCT) to treat autoimmune diseases (AID) is thought to reset immunological memory directed against autoantigens. This hypothesis can only be studied indirectly because the exact nature of the pathogenetic autoantigens is unknown in most AID. Therefore, 19 children with juvenile idiopathic arthritis (JIA) or systemic lupus erythematodes (SLE) and 10 adults with multiple sclerosis (MS) were vaccinated with the T-cell-dependent neoantigen rabies and the recall antigen tetanus toxoid after, respectively before, bone marrow harvest. Both vaccinations were repeated after ASCT. All except two of the responders mounted a primary antibody response to rabies after revaccination, and 44% of the responders mounted a primary antibody response to tetanus boost after ASCT. These data show that immunological memory to a neoantigen is lost in most patients with AID after immunoablative pretreatment; however, memory to a recall antigen boosted before bone marrow harvest is only lost in part of the patients. Disease progression was arrested in all patients with JIA/SLE except one, but only in a minority of MS patients. Clinical outcome on a per case basis was not associated with the profile of the immune response toward the vaccination antigens after ASCT.
Keyword: Rabies, tetanus, vaccination, autologous stem cell transplantation, autoimmune diseases, antibody response, in vitro proliferation
Introduction
Autologous stem cell transplantation (ASCT) is studied since 1996 as a therapeutical approach for several refractory autoimmune diseases (AID). The efficacy of this treatment has been demonstrated in phase II studies [1–5]. Nowadays, more than 800 ASCTs have been performed worldwide for the treatment of progressive AID [1]. Although the pathogenesis of AID such as juvenile idiopathic arthritis (JIA) and multiple sclerosis (MS) is not fully understood, there is indirect evidence that a T cell dysregulation plays a key role in the onset and probably also in the perpetuation of the disease [6–8]. The therapeutical concept of ASCT is based on the eradication of adaptive immunity, including autoimmunity, by an immunoablative pretreatment of the patient, followed by the reinstitution of an equilibrated adaptive immunity without autoimmunity. Different treatment protocols have been practiced [2, 9]. The majority of recent studies consist of intensive immunosuppressive (also called immuno- or lymphoablative) pretreatment, followed by reinfusion of a T cell depleted autograft. These protocols are based on results obtained in animal studies [10]. At least two separate events are considered to contribute to the overall effect of ASCT for AID. First, the effect of in vivo conditioning and ex vivo T cell depletion on the eradication of immunological memory, and second, the reinstitution of a well-regulated adaptive immunity with a repertoire not causing AID. The first effect can only be evaluated indirectly, i.e., by investigating the characteristics of the immunological response to a test antigen, as proven relevant autoantigens are not exactly known. The second effect can only be studied by long-term follow-up of the patient’s disease status. Therefore, 19 children with JIA or systemic lupus erythematodes (SLE) and 10 adults with MS were vaccinated with a T-cell-dependent neoantigen (rabies) immediately after and with the recall antigen tetanus toxoid (TT) before bone marrow harvest. After immunoablative conditioning and T cell depleted ASCT patients were revaccinated from 3 months after ASCT onwards with TT, and at 6 months after ASCT with rabies. We tested the hypothesis that an effective conditioning regimen and T cell depletion of the graft would result in the elimination of antigen-specific memory T and B cells producing high-avidity IgG antibodies. Consequently, the immune response to rabies would show characteristics of a primary response before and after ASCT. Finally, the course of the original AID after ASCT was followed, and the therapeutical effect of ASCT was correlated with the profile of the immune response toward the vaccination antigens.
Materials and Methods
Study Population
Between March 1997 and July 2002, 19 children [17 JIA and 2 SLE; 7 girls; median age 9 years (range 4–15)] and 10 adults with MS [7 women; median age 37 years (range 23–50)] from three different centers, were prospectively enrolled into this study [4, 5]. Patient characteristics are given in Table I. Median disease duration before transplantation was 70 months (range 13–179) in JIA/SLE and 60 months (range 24–144) in MS patients. Immunomodulating drugs were stopped 1 month before the bone marrow harvest and the first rabies immunization. A written informed consent was obtained from all patients and/or their parents, and the Local Committee on Medical Ethics approved the study. Furthermore, in 18 healthy volunteers [9 women; median age 31 years (range 19–49)], both the B and T cell immune responses to a single rabies vaccination and a boost 3 months later were investigated. These results served as reference data [11]. Before this study, neither the patients nor the healthy controls had been exposed to rabies or had been vaccinated with (inactivated) rabies vaccine.
Table I.
Patient Characteristics
| Patient | Diagnosis | Sex | Age at ASCT (years) | Medication history before ASCT | Dx–Tx (months) |
|---|---|---|---|---|---|
| 1 | MS | F | 48 | MP | 60 |
| 2 | MS | M | 47 | MP | 60 |
| 3 | MS | F | 44 | MP, IFNβ | 48 |
| 4 | MS | F | 31 | MP, IFNβ | 60 |
| 5 | MS | F | 37 | MP | 60 |
| 6 | MS | F | 50 | MP | 48 |
| 7 | MS | F | 41 | MP, IFNβ, IVIG | 24 |
| 8 | MS | M | 23 | MP, IFNβ, IVIG | 72 |
| 9 | MS | F | 34 | MP, IFNβ | 36 |
| 10 | MS | M | 34 | MP, IFNβ | 144 |
| 11 | sJIA | F | 7 | NSAID, St, MTX, CsA, AZA, IVIG | 69 |
| 12 | pJIA | F | 8 | NSAID, St, MTX, CsA, | 47 |
| 13 | sJIA | M | 11 | NSAID, St, MTX, CsA, | 108 |
| 14 | sJIA | F | 11 | NSAID, St, MTX, CsA, AZA, IVIG, SSZ | 73 |
| 15 | sJIA | M | 14 | NSAID, St, MTX, AZA, SSZ, AURO | 117 |
| 16 | pJIA | M | 6 | NSAID, St, MTX, SSZ | 31 |
| 17 | sJIA | M | 10 | NSAID, St, MTX, CsA, AZA, CYC, HCQ, SSZ | 71 |
| 18 | sJIA | M | 9 | NSAID, St, MTX, CsA, AZA, IVIG | 55 |
| 19 | sJIA | F | 15 | NSAID, St, MTX, CsA, AZA, HCQ | 135 |
| 20 | pJIA | M | 12 | NSAID, St, MTX, CsA, HCQ, SSZ, AURO | 86 |
| 21 | sJIA | F | 4 | NSAID, St, MTX, CsA | 36 |
| 22 | sJIA | F | 5 | NSAID, St, MTX, CsA | 27 |
| 23 | pJIA | M | 5 | NSAID, St, MTX, CsA, SSZ | 75 |
| 24 | sJIA | M | 8 | NSAID, St, MTX, CsA, anti-TNF-α | 27 |
| 25 | sJIA | M | 8 | NSAID, St, MTX, CsA | 80 |
| 26 | sJIA | M | 4 | NSAID, St, MTX, CsA | 13 |
| 27 | sJIA | M | 12 | NSAID, St, MTX, CsA, anti-TNF-α | 106 |
| 28 | SLE | M | 15 | NSAID, St, AZA, CYC, HCQ | 175 |
| 29 | SLE | F | 15 | NSAID, St, AZA, IVIG, CYC | 179 |
Anti-TNF-α anti-tumor necrosis factor alpha therapy, AURO auromyosine, AZA azathioprine, CsA cyclosporine A, CYC cyclophosphamide, Dx–Tx months from diagnosis to ASCT, F female, HCQ hydroxychloroquine, INF β interferon β, IVIG intravenous immunoglobulins, M male, MP methylprednisolone, MS multiple sclerosis, MTX methotrexate, NSAID non steroidal anti-inflammatory drugs, pJIA\ polyarticular JIA, sJIA systemic JIA, St steroids, SLE systemic lupus erythematodes, SSZ sulfasalazine
Autologous Stem Cell Transplantation
ASCT was performed according to the European League against Rheumatism (EULAR) and the European Group for Blood and Marrow Transplantation (EBMT) guidelines for bone marrow transplantation in AID [12–14]. In short, in JIA and SLE patients, the conditioning regimen consisted of antithymocyte globulin i.v. (rabbit-ATG, Imtix-SangStat, Lyon, France) at days −9 to −6 (cumulative dose 20 mg/kg), cyclophosphamide i.v. at days −5 to −2 (cumulative dose 200 mg/kg) and low dose total body irradiation at day −1 (4 Gy, single dose), followed by reinfusion of T cell depleted autologous stem cells at day 0 [5, 15]. In MS patients, the conditioning regimen consisted of horse antithymocyte globulin i.v. (ATG, Mérieux Marcy L’Etoile, France) at days −7 to −3 (cumulative dose 75 mg/kg), cyclophosphamide i.v. at days −4 and −3 (cumulative dose 120 mg/kg) and high-dose total body irradiation at days −2 and −1 in two fractions of 5 Gy daily, followed by reinfusion of the T cell depleted autograft at day 0 [4]. Aspirates of unprimed bone marrow were the source of autologous hematopoietic stem cells in all patients. The bone marrow graft was depleted of mature T cells by two different methods, i.e., either by T cell depletion using immunorosette-sedimentation with specific monoclonals (anti-CD2 and anti-CD3) coupled to autologous red blood cells (n = 16) or by positive CD34+ selection by CliniMACS (Miltenyi Biotec, Munich, Germany; n = 13), performed at Sanquin Pharmaceutical Services (Sanquin, Amsterdam) under Good Manufacturing Practice (GMP) conditions [16]. The T cell depleted graft was cryopreserved until further use.
Vaccines
The rabies vaccine used was the human diploid cell vaccine (HDCV), manufactured by the Institute Pasteur Mérieux MSD, Lyon, France (commercial lot number NO 976-8). It is a safe and effective antigen for measurement of both B and T cell immune responses to a neoantigen in healthy individuals [11]. There are no contra-indications for vaccination of immunocompromised patients [17, 18]. For TT vaccination, DTP [National Institute for Public Health and the Environment (RIVM), Bilthoven, The Netherlands] was used, which contains diphtheria toxoid, TT, and inactivated poliovirus types 1, 2, and 3. The DTP vaccination before bone marrow harvest and ASCT was considered to be a booster vaccination because all patients have been vaccinated in the past as part of the national vaccination program for children in The Netherlands.
Vaccination and Sampling Protocol
One milliliter of HDCV was given intramuscularly directly after the bone marrow harvest (≥4 weeks before conditioning) in the patients; this was repeated at 6 months after ASCT. DTP vaccination was given at least 1 month before bone marrow harvest and three subsequent DTP vaccinations were given at 3, 4, and 5 months, respectively, after ASCT (see Fig. 1). Blood samples were drawn at days 0 and 28 after each vaccination. Any adverse events were registered. Sera were stored at −20°C and PBMC, obtained by Ficoll-Isopaque gradient centrifugation, were viably stored in liquid nitrogen. One MS patient turned out to have never been vaccinated with TT in the past; her primary anti-TT responses were not included in the analysis. Two MS patients and five JIA patients were excluded from analysis of antirabies immune responses because of incomplete vaccination or sampling.
Fig. 1.
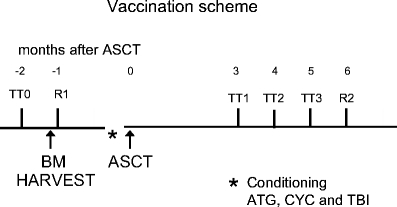
Vaccination scheme. ASCT, autologous stem cell transplantation; TT0, tetanus vaccination pre ASCT (and before bone marrow harvest); TT1, tetanus vaccination at 3 months post ASCT; TT2, tetanus vaccination at 4 months post ASCT; TT3, tetanus vaccination at 5 months post ASCT; R1, rabies vaccination after bone marrow harvest; R2, rabies vaccination 6 months post ASCT. Conditioning: ATG, anti-thymocyte globulins, CYC, cyclophosphamide, TBI, total body irradiation.
Antigen for in Vitro Assays
Rabies virus Pitman Moore (PM) strain (Wistar PM/WI-38-1503-3 M) was propagated in primary dog kidney cells (DKCV), concentrated and purified by ultrafiltration and inactivated with β-propiolactone [19]. DKCV was used as coating antigen in ELISA, and compared with the fifth International Standard for Rabies Vaccine (Statens Serum Institut, Copenhagen, Denmark, supplied by NIBSC, Potters Bar, UK) [11, 20, 21]. The results obtained from ELISA assays using either the fifth International Standard for Rabies Vaccine or DKCV as coating material were comparable (data not shown). The TT antigen stock contained 150 Lf/ml (RIVM, [22]).
Antibody Quantification
Rabies
The concentrations of IgG, IgG subclasses, IgA, and IgM antirabies antibodies were measured by sandwich ELISA technique as described in detail previously [11]. The second International Standard reference serum of the WHO containing 30 IU/ml of total IgG antirabies [23] (Statens Serum Institut) and an in house prepared secondary reference serum (total IgG antirabies, 3.3 IU/ml) were used to express the IgG data in IU/ml. For IgG subclass antirabies and IgM and IgA antirabies, no reference sera are available; therefore, our secondary reference serum was used as standard, as described previously [11]. Indirectly, IgG subclass antirabies could be expressed in IU/ml by determination of the relative contribution of the IgG subclasses to the total IgG antirabies response [11]. The responder criteria for rabies are defined as a twofold increase in antirabies antibody titer, either in IgM or IgG, postvaccination. Criteria for a secondary response are the concentration of antirabies antibodies and occurrence of an isotype switch and avidity maturation [11].
Tetanus Toxoid
Quantification of IgG and IgG subclasses antibodies against TT was performed with an antibody-capture enzyme-linked immunosorbent assay, as previously described [22, 24]. In short, 96-well polystyrene microtiter plates (Costar, Cambridge, MA, USA) were coated with TT in a concentration of 1.5 Lf/ml in carbonate buffer pH 9.6 at 37°C for 3 h. After a 1-h blocking step with 1% bovine serum albumin (BSA, Sigma, St Louis, MO, USA) in PBS, serum samples and standard sera diluted in PBS/0.05% Tween 20 /1% BSA were added and incubated at 37°C for 2 h. The quantification of IgG and IgG subclass anti-TT antibodies was according to the method used for antirabies antibodies [11]. Reference sera for TT were in house prepared secondary standards calibrated on international standards [total IgG anti-TT 24 IU/ml (=122 μg/ml)]. The criteria for a tetanus response after booster vaccination are defined as ≥1.25-fold increase in anti-TT antibodies and a minimum titer of 5 μg/ml anti-TT antibodies or a twofold increase in the antibody concentration and a minimum titer of 1 μg/ml anti-TT antibodies [22]. A primary response to tetanus after ASCT is defined as an increase in antibody concentration as mentioned above in combination with at least a 10% decrease in avidity index, compared to the response upon TT vaccination before ASCT.
Antibody Avidity
In relevant cases, the avidity of IgG1 antirabies and IgG1 anti-TT was measured by a modified elution ELISA as described in detail previously [11, 25]. In this assay, antigen-bound antibodies were eluted with a range of concentrations (range 0.5–4.5 M) of the chaotropic agent sodium thiocyanate (NaSCN). The relative avidity index is defined as the molarity of NaSCN at which 50% of the amount of IgG subclass antibodies remains bound to the coated rabies or tetanus antigen.
Lymphocyte Proliferation
Triplicate cultures of 1 × 105 PBMC per well were performed in 96-well round-bottom microtiter plates (Costar) in a final volume of 200 μl RPMI 1640 glutamax 1 (Invitrogen, Carlsbad, CA, USA) supplemented with 10% heat-inactivated pooled human AB serum, penicillin (100 U/ml; Invitrogen) and streptomycin (100 μg/ml; Invitrogen). Cells were stimulated with DKCV (0.5 μg/ml) or TT (5 Lf/ml) for 5 days at 37°C and 5% CO2. Subsequently, 1 μCi/well 3H-thymidine (Amersham, San Francisco, CA, USA) was added 18 h before harvesting. 3H-thymidine uptake of cultured PBMC was measured as counts per minute (cpm) of triplicate cultures by liquid scintillation counter (Wallac, Turku, Finland) and expressed as a stimulation index (SI; ratio mean cpm antigen/mean cpm medium). SI ≥ 3.0 was considered as evidence of antigen-induced proliferation. As positive control, the proliferative capacity of the PBMC was tested after mitogenic or polyclonal stimulation, using phytohemagglutinin (PHA, 5 μg/ml Murex, Dartford, England) or anti-CD3 monoclonal antibody (coat 0.1 μg/ml OKT3), followed by 3H-thymidine incorporation at day 4 [11].
Statistical Analysis
The geometrical mean titer (GMT), with its 95% confidence interval (CI), expressed in IU/ml for IgG and IgG subclasses and in U/ml for IgA and IgM antirabies, and in μg/ml for IgG and IgG subclasses anti-TT antibodies, were calculated for each time-point of sampling. Differences between time-points with respect to GMT values for all parameters were assessed by the paired Student’s t test. The differences between a primary and secondary immune response, i.e., concentration of antibody levels, isotype switch and avidity maturation, were evaluated using the paired t test. Differences between groups were determined with an independent t test. Lymphocyte proliferative responses after first and booster vaccination were compared by the paired t test. Statistical analysis was carried out using SPSS 12.0.1; p < 0.05 was considered to be statistically significant.
Results
Transplant Procedure and Outcome
Data on the immunological responses (B and T cellular) after vaccination and on the clinical outcome of the patients at 2 years post-ASCT are given in Table II. More details on the clinical course of the patients with JIA and MS were published previously [4, 5]. The graft contained a median number of 1.5 × 106 CD34+ cells/kg body weight (range 0.4 to 6.0 × 106) in JIA/SLE patients and a median number of 1.1 × 106 CD34+ cells/kg body weight (range 0.4 to 1.9 × 106) in MS patients. T cell depletion was achieved, leaving a median of 2.2 × 104 CD3+ cells/kg body weight (range 0.2 to 35.4 × 104) in the graft of JIA/SLE patients and about 0.4 × 104 CD3+ cells/kg body weight (range between the detection level of 0.3 and 3.3 × 104) in MS patients. The immunoablative conditioning resulted in loss of virus-specific immunological memory, as evidenced by a high frequency of reactivation of herpetic viruses, as published previously [4, 5]. Early after transplant two JIA patients died due to a fatal macrophage activation syndrome (MAS) at 18 days and 4 months post-ASCT [3, 26]; only their TT and rabies data pre-ASCT are included for analysis. No adverse effects were reported in patients after rabies and DTP vaccinations. Reconstitution of different lymphoid subsets, including CD3+ T cells and CD19+ B lymphocytes, was determined in absolute cell counts per microliter blood by flow cytometry. At time of TT revaccination, 3 months post-ASCT, median CD3+ T cells counts were 215/μl (range 22 to 3,363) for JIA/SLE patients and 354/μl (range 100 to 1,040) for MS patients. Three months post-ASCT median CD19+ B cell counts were 129/μl (range 2 to 690) for JIA/SLE patients and 143/μl (range 59 to 424) for MS patients. At the time of revaccinations post-ASCT all patients showed normal in vitro proliferative responses of PBMC after mitogenic or polyclonal stimulation (data not shown).
Table II.
Immune Response to TT and Rabies and Clinical Outcome After ASCT
| Patient | Diagnosis | Humoral responders post-ASCT | Clinical outcome 2 years post ASCT | Follow-up (months) | |||
|---|---|---|---|---|---|---|---|
| TT | Primary reponse TT | Rabies | Primary response rabies | ||||
| 1 | MS | + | − | nv | nv | Progressive | 97 |
| 2 | MS | + | + | − | ne | Stable | 94 |
| 3 | MS | pv | ne | + | + | Progressive | 86 |
| 4 | MS | − | ne | + | + | Improved | 81 |
| 5 | MS | − | ne | + | + | Improved | 79 |
| 6 | MS | − | ne | + | + | Progressive | 76 |
| 7 | MS | − | ne | + | + | Progressive | 61 |
| 8 | MS | + | + | nv | nv | Progressive | 59 |
| 9 | MS | − | ne | − | ne | Progressive | 55 |
| 10 | MS | − | ne | − | ne | Progressive | 54 |
| 11 | sJIA | + | + | nv | nv | Remission | 118 |
| 12 | pJIA | + | − | nv | nv | Remission | 116 |
| 13 | sJIA | + | − | nv | nv | Partial responder | 111 |
| 14 | sJIA | + | − | + | + | Remission | 107 |
| 15 | sJIA | + | − | + | + | Remission | 106 |
| 16 | pJIA | + | − | + | − | Partial responder | 104 |
| 17 | sJIA | + | − | + | + | Partial responder | 103 |
| 18 | sJIA | + | + | + | + | Remission | 99 |
| 19 | sJIA | Deceased | Deceased | Deceased | Deceased | Deceased (MAS) | 4 |
| 20 | pJIA | + | − | + | + | Remission | 95 |
| 21 | sJIA | + | + | + | + | Remission | 87 |
| 22 | sJIA | + | + | + | −a | Partial responder | 79 |
| 23 | pJIA | + | − | + | + | Partial responder | 79 |
| 24 | sJIA | + | − | + | + | Remission | 77 |
| 25 | sJIA | + | + | nv | nv | Failure/ongoing disease | 72 |
| 26 | sJIA | Deceased | Deceased | Deceased | Deceased | Deceased (MAS) | 0.6 |
| 27 | sJIA | + | + | + | + | Remission | 67 |
| 28 | SLE | + | ne | + | + | Partial responder | 92 |
| 29 | SLE | + | ne | + | + | Remission | 98 |
Partial responder: partial response was defined as improvement of disease activity according to the core set variables after reinstitution on low dose DMARDS or steroids after a relapse [5]. Clinical outcome at 24 months after ASCT is indicated
MAS macrophage activation syndrome, MS multiple sclerosis, ne not evaluable because of no response or too low antigen-specific antibody titers for measurement of avidity, nv not vaccinated, pJIA polyarticular JIA, pv primary vaccination, sJIA systemic JIA, TT tetanus toxoid
aParticular patient with a second BM harvest after rabies vaccination (see Fig. 6)
Humoral Response to Vaccinations
Tetanus Toxoid
In Fig. 2 (upper part), the humoral response (total IgG) to TT upon vaccination before bone marrow harvest and after ASCT for children with JIA/SLE and adults with MS is shown. All but one JIA and all MS patients responded to the TT vaccination pre-ASCT (p = 0.01 and p = 0.04, respectively). The antibody concentrations after vaccination of most patients are within the range of TT booster responses in healthy adult controls (gray shaded area upper part of Fig. 2) [22]. After the conditioning regimen, the concentration of total IgG anti-TT in JIA/SLE patients decreased to the same level as before vaccination before the bone marrow harvest (p = 0.74). A significant and increasing response to TT was found after subsequent vaccinations 3, 4, and 5 months post-ASCT (TT3 versus TT1 p = 0.01). In the pediatric cohort, all 17 evaluable patients could be classified as responders after one to three booster vaccinations post-ASCT (Table II and Fig. 3). In two SLE responders, the avidity index could not be measured due to low antigen-specific antibody titers. Based on the avidity index, 6 of the remaining 15 JIA/SLE responders showed characteristics of a primary antibody response (40%), 9 showed characteristics of a secondary response (60%, data not shown).
Fig. 2.
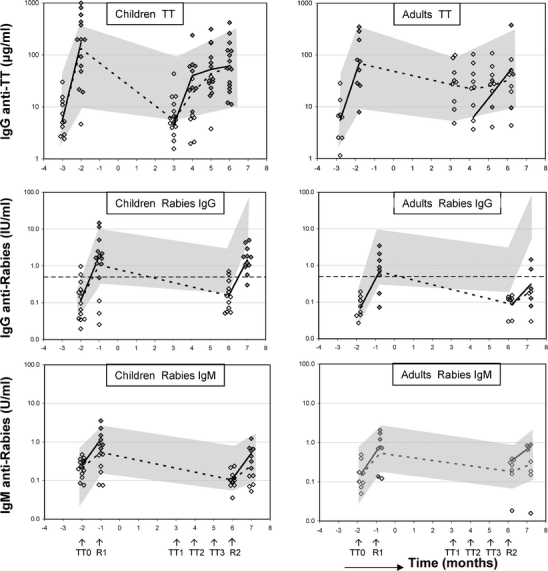
Total IgG response to tetanus and rabies in JIA/SLE and MS patients. Individual data and geometric mean titer (GMT responders, solid lines; GMT whole cohort, dashed lines) of IgG anti-TT (ig/ml) and IgG and IgM antirabies (IU/ml and U/ml, respectively) for children (JIA/SLE) and adult (MS) patients are depicted. Gray-shaded area in upper part of figure represents the total IgG anti-TT response in healthy adults to a TT booster vaccination with its 95% confidence interval (CI). TT0, tetanus vaccination pre ASCT (and before bone marrow harvest); TT1, tetanus vaccination at 3 months post ASCT; TT2, tetanus vaccination at 4 months post ASCT; TT3, tetanus vaccination at 5 months post ASCT. Gray-shaded area in middle and lower part of figure represents the 95% CI for the primary and secondary IgG and IgM response to rabies and the 95% CI in healthy adults, respectively. R1, rabies vaccination after bone marrow harvest; R2, rabies vaccination 6 months post ASCT. Horizontal dashed line represents WHO seroconversion level (IgG antirabies 0.5 IU/ml) [11], open diamonds represent non-responders, filled diamonds represent responders.
Fig. 3.
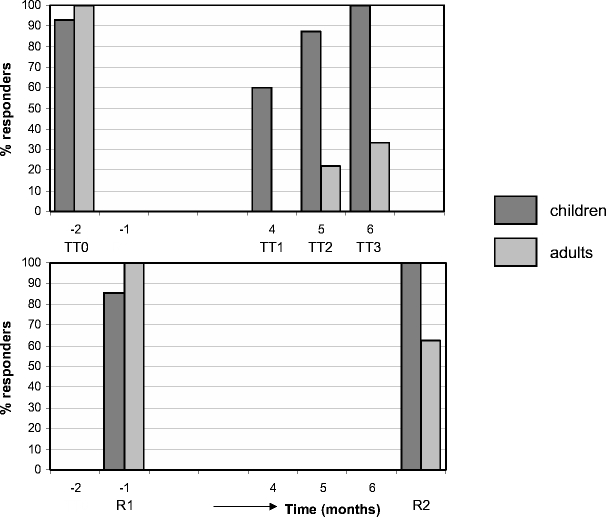
Cumulative response rate to tetanus and rabies in JIA/SLE and MS post-ASCT. The cumulative percentage of responders is indicated for both patient cohorts (dark gray represent children, light gray represent adults). TT0, tetanus vaccination pre ASCT (and before bone marrow harvest); TT1, tetanus vaccination at 3 months post ASCT; TT2, tetanus vaccination at 4 months post ASCT; TT3, tetanus vaccination at 5 months post ASCT. R1, rabies vaccination after bone marrow harvest; R2, rabies vaccination 6 months post ASCT.
In MS patients, the concentration of total IgG anti-TT antibodies after conditioning and before revaccination post-ASCT decreased to 39% (p = 0.02) of the amount measured post-TT vaccination before ASCT. This is comparable to what has been reported in healthy adults 12 months after TT booster vaccination [22]. In contrast to the JIA/SLE cohort, only three out of nine evaluable MS patients (33%) could be classified as responders after two to three booster vaccinations (Table II and Fig. 3). Two of them showed characteristics of a primary antibody response based on a decrease in avidity index.
Rabies
IgG and IgM antirabies antibody responses of JIA/SLE and MS patients are shown in Fig. 2 (middle part and lower part, respectively). As in healthy controls [11], IgG antirabies antibodies were mostly of the IgG1 subclass, less were found in IgG3, and no antibodies were detectable in IgG2 and IgG4 (data not shown). After the first rabies vaccination before ASCT 86% (12/14) of JIA/SLE patients and 100% (8/8) of MS patients could be classified as responder (p = 0.03 and p = 0.04, respectively; Figs. 2 and 3), compared with 100% of healthy controls [11]. After conditioning, the antibody titers decreased to prevaccination levels in all patients. After a second rabies vaccination at 6 months post-ASCT, all evaluable JIA/SLE patients fulfilled the criteria for responder, compared to five of eight MS patients (63%; Fig. 2, middle part, and Fig. 3). In only 1 of the 12 responders of the JIA/SLE cohort and in none of the responders of the MS cohort, a significant increase in avidity index of IgG1 antirabies after secondary vaccination post-ASCT was observed (Fig. 4). The median avidity index of IgG1 antirabies pre- and post-ASCT for the JIA/SLE cohort was 0.7, median avidity index was 0.6 pre-ASCT and 0.7 post-ASCT for the MS group, comparable to the avidity index in healthy adults after a first vaccination [11]. In most JIA/SLE patients and MS patients an IgM response to rabies was present pre- and post-ASCT (Fig. 2, lower part). An isotype switch from IgM to IgG was not observed after ASCT.
Fig. 4.
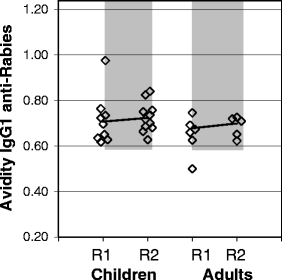
Relative avidity index of IgG1 antirabies in JIA/SLE and MS patients. Relative avidity index (M NaSCN) of IgG1 antirabies in JIA/SLE (children) and MS (adults) patients after a primary and booster rabies vaccination pre- and post-ASCT, respectively. Gray-shaded area represents the 95% CI for the avidity index for IgG1 antirabies measured after a primary and booster vaccination with rabies in healthy adult controls [11].
T cell Response to Vaccinations
PBMC from six JIA and all MS patients were available and could be tested for in vitro proliferative T cell responses to rabies and TT. Results are shown in Fig. 5. Before TT vaccination pre-ASCT, a proliferative response to tetanus (SI > 3) could be elicited in all JIA patients [SI JIA cohort geometric mean (GM) 33.2, range 19.7 to 59.4] and MS patients (SI MS cohort GM 14.1, range 5.7 to 107.7). After conditioning a significant decrease in SI was found at 3 months post-ASCT in both patient groups (SI JIA cohort GM 1.7, range 0.8 to 3.9, SI MS cohort GM 2.6, range 1.0 to 8.6; total patient cohort p = 0.01). Before revaccination post-ASCT in two of four JIA patients and in seven of ten MS patients, no antigen-specific proliferative response could be detected. After one TT revaccination, a proliferative response was found in all patients, except one MS patient (Fig. 5; p = 0.02).
Fig. 5.
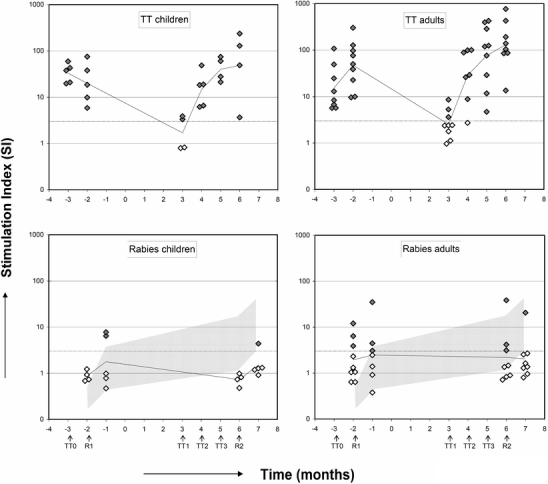
Stimulation index (SI) of rabies and tetanus induced proliferation in cultures of PBMC from JIA patients, MS patients. Arrows indicate time-points of vaccinations. Gray-shaded area in lower panel represents the SI after a single and booster rabies vaccination in healthy adults with its 95% confidence interval (CI) (11). Open diamonds represent nonresponders, closed diamonds represent responders (SI ≥ 3).
At 4 weeks after primary rabies vaccination before ASCT, two of five JIA patients (SI JIA cohort GM 1.8, range 0.5 to 7.8) and three of seven MS patients (SI MS cohort GM 2.0, range 0.4 to 35) showed a SI ≥ 3. Four weeks after a rabies revaccination post-ASCT, one of five JIA patients (SI JIA cohort: GM 1.5, range 0.9 to 4.4) and one of nine MS patients (SI MS cohort: GM 2.1, range 0.8 to 20.6) showed a proliferative response (Fig. 5). No significant increase in the proliferative response after rabies revaccination was observed (p = 0.57).
Course of the Disease and Responses to TT and Rabies Vaccination
No clear relation could be found between clinical outcome at 24 months after ASCT and characteristics of the immune response to vaccinations in the JIA/SLE and MS cohorts (see Table II). In the JIA/SLE cohort, six patients showed a primary anti-TT humoral response after ASCT; four of them were in remission, one was a partial responder, and one had ongoing disease activity. Nine JIA/SLE patients showed a memory anti-TT response; five of them were in remission, four were classified as partial responders. After ASCT, 11 JIA/SLE patients showed a primary humoral antirabies response; 8 of them were in remission, and 3 were partial responders. One JIA patient showed an increase in avidity index after rabies booster post-ASCT; this patient was classified as a partial responder. In another particular JIA patient (patient 22, Tables I and II) a memory response upon rabies vaccination after ASCT, characterized by a fast rise of antibody titer at 1 week combined with a clear increase in avidity of antirabies antibodies, could be observed (Fig. 6). In this case, bone marrow had to be harvested twice because of an intermediate change of the strategy of T cell depletion of the graft, as described in an amended protocol after the occurrence of lethal MAS in two cases [5, 26]. However, in this particular patient, the first rabies vaccination had already been given after the first harvest. The second graft, theoretically containing rabies-specific memory B and T cells, was used for transplantation. Three of nine MS patients were TT responders post-ASCT (Table II); two of them showed a primary anti-TT humoral response (one with progressive disease and one with stable disease). The other MS patient showing a memory anti-TT response post-ASCT had progressive disease. All five rabies responders post-ASCT in the MS cohort showed a primary response; three with progressive disease and two with an improved course. Despite a profound lymphopenia of CD4+CD45RA+ T cells for at least 6–12 months in the whole group of patients [5, 27], no correlation was observed between the number of CD4+ (CD4+CD45RO+ and CD4+CD45RA+) T cells at the time points of vaccination post-ASCT and the level of antibody responses elicited to tetanus and rabies vaccination (data not shown). In addition, no relation could be detected between the number of CD3+ cells infused with the graft and the clinical outcome.
Fig. 6.
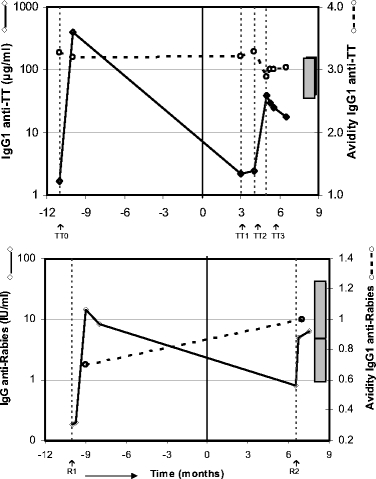
Antirabies and antitetanus response in a particular JIA patient. One JIA patient was grafted with a second bone marrow harvest at 5 months before transplantation and after rabies as well as tetanus vaccination. Arrows represent the time points of vaccination. The concentrations of total IgG antitetanus antibodies (μg/ml) and total IgG antirabies antibodies (IU/ml) are shown by diamonds and solid lines. The relative avidity index of IgG1 anti-TT and antirabies antibodies are depicted by open circles and dotted lines. The bars represent the 95% confidence interval of the relative avidity of IgG1 antirabies after primary (R1) and secondary vaccination (R2) with rabies and IgG1 anti-TT after booster vaccination with TT of healthy adults.
Discussion
This study was performed to investigate the effect of conditioning in vivo and graft manipulation ex vivo on the elimination of adaptive immunological memory in the setting of ASCT [15]. This was tested using two T-cell-dependent vaccine antigens, the neoantigen rabies (HDCV) [11] and the recall antigen TT, and specific antibody production and antigen-stimulated T cell proliferation as read-outs. Before ASCT, nearly all evaluable children (JIA/SLE cohort) and all adult patients (MS cohort) mounted a primary humoral response to rabies and a memory response to TT and all patients mounted a cellular response to TT. After ASCT, striking differences between the pediatric and adult patient groups were observed: 100% of JIA/SLE patients, but only 33% of the MS patients reached the humoral responder status after the third tetanus revaccination (see Fig. 3). Based on avidity testing, about 56% of the responders showed a memory response. All JIA/SLE patients and 63% of the MS patients were able to respond to the rabies vaccination at 6 months after ASCT. In all but two the response to the rabies revaccination had characteristics of a primary response, i.e., neither an isotype switch from IgM to IgG nor avidity maturation had occured. These data clearly show that in most cases, the immunological memory to a T-cell-dependent neoantigen was eradicated after conditioning. A vaccination with the T-cell-dependent recall antigen TT was given shortly before bone marrow harvest. Therefore, the response to TT after ASCT not only reflects the effect of the conditioning, but also the potential transfer of adaptive immunity by the graft. The humoral response to TT was severely suppressed for a prolonged period of time in MS patients; it was present in 60% in JIA/SLE patients after the first revaccination and restored rapidly after repeated vaccinations (see Fig. 3). The deficient anti-TT antibody production in adult patients may in part be explained by a higher IgG anti-TT titer remaining present after ASCT and before revaccination, compatible with an ongoing production of specific IgG antibodies by radioresistent plasma cells [28–30]. This phenomenon has also been observed in healthy multivaccinated adults [22] and after ASCT for malignant diseases in adults [31, 32]. Consistent with this is the finding that preexisting oligoclonal IgG-bands remain present for a long time in the cerebrospinal fluid in four out of five of the included MS patients after ASCT [4]. This was also observed by others [33, 34]. The meaning of the latter finding for the course of the pathological process is unclear. Two other factors possibly relevant for the obvious difference in responses to a recall antigen in adults versus children are the more intense conditioning and the more rigorous T cell depletion of the graft in the MS cohort. The biological effective dose (BED) of the TBI can be calculated [35] to be 15 Gy in adults versus 5.6 Gy in children. The former equals the intensity of myeloablative pretreatment protocols for SCT in hematological malignancies. The T cell depletion of the autograft in MS patients was at least five times more rigorous than in children, and probably prohibited any transfer of T cell memory. In fact, similar findings were previously reported by our group and others [36–41], after myeloablative conditioning, in vivo T cell depletion and allogeneic or autologous SCT. The transfer of a T-cell-dependent humoral memory response after immunoablative conditioning and SCT with a not rigorously T cell depleted graft (leaving 1.0 × 104 CD3+ cells/kg body weight) was demonstrated by chance in the present study by the particular JIA case who was vaccinated with rabies before the (second) bone marrow harvest: this child mounted a secondary response after rabies revaccination post-ASCT (see Fig. 6). In conclusion, the results of the present study indicate that immunoablative conditioning may be sufficient to eliminate immunological memory generated against a neoantigen given after graft harvest and before conditioning. On the other hand, as illustrated by the secondary humoral response to TT in 60% of the children after ASCT, the same transplant procedure including moderately stringent T cell depletion of the graft was insufficient to eliminate immunological memory for a recall antigen boosted before graft harvest. The therapeutical effect on the disease was quite different between children and adults: 16 out of 17 evaluable children were cured or remitted of disease progression, whereas only 3 out of 10 evaluable adults improved or had stable disease during follow-up. Whether the difference in kinetics of T-cell-mediated immunological recovery after a transient suppression, i.e., more rapid in children than in adults, influenced the post-ASCT course of the autoimmune disease cannot be substantiated in this evaluative study. Apart from the obvious differences in etiopathology of JIA/SLE and MS other transplant-related factors may have contributed to the disappointing therapeutical effect in MS patients. For instance, the graft of children contained a higher number of T cells than the graft of adults, due to differences in depletion techniques. Although it is generally thought that the recurrence of disease post-ASCT either reflects the presence of autoagressive cells in the stem cell graft or the persistence of these cells in the host, evidence is increasing that further depletion of T cells is not the way to improve the outcome of ASCT and can possibly even lead to more relapses, as seen in our MS patients; the lack of therapeutical effect may be the result of the depletion of regulatory T cells [3, 42, 43]. Autoantigen-specific regulatory T cells have so far not been studied in AID; the reappearance of nonspecific CD25+FoxP3+ T cells after ASCT has been described by de Kleer et al. [44], but their exact role in controlling JIA is yet unknown. In MS, such studies have not been performed. The severe and prolonged B and T cell immune dysfunction following the intensive (rather myeloablative) pretreatment of MS patients in this study, as shown by their slow immune recovery for vaccine antigens following ASCT, may have been inappropriate for a regulated and equilibrated nonautoimmune restoration of their adaptive immunity. Further study on autoantigen-specific regulatory T cells before and after ASCT in T-cell-dependent AID may throw more light onto the mechanism behind dysregulated immunity in these patients.
Concluding Remarks
Strong reduction in mature lymphocytes, including memory cells, is likely to reduce the number of autoagressive lymphocytes in patients with an immune-mediated disorder. In the setting of ASCT, immunoablative conditioning eliminated immunological memory for a neoantigen given after the graft harvest, but did not consistently eliminate immunological memory for a recall antigen, boosted before harvest following nonrigorous T cell depletion of the autograft. The clinical benefit of ASCT goes beyond mere temporary suppression of adaptive immunity. It is hypothesized that a relatively rapid recovery of the T cell immune capacity, directed by a still adequately functioning thymus, will lead to a diverse T cell repertoire, capable of differentiating between foreign and own antigens, i.e., including newly presented (formerly sequestered) autoantigens. On the other hand, a delayed repopulation of the heavily depleted host tissues by naïve selected T cells, including regulatory T cells, may have missed the window of opportunity to regain tolerance to autoantigens.
Acknowledgment
This work was supported by research grants from NWO, The Netherlands Organisation for Scientific Research (Grant Number 940-37-004) and the National League against Rheumatism (National Reumafonds Grant Number NR 901). We are grateful to Prof. Dr. A.D.M.E. Osterhaus for providing DKCV. We thank G. Ruijgrok for technical assistance with the ELISA.
Reference
- 1.Hough RE, Snowden JA, Wulffraat NM. Haemopoietic stem cell transplantation in autoimmune diseases: a European perspective. Br J Haematol 2005;128:432–59. [DOI] [PubMed]
- 2.Tyndall A, Daikeler T. Autologous hematopoietic stem cell transplantation for autoimmune diseases. Acta Haematol 2005;114:239–47. [DOI] [PubMed]
- 3.de Kleer IM, Brinkman DM, Ferster A, Abinun M, Quartier P, van der NJ, et al. Autologous stem cell transplantation for refractory juvenile idiopathic arthritis: analysis of clinical effects, mortality, and transplant related morbidity. Ann Rheum Dis 2004;63:1318–26. [DOI] [PMC free article] [PubMed]
- 4.Samijn JP, te Boekhorst PA, Mondria T, van Doorn PA, Flach HZ, van der Meche FG, et al. Intense T cell depletion followed by autologous bone marrow transplantation for severe multiple sclerosis. J Neurol Neurosurg Psychiatry 2006;77:46–50. [DOI] [PMC free article] [PubMed]
- 5.Brinkman DMC, de Kleer IM, Ten Cate R, van Rossum MAJ, Bekkering WP, Fasth A, van Tol MJD, Kuis W, Wulffraat NM,Vossen JM. Autologous stem cell transplantation in children with severe progressive systemic or polyarticular juvenile idiopathic arthritis: long-term follow-up of a prospective clinical trial. Arthritis Rheum 2007 56(7):2410–2421. [DOI] [PubMed]
- 6.Grom AA, Giannini EH, Glass DN. Juvenile rheumatoid arthritis and the trimolecular complex (HLA, T cell receptor, and antigen). Differences from rheumatoid arthritis. Arthritis Rheum 1994;37:601–7. [DOI] [PubMed]
- 7.Grom AA, Hirsch R. T-cell and T-cell receptor abnormalities in the immunopathogenesis of juvenile rheumatoid arthritis. Curr Opin Rheumatol 2000;12:420–4. [DOI] [PubMed]
- 8.Compston A, Coles A. Multiple sclerosis. Lancet 2002;359:1221–31. [DOI] [PubMed]
- 9.Burt RK, Marmont A, Oyama Y, Slavin S, Arnold R, Hiepe F, et al. Randomized controlled trials of autologous hematopoietic stem cell transplantation for autoimmune diseases: the evolution from myeloablative to lymphoablative transplant regimens. Arthritis Rheum 2006;54:3750–60. [DOI] [PubMed]
- 10.van Bekkum DW. Stem cell transplantation for autoimmune disorders. Preclinical experiments. Best Pract Res Clin Haematol 2004;17:201–22. [DOI] [PubMed]
- 11.Brinkman DM, Jol-van der Zijde CM, ten Dam MM, Vossen JM, Osterhaus AD, Kroon FP, et al. Vaccination with rabies to study the humoral and cellular immune response to a T-cell dependent neoantigen in man. J Clin Immunol 2003;23:528–38. [DOI] [PubMed]
- 12.Wulffraat NM, Kuis W, Petty R. Addendum: proposed guidelines for autologous stem cell transplantation in juvenile chronic arthritis. Paediatric Rheumatology Workshop. Rheumatology (Oxford) 1999;38:777–8. [DOI] [PubMed]
- 13.Tyndall A, Gratwohl A. Blood and marrow stem cell transplants in autoimmune disease. A consensus report written on behalf of the European League Against Rheumatism (EULAR) and the European Group for Blood and Marrow Transplantation (EBMT). Br J Rheumatol 1997;36:390–2. [DOI] [PubMed]
- 14.Comi G, Kappos L, Clanet M, Ebers G, Fassas A, Fazekas F, et al. Guidelines for autologous blood and marrow stem cell transplantation in multiple sclerosis: a consensus report written on behalf of the European Group for Blood and Marrow Transplantation and the European Charcot Foundation. BMT-MS Study Group. J Neurol 2000;247:376–82. [DOI] [PubMed]
- 15.Vossen JM, Brinkman DM, Bakker B, Hoogerbrugge PM, ten Cate R. Rationale for high-dose cyclophosphamide and medium-dose total body irradiation in the conditioning of children with progressive systemic and polyarticular juvenile chronic arthritis before autologous stem cell transplantation. Rheumatology (Oxford) 1999;38:762–3. [DOI] [PubMed]
- 16.Slaper-Cortenbach IC, Wijngaarden-du Bois MJ, Vries-van Rossen A, Borst HP, van der LH, van Heugten HG, et al. The depletion of T cells from haematopoietic stem cell transplants. Rheumatology (Oxford) 1999;38:751–4. [DOI] [PubMed]
- 17.Thisyakorn U, Pancharoen C, Ruxrungtham K, Ubolyam S, Khawplod P, Tantawichien T, et al. Safety and immunogenicity of preexposure rabies vaccination in children infected with human immunodeficiency virus type 1. Clin Infect Dis 2000;30:218. [DOI] [PubMed]
- 18.Chutivongse S, Wilde H, Benjavongkulchai M, Chomchey P, Punthawong S. Postexposure rabies vaccination during pregnancy: effect on 202 women and their infants. Clin Infect Dis 1995;20:818–20. [DOI] [PubMed]
- 19.van Wezel AL, van Steenis G, Hannik CA, Cohen H. New approach to the production of concentrated and purified inactivated polio and rabies tissue culture vaccines. Dev Biol Stand 1978;41:159–68. [PubMed]
- 20.Lyng J, Bentzon MW, Ferguson M, Fitzgerald EA. Rabies vaccine standardization: International Collaborative Study for the Characterization of the fifth International Standard for Rabies Vaccine. Biologicals 1992;20:301–13. [DOI] [PubMed]
- 21.van Wezel AL, van Steenis G. Production of an inactivated rabies vaccine in primary dog kidney cells. Dev Biol Stand 1978;40:69–75. [PubMed]
- 22.Jol-van der Zijde CM, van der Kaaden M, Rümke HC, Gerritsen EJA, Vossen JM, van Tol MJD. The antibody response against tetanus toxoid: a longitudinal study in healthy infants and adults, abstract in Progress in immune deficiemcy III, HM Chapel, RJ Levinsky, ADB Webster. Royal Society of Medicine Services International Congress and Symposium,1991, p. 238–40.
- 23.Lyng J. Calibration of a replacement preparation for the International Standard for Rabies Immunoglobulin. Biologicals 1994;22:249–55. [DOI] [PubMed]
- 24.Gerritsen EJ, van Tol MJ, ’t Veer MB, Wels JM, Khouw IM, Touw CR, et al. Clonal dysregulation of the antibody response to tetanus-toxoid after bone marrow transplantation. Blood 1994;84:4374–82. [PubMed]
- 25.Pullen GR, Fitzgerald MG, Hosking CS. Antibody avidity determination by ELISA using thiocyanate elution. J Immunol Methods 1986;86:83–7. [DOI] [PubMed]
- 26.ten Cate R, Brinkman DM, van Rossum MA, Lankester AC, Bredius RG, Egeler MR, et al. Macrophage activation syndrome after autologous stem cell transplantation for systemic juvenile idiopathic arthritis. Eur J Pediatr 2002;161:686. [DOI] [PubMed]
- 27.te Boekhorst PA, Lamers CH, Schipperus MR, Hintzen RQ, van der HB, Cornelissen JJ, et al. T-lymphocyte reconstitution following rigorously T-cell-depleted versus unmodified autologous stem cell transplants. Bone Marrow Transplant 2006;37:763–72. [DOI] [PubMed]
- 28.Miller JJ, Cole LJ. The radiation resistance of long-lived lymphocytes and plasma cells in mouse and rat lymph nodes. J Immunol 1967;98:982–90. [PubMed]
- 29.Moser K, Muehlinghaus G, Manz R, Mei H, Voigt C, Yoshida T, et al. Long-lived plasma cells in immunity and immunopathology. Immunol Lett 2006;103:83–5. [DOI] [PubMed]
- 30.Hoyer BF, Manz RA, Radbruch A, Hiepe F. Long-lived plasma cells and their contribution to autoimmunity. Ann NY Acad Sci 2005;1050:124–33. [DOI] [PubMed]
- 31.Gandhi MK, Egner W, Sizer L, Inman I, Zambon M, Craig JIO, et al. Antibody responses to vaccinations given within the first two years after transplant are similar between autologous peripheral blood stem cell and bone marrow transplant recipients 2001;28:775–81. [DOI] [PubMed]
- 32.Chan CY, Molrine DC, Antin JH, Wheeler C, Guinan EC, Weinstein HJ, et al. Antibody responses to tetanus toxoid and Haemophilus influenzae type b conjugate vaccines following autologous peripheral blood stem cell transplantation (PBSCT). Bone Marrow Transplant 1997;20:33–8. [DOI] [PubMed]
- 33.Storek J, Zhao Z, Lin E, Berger T, McSweeney PA, Nash RA, et al. Recovery from and consequences of severe iatrogenic lymphopenia (induced to treat autoimmune diseases). Clin Immunol 2004;113:285–98. [DOI] [PMC free article] [PubMed]
- 34.Saiz A, Carreras E, Berenguer J, Yague J, Martinez C, Marin P, et al. MRI and CSF oligoclonal bands after autologous hematopoietic stem cell transplantation in MS. Neurology 2001;56:1084–9. [DOI] [PubMed]
- 35.Kal HB, Loes vK-H, Heijenbrok-Kal MH, Struikmans H. Biologically effective dose in total-body irradiation and hematopoietic stem cell transplantation. Strahlenther Onkol 2006;182:672–9. [DOI] [PubMed]
- 36.Labadie J, van Tol MJ, Dijkstra NH, Zwaan FE, Vossen JM. Transfer of specific immunity from donor to recipient of an allogeneic bone marrow graft: effect of conditioning on the specific immune response of the graft recipient. Br J Haematol 1992;80:381–90. [DOI] [PubMed]
- 37.Labadie J, van Tol MJ, Dijkstra NH, van der KM, Jol-van der Zijde CM, de Lange GG, et al. Transfer of specific immunity from donor to recipient of an allogeneic bone marrow graft: evidence for donor origin of the antibody producing cells. Br J Haematol 1992;82:437–44. [DOI] [PubMed]
- 38.Storek J, Dawson MA, Lim LCL, Burman BE, Stevens-Ayers T, Viganego F, et al. Efficacy of donor vaccination before hematopoietic cell transplantation and recipient vaccination both before and early after transplantation. 2004;33:337–46. [DOI] [PubMed]
- 39.Hammarstrom V, Pauksen K, Bjorkstrand B, Simonsson B, Oberg G, Ljungman P. Tetanus immunity in autologous bone marrow and blood stem cell transplant recipients 1998;22:67–71. [DOI] [PubMed]
- 40.Lausen BF, Hougs L, Schejbel L, Heilmann C, Barington T. Human memory B cells transferred by allogenic bone marrow transplantation contribute significantly to the antibody repertoire of the recipient 2004;172:3305–18. [DOI] [PubMed]
- 41.Wimperis JZ, Gottlieb D, Duncombe AS, Heslop HE, Prentice HG, Brenner MK. Requirements for the adoptive transfer of antibody responses to a priming antigen in man. J Immunol 1990;144:541–7. [PubMed]
- 42.Bourgeois C, Stockinger B. CD25(+)CD4(+) regulatory T cells and memory T cells prevent lymphopenia-induced proliferation of naive T cells in transient states of lymphopenia 2006;4558–66. [DOI] [PubMed]
- 43.Moore J, Brooks P, Milliken S, Biggs J, Ma D, Handel M, et al. A pilot randomized trial comparing CD34-selected versus unmanipulated hemopoietic stem cell transplantation for severe, refractory rheumatoid arthritis. Arthritis Rheum 2002;46:2301–9. [DOI] [PubMed]
- 44.de Kleer I, Vastert B, Klein M, Teklenburg G, Arkesteijn G, Yung GP, et al. Autologous stem cell transplantation for autoimmunity induces immunologic self-tolerance by reprogramming autoreactive T cells and restoring the CD4+CD25+ immune regulatory network. Blood 2006;107:1696–702. [DOI] [PubMed]