

Abstract
Peptides derived from the membrane proximal region of fusion proteins of human immunodeficiency viruses 1 and 2, Coronavirus 229 E, severe acute respiratory syndrome coronavirus and Ebola virus were all potent antagonists of the formyl peptide receptor expressed in Chinese hamster ovary cells. Binding of viral peptides was affected by the naturally occurring polymorphisms at residues 190 and 192, which are located at second extracellular loop–transmembrane helix 5 interface. Substitution of R190 with W190 enhanced the affinity for a severe acute respiratory syndrome coronavirus peptide 6 fold but reduced the affinity for N-formyl-Nle–Leu–Phe by 2.5 fold. A 12 mer peptide derived from coronavirus 229E (ETYIKPWWVWL) was the most potent antagonist of the formyl peptide receptor W190 with a Ki of 230 nM. Fluorescently labeled ETYIKPWWVWL was effectively internalized by all three variants with EC50 of ∼25 nM. An HKU-1 coronavirus peptide, MYVKWPWYVWL, was a potent antagonist but N-formyl-MYVKWPWYVWL was a potent agonist. ETYIKPWWVWL did not stimulate GTPγS binding but inhibited the stimulation by formyl-NleLeuPhe. It also blocked β arrestin translocation and receptor downregulation induced by formyl-Nle–Leu–Phe. This indicates that formyl peptide receptor may be important in viral infections and that variations in its sequence among individuals may affect their likelihood of viral and bacterial infections.
Abbreviations: FPR, formyl peptide receptor; CHO S, Chinese hamster ovary cells designed for suspension culture; HRSV, human respiratory syncytial virus; FIV, feline immunodeficiency virus; fMLF, N-formyl-methionyl-leucyl-phenylalanine; AIDS, Acquired Immunodeficiency Syndrome; SIV, Simian Immunodeficiency Virus; HIV, human immunodeficiency virus; SARS, severe acute respiratory syndrome; GP-41, 41 kilodalton glycoprotein; HR, Heptade Repeat; FITC, Fluorescein isothiocyanate; formyl-Nle–Leu–Phe–Nle–Tyr–Lys–FITC, formyl-Nle–Leu–Phe–Nle–Tyr–Lys labeled at the Lys residue with Fluorescein isothiocyanate; formyl-Nle–Leu–Phe–Nle–Tyr–Lys–Alexa Fluor, formyl-Nle–Leu–Phe–Nle–Tyr–Lys labeled at the Lys residue with Alexa Fluor N-hydroxy-succinimide; ETYIK-(Alexa Fluor)WPWWVWL, ETYIKWPWWVWL labeled with Alexa Fluor 488 N-hydroxy-succinimide; GTPγS, guanosine 5′-3-O-(thio)triphosphate; TMH, transmembrane helix; FPRL1, formyl peptide like receptor 1; FPRL2, formyl peptide receptor like 2
Keywords: Virus, Signal Transduction, G protein coupled receptor, Polymorphism
1. Introduction
Neutrophils play an essential role in innate immunity. In addition to activating phagocytosis and secreting superoxides and hydrolytic enzymes, the neutrophil must be able to chemotax, or migrate toward the source of a chemoattractant. Upon activation by a chemoattractant, G-protein coupled receptors on the membrane of the neutrophil change conformation and bind to G proteins, activating a signal transduction pathway that results in intracellular actin polymerization, the formation of pseudopodia, reorientation toward a chemoattractant gradient, and adhesion to and infiltration through the blood vessel endothelium [1], [2]
The formyl peptide receptor (FPR) is a chemoattractant G protein-coupled receptor found on the surface of phagocytes. It is thought to play an important role in allowing phagocytic cells to recognize the presence of bacteria, which are believed to be the source of formyl peptides [3]. FPR is activated by other ligands in addition to fMLF. FPR binds peptides derived from the GP-41 envelope protein of HIV-1 [4], [5]. FPR activation is inhibited by a polypeptide secreted by S. aureus [6], [7], an undecapeptide (Cyclosporin H) derived from the fungus Tolypocladium inflatum [8], and peptides derived from murine leukemia virus and human T cell leukemia virus [9]. A peptide derived from herpes simplex virus type 2 elicited chemotaxis and superoxide production in neutrophils in a process that appeared to involve FPR [10]. Thus FPR appears to interact with peptides derived from a variety of sources.
Lentiviruses are associated with immunological impairment in their respective hosts and both human immunodeficiency and feline immunodeficiency viral infections increase the likelihood of secondary bacterial infections [11]. Recently, Kubes et al. [12] demonstrated that feline neutrophils exhibited a marked (> 90%) reduction of neutrophil chemotaxis toward fMLF following infection with FIV. It is possible that viral infections may release factors which impair chemotaxis in vivo, which would be consistent with the observations of virally derived peptides on FPR.
Polymorphisms in the FPR appear to be very common. Sahagun-Ruiz et al. [13] did an extensive haplotype investigation of FPR and reported finding at least 23 haplotypes for FPR. No polymorphisms were found in the closely related receptor FPRL1 [13]. One possible reason for the existence of polymorphisms is that polymorphisms might prevent pathogens from binding to receptors and using them as an entry point into cells. Several G protein-coupled receptors have been used by pathogens as a point of entry into cells and the use of CCR5 and CXCR4 as co-receptors for HIV-1 and CXCR4 as a co-receptor for FIV are notable examples [14], [15], [16]. The CCR5 receptor in African green monkeys [17] has been shown to be polymorphic and these polymorphisms provide some protection against SIV infection.
Numerous studies have indicated that patients with aggressive periodontitis exhibit a ∼ 2 fold reduction in chemotaxis toward fMLF [18], [19], [20], [21], [22], [23]. Zhang et al. [24] found that aggressive periodontitis patients of African American decent were more likely (P = 0.002) to have at least one allele as the R190/K192 polymorphism when compared to African American controls (R/W190 and N/K192 are located at the extracellular loop 2–TMH5 interface). They also found that aggressive periodontitis patients of African American decent were much less likely (P = 0.003) to have one of their alleles as the W190/N192 polymorphism. R190/K192 was not associated with an increased risk of aggressive periodontitis among either Brazilians or Turks nor was W190/N192 associated with reduced incidence of aggressive periodontal disease in these two populations [24]. It is unclear at this point why FPR W190/R190 and N192/K192 are only associated with altered risk of aggressive periodontitis in African Americans but the effects of these polymorphisms on receptor function clearly needs to be determined.
We previously observed that a single amino acid mutation, D106N in TMH3, markedly enhanced the ability of FPR to bind a peptide derived from HIV-1 gp41 but also markedly reduced its affinity for fMLF [25]. Thus, determination of the affinity of these two very different ligands provides a way to observe changes in ligand specificity associated with changes in FPR. In order to determine whether polymorphisms W190 or K192 might similarly affect ligand binding and possibly explain why K192 is associated with an increased risk of periodontal disease and W190 is associated with reduced risk of disease, we expressed R190/N192, W190/N192, and R190/N192 FPR variants in CHO S cells and determined their affinity for numerous ligands, including several formyl peptides and peptides derived from HIV-1 gp-41 and analogous peptides derived from HIV-2, SARS coronavirus, coronavirus 229E, coronavirus OC43, and Ebola virus.
2. Materials and methods
2.1. Cell transfection
Geneticin- and ampicillin-resistant and FPR-expressing DNA plasmid PBGSA was prepared as previously described [26]. Mutagenesis was performed according to the Quickchange Mutagenesis Kit instructions using pfu DNA polymerase essentially as described previously [27]. The plasmids were cut with BspEI and XboI and ligated back into the original FPR-expressing DNA plasmid PBGSA. This step was necessary to obtain good expression levels. DNA was purified, sequenced (Davis Sequencing) and transfected into CHO S cells (Invitrogen) using Lipofectamine 2000, cloned, and maintained in Gibco CHO-S-SFMII media containing 100 μg/ml G418. Numerous clones expressing R190/N192, R190/K192, or W190/N192 were evaluated for their expression by FACScan analysis using 10 nM formyl-Nle–Leu–Phe–Nle–Tyr–Lys–FITC . A clone was chosen for each sequence that exhibited nearly identical levels of surface expression; less than ∼ 20% difference between the three variants.
2.2. Peptide synthesis
Peptides were obtained from either Genscript or Macromolecular Resources and purified by reverse phase HPLC. Peptides were synthesized with unmodified N and C termini. Mass spectral data was provided with each peptide and the major mass peak matched the expected mass of the peptide in all cases. Formyl-Nle–Leu–Phe was obtained from Sigma and used without further purification.
2.3. Fluorescent labeling of peptides and anti β-arrestin antibody
Peptides were reacted with Alexa Fluor 488 n-hydroxysuccinimidylester (Molecular Probes) at a 1.5 to 1 mole ratio (peptide to fluorophore) in DMSO/1% triethylamine at 30 °C for ∼ 4 h and purified by reversed phase HPLC. For formyl-Nle–Leu–Phe–Nle–Tyr–Lys the only site of labeling is the Lys. For ETYIKWPWWVWL either the Lys could be labeled or the N-terminus could be labeled. However, the intrinsic reactivity of the ε amino of Lys is generally ∼ 100 fold greater than the N-terminal amino group so it is likely that almost all labeling occurs at the lys when the peptide is used in excess to the fluorophore. Anti β-arrestin antibody (BD Transduction Laboratories) at 250 μg/ml in PBS was incubated with 5 mM 6-(fluorescein-5-carboxamido)hexanoic acid, succinimidyl ester (Molecular Probes) for 1 h at 37 °C and the labeled antibody was purified by HPLC gel filtration on a Sigmachrome 100 column. The antibody contained approximately 1.5 moles of fluorescein/mole of antibody.
2.4. Determination of GTPγS binding
Membranes from CHO S cells expressing FPR were prepared as follows: The cells were centrifuged at 100 g for 5 min. The cell pellets were resuspended in 10 mM phosphate, pH 7.4, 10 mM KCl, 100 mM NaCl, 0.1 μM GDP and 5 μg/ml leupeptin. The cells were sonicated for 30 s at 20% power and stored as aliquots of equal amounts of protein at −70 °C. Binding assays were done in 0.2 ml of 10 mM HEPES pH 7.4, 100 mM NaCl, 1 mM Mg++, 1 μM GDP. Samples containing 30 μg of protein were preincubated with ligand at 30 °C for 10 min, 0.05 μCi of [35S] GTPγS was added and the reaction continued for 10 min. The samples were filtered through a GF/C glass microfiber filter. Filters were washed with 15 ml of 10 mM phosphate, pH 7.4, 3 mM KCl, 100 mM NaCl , and the bound [35S] GTPγS was determined by liquid scintillation counting.
2.5. FACScan analysis of ligand binding to FPR variants expressed in CHO S cells
CHO S cells (100 μl in Gibco CHO-S-SFMII) were added to 3 mM KCl, 100 mM NaCl, 10 mM sodium phosphate, pH 7.4, 1 mM Mg++, 1 mM Ca++ containing 5% fetal bovine serum and incubated at 4 °C for 60 min with varying concentrations of fluorescent peptide. The mean fluorescence of the cells was determined, and the mean fluorescence observed in non-expressing CHO S cells (non-specific binding) was subtracted. The data were then analyzed by nonlinear least squares analysis and the K d and B max determined. The affinity of peptides was determined using increasing concentrations of peptide in the presence of constant formyl-Nle–Leu–Phe–Nle–Tyr–Lys–FITC. The K i was then determined by nonlinear least squares analysis using the known K d for formyl-Nle–Leu–Phe–Nle–Tyr–Lys–FITC for each FPR variant and the observed EC50 of each peptide. Formyl-Nle–Leu–Phe–Nle–Tyr–Lys–FITC concentrations of 0.3 nM, were used for all three FPR variants, and the K i was determined from the equation K i = EC50 / (1 + (ligand/K d) using the graphics/statistics program Graphpad Prizm.
2.6. Translocation of arrestin
CHO S cells were incubated in Gibco CHO-S-SFMII at 37 °C for 1 h with 10 μM fNle–Leu–Phe, 10 μM ETYIKWPWWVWL , 10 μM each of both peptides or no peptides. DMSO was the same concentration in all cases. The cells were washed with 10 ml 10 mM phosphate, pH 7.4, 3 mM KCl, 100 mM NaCl. Cells were resuspended in 10 mM phosphate, pH 7.4, 3 mM KCl, 100 mM NaCl , 0.02% saponin, 60 μg/ml leupeptin, protein phosphatase inhibitors (Sigma) 1/100, and 8 nM of fluorescein labeled anti arrestin. The cells were incubated for 15 h at 4 °C, diluted 1 to 3 with 10 mM phosphate, pH 7.4, 3 mM KCl, 100 mM NaCl, and then analyzed by FACScan. Background was determined by adding unlabeled anti-arrestin in 10 fold molar excess to the fluorescein labeled anti arrestin.
2.7. Downregulation of surface FPR
CHO S cells expressing FPR were incubated in Gibco CHO-S-SFMII at 37 °C for 1 h with 10 μM fNle–Leu–Phe, 10 μM ETYIKWPWWVWL, 10 μM each of both peptides or no peptides. DMSO was the same concentration in all cases. The cells were washed 2× with 10 ml 10 mM phosphate, pH 7.4, 3 mM KCl, 100 mM NaCl at 4 °C. The cells were resuspended in 100 mM NaCl, 3 mM KCl, 10 mM sodium phosphate, pH 7.4, 1 mM Mg++, 1 mM Ca++, 5% fetal bovine serum and 50 nM formyl-Nle–Leu–Phe–Nle–Tyr–Lys–FITC and incubated for 1 h at 4 °C and analyzed by FACScan as describe above. Background observed with non-expressing CHO S cells was subtracted.
2.8. Internalization of ETYIK-(Alexa Fluor)WPWWVWL
CHO S cells expressing FPR were incubated in Gibco CHO-S-SFMII at 37 °C for 1 h with varying concentrations ETYIK-(Alexa Fluor)WPWWVWL, The cells were washed 2× with 10 ml 10 mM phosphate, pH 7.4, 3 mM KCl, 100 mM NaCl at 4 °C. The cells were resuspended in 100 mM NaCl, 3 mM KCl, 10 mM sodium phosphate, pH 7.4, 1 mM Mg++, 1 mM Ca++, 5% fetal bovine serum and 10 μM formyl-Nle–Leu–Phe and incubated for 1 h at 4 °C to displace any ligand bound to surface receptor. Internalized ligand was determined by FACScan as describe above. Background observed with non-expressing CHO S cells (< 20% observed with expressing cells) was subtracted.
2.9. Statistical analysis
Experimental results are expressed as the mean ± standard error of the mean. The significance was evaluated by unpaired t test (2 tailed) using the computer program Graphpad Prizm.
3. Results
3.1. Choice of viral peptides for analysis
Peptides derived from HIV-1 GP-41 have previously been shown to interact with FPR [4] and we previously observed that a gp41 36 mer (T20/DP178) inhibited formyl-Nle–Leu–Phe–Nle–Tyr–Lys–FITC binding to FPR [25]. Since gp41 is a fusion glycoprotein which contains two heptad repeat regions, we chose viral glycoproteins with this property. T20/DP178 is derived from the second heptad repeat region of HIV-1 gp41 but also includes residues C-terminal to the end of the heptad 2 repeat region [28]. Sainz et al. called these residues in HIV-1 and SARS coronavirus the “aromatic rich region” and they noted a similar region in Ebola virus [29]. This region has also been called the membrane proximal region. We therefore chose to synthesize peptides from HIV-2 and several human coronaviruses (SARS, OC43, and 229E) that corresponded to T20/DP178 in HIV-1. An alignment of sequences is shown in Table 1 , which also includes the newly discovered coronaviruses HKU-1 and NL-63 [30], [31], [32] and the aromatic rich region from the Ebola spike protein.
Table 1.
Alignment of sequences of viral fusion proteins
| HIV-1 | (T20/DP-178) | YTSLIHSLIEESQNQQEKNEQELLELDKWASLWNWF |
| HIV-2 | YLEANISELLEQAQIQQEKNMYELQKLNSWDVFGNWF | |
| CORONAVIRUS | (HUMAN-SARS) | IDRLNEVAKNLNESLIDLQELGKYEQYIKWPWYVWL |
| CORONAVIRUS | OC43 | MNRLQEAIKVLNQSYINLKDIGTYEYYVKWPWYVWL |
| CORONAVIRUS | HKU-1 | MNLIQESIKSLNNSYINLKDIGTYEMYVKWPWYVWL |
| CORONAVIRUS | 229E | VQKLQTLIDNINSTLVDLKWLNRVETYIKWPWWVWL |
| CORONAVIRUS | NL63 | TVELQGLIDQINSTYVDLKLLNRFENYIKWPWWVWL |
| ZAIRE EBOLA SPIKE PROTEIN | NDNWWTGWRQWI |
The alignments show that all these viral sequences contain aromatic rich C-termini which are very rich in tryptophan. The 12 C-terminal residues of the coronaviruses are highly conserved. The 12 c-terminal residues of HIV-1 T20/DP178 are also highly conserved among known HIV-1 sequences and this 12 amino acid region overlaps the epitopes of 2 broadly protective anti-HIV antibodies 2F5 and 4E10 [33], [34].12 mer peptides corresponding to C-termini of the sequences for HIV-1, HIV-2, SARS coronavirus, and coronavirus 229E were synthesized (residues are underlined in Table 1) as well as a 12 residue peptide corresponding to the aromatic rich region from the Ebola spike protein. We also synthesized smaller peptides of coronavirus 229E to localize residues which might be important in binding (ETYIKWP, WPWWVWL, and WWVWL).
3.2. Ligand specificity and affinity determination of R190/N192, W190/N192, and R190/K192 variants
We transformed CHO S cells with FPR variants R190/N192, W190/N192, and R190/K192. A schematic figure showing the topology of FPR is shown in Fig. 1 . The location of R/W 190 and N/K 192 at the extracellular loop 2–TMH5 interface is indicated in the figure. Residues that have been shown in previous crosslinking and mutagenesis studies to be important in ligand binding are V83 (TMH2), R84 (TMH2), K85 (TMH2) [35]; L78A (TMH2), D106N (TMH3), L109A (TMH3), T157A (TMH4), R201A (TMH5), I204Y (TMH5), R205A (TMH2), W254A (TMH6), Y257A (TMH6) and F291A (TMH7) [26]. D106 and R201 are the sites of formyl group binding and R205 interacts with the C-terminus of fMLF [25]. The beginning of TMH5 was assigned to residue 191 and the end to residue 228 by hydrophobicity analysis according to Kyte and Doolittle [36]. We analyzed CHO S cells expressing R190/N192, W190/N192, and R190/K192 FPR for their ability to bind formyl-Nle–Leu–Phe–Nle–Tyr–Lys–FITC. We observed that the polymorphisms in FPR result in significantly altered formyl-Nle–Leu–Phe–Nle–Tyr–Lys–FITC binding affinity (Fig. 2 ) with R190/K192 having the highest affinity (K d = 0.71 ± 0.10 nM), R190/N192 intermediate (K d = 1.06 ± 0.09 nM), and W190/N192 having the lowest affinity (K d = 1.68 ± 0.13 nM); P = 0.02; R190/N192 vs. R190/K192 : P = 0.001; R190/N192 vs. W190/N192, and P < 0.0001 R190/K192 vs. W190/N192, respectively). We also synthesized formyl-Nle–Leu–Phe–Nle–Tyr–Lys–Alexa Fluor and determined the K ds: R190/K192 (K d = 0.76 ± 0.05 nM), R190/N192 (K d = 1.20 ± 0.14 nM), and W190/N192 (K d = 2.0 ± 0.10 nM), the results being essentially identical as that observed with formyl-Nle–Leu–Phe–Nle–Tyr–Lys–FITC. We then determined the affinities of a broad range of peptides for these three polymorphisms based on their ability to compete with formyl-Nle–Leu–Phe–Nle–Tyr–Lys–FITC (Table 2 ).
Fig. 1.
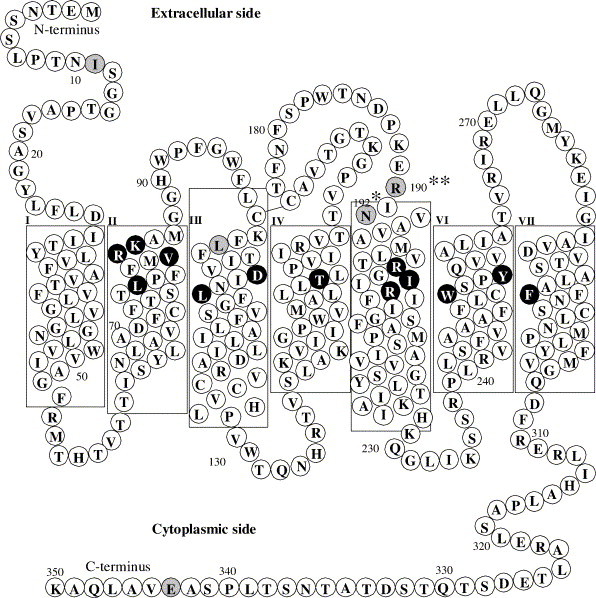
Transmembrane model of FPR. Residues in black are residues that have been shown in previous crosslinking and mutagenesis studies to be important in ligand binding [25], [26], [35]. Residues in gray are sites of known polymorphisms [13].**R or W, *N or K.
Fig. 2.
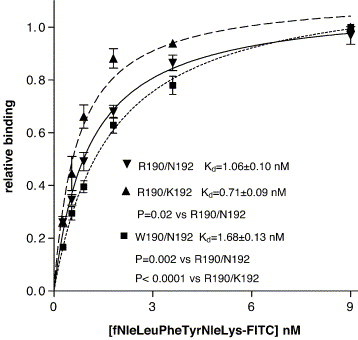
Determination of affinity of ligand binding. Binding of formyl-Nle–Leu–Phe–Nle–Tyr–Lys–FITC to FPR variants R190/N192, R190/K192, or W190/N192 was determined as a function of ligand concentration.
Table 2.
Ki (nM ± SEM) for binding of peptides to R190/N192, R190/K192, or W190/N192 FPR
| R190/N192 | R190/K192 | W190/N192 | |
|---|---|---|---|
| Formyl-NleLeuPhe | 67 ± 16 | 62 ± 8 | 160 ± 25⁎⁎ |
| MMWLL | 330 ± 50 | 190 ± 30 | 840 ± 200⁎ |
| HKU coronavirus peptides | |||
| MYVKWPWYVWL | 783 ± 50 | 837 ± 32 | 263 ± 9⁎⁎ |
| Formyl-MYVKWPWYVWL | 134 ± 9 | 134 ± 12 | 133 ± 8 |
| SARS coronavirus peptides | |||
| 36mer | 3,270 ± 250 | 3,140 ± 150 | 770 ± 25⁎⁎ |
| 12 mer (EQYIKWPWYVWL) | 2,050 ± 107 | 2,400 ± 170 | 390 ± 30⁎⁎ |
| Coronavirus OC43 peptide | |||
| 36mer | 850 ± 80 | 740 ± 80 | 460 ± 50⁎⁎ |
| Coronavirus 229E peptides | |||
| 36 mer | 10,000 ± 1400 | 6,800 ± 670 | 1300 ± 60⁎⁎ |
| 12 mer (ETYIKWPWWVWL) | 533 ± 30 | 370 ± 30 | 270 ± 10⁎⁎ |
| LWVWWPWKIYTE (reverse) | > 50,000 | > 50,000 | > 50,000 |
| WPWWVWL | ∼ 600 | ∼ 600 | ∼ 600 |
| WWVWL | ∼ 1000 | ∼ 1400 | ∼ 600 |
| ETYIKWP | > 260,000 | > 260,000 | > 260,000 |
| HIV-1 peptides | |||
| 36 mer | 1500 ± 80 | 820 ± 50 | 960 ± 200 |
| 12 mer (ELDKWASLWNWF) | 1200 ± 100 | 940 ± 150 | 1050 ± 100 |
| HIV-2 peptides | |||
| 37 mer | 15,700 ± 1000 | 10,000 ± 1200 | 4000 ± 1000⁎⁎ |
| 12 mer (KLNSWDVFGNWF) | 15,000 ± 2000 | 9,500 ± 2000 | 4700 ± 290⁎⁎ |
| Ebola peptide | |||
| 12 mer (NDNWWTGWRQWI) | 1400 ± 200 | 1200 ± 90 | 1200 ± 150 |
P < 0.05 W190/N192 vs. either R190/N192 or R190/K192.
P < 0.01 W190/N192 vs. either R190/N192 or R190/K192.
We observed that R190/K192 exhibited the highest affinity for formyl-Nle–Leu–Phe (chosen over fMLF in order to prevent possible oxidation of methionine) and MMWLL, a potent non-formylated chemoattract for FPR, [25], [37]. Both formyl-Nle–Leu–Phe and MMWLL bound with ∼3 fold higher affinities to R190/K192 than to W190/N192. W190/N192 had the highest affinity for most virally derived peptides, especially the coronavirus peptides. W190/N192 bound the SARS coronavirus peptides with 4–6 fold higher affinity than either R190/N192 or R190/K192. This indicates that changes in these two positions affected ligand specificity, but most of the change in specificity was associated with the R to W change at residue 190. Most of the binding affinity of the coronavirus 229E peptides appears to reside in the 5 C-terminal residues since WWVWL exhibited only a modest reduction in affinity compared to ETYIKWPWWVWL. ETYIKWP was completely devoid of activity. WPWWVWL had similar affinity to WWVWL. ETYIKWPWWVWL bound to all three FPR variants with 15 to 30 fold higher affinity than the HIV-2 12 mer (KLNSWDVFGNWF) indicating a preference of coronavirus peptides over HIV-2 peptides. The reverse peptide, LWVWWPWKIYTE, exhibited very little inhibition at concentrations up to 40,000 nM. Formylation of MYVKWPWYVWL enhanced its affinity 6 fold for both R190/N192 and R190/K192 but had only a very modest 2 fold affect on affinity for W190/N192.
In order to determine whether the binding of coronavirus peptides was specific for FPR, we labeled ETYIKWPWWVWL with Alexa Fluor 488, purified it by HPLC, and compared the binding to CHO S cells expressing R190/N192, W190/N192, R190/K192 or cells not expressing FPR. All three FPR variants exhibited binding of this Alexa Fluor 488 labeled peptide, compared with non-expressing cells (Fig. 3 ). W190/N192 exhibited the greatest binding at saturation, but the K ds for R190/N192, R190/K192, and W190/N192 were similar: 121 ± 18 nM, 115 ± 10 nM, and 99 ± 10 nM, respectively. When ETYIK-(Alexa Fluor)WPWWVWL and formyl-Nle–Leu–Phe–Nle–Tyr–Lys–Alexa Fluor binding to R190/N192, R190/K192, and W190/N192 were determined at saturating concentrations of either ligand, Alexa Fluor- ETYIKWPWWVWL exhibited twice the fluorescence of formyl-Nle–Leu–Phe–Nle–Tyr–Lys–Alexa Fluor when bound to W190/N192, but only about 60% the fluorescence of formyl-Nle–Leu–Phe–Nle–Tyr–Lys–Alexa Fluor bound to either R190/N192 or R190/K192 (data not shown, but see below for an explanation).
Fig. 3.
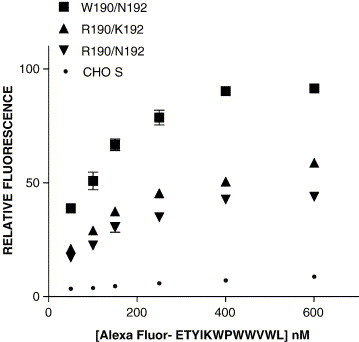
Determination of affinity of Alexa Fluor labeled ETYIKWPWWVWL. Binding of Alexa Fluor labeled ETYIKWPWWVWL to W190/N192 FPR, R190/K192 FPR, and R190/N192 FPR and to CHO-S cells not expressing FPR are shown as a function of concentration. Kds for W190/N192, R190/K192, and R190/N192 were 99 ± 10 nM, 115 ± 10 nM, and 121 ± 18 nM, respectively.
We also investigated the ability of ETYIK-(Alexa Fluor)WPWWVWL to be internalized at 37 °C. The results are shown in Fig. 4 . The EC50s for internalization were ∼4 fold lower than the K d for binding (R190/N192, R190/K192, and W190/N192; 29 ± 5 nM, 27 ± 8 nM, and 20 ± 3 nM, respectively). This indicates that either the affinity of ETYIK-(Alexa Fluor)WPWWVWL for FPR is greater at 37 °C used for internalization than at 4 °C used in the binding experiments to prevent internalization or that saturation of the internalization process occurs at concentrations well below those required for receptor saturation. Since the peptides behaved as antagonists, we were somewhat surprised that they were so readily internalized. This probably reflects turnover of FPR on the cell surface, since treatment of the cells with ETYIKWPWWVWL did not result in net receptor down regulation (see Fig. 9) below. Nonetheless, it indicates that if a virus interacts with FPR, it could be effectively internalized even if it is unable to activate the receptor.
Fig. 4.
ETYIKWPWWVWL labeled with Alexa Fluor 488 is internalized in CHO cells expressing FPR. CHO cells expressing FPR W190/N192, FPR R190/N192 or FPR R190/ K192 were incubated with indicated concentrations of Alexa Fluor labeled peptide for 37 °C for 1 h and the amount of internalized ligand was determined. The very low internalization of CHO cells not expressing FPR has been subtracted.
Fig. 9.
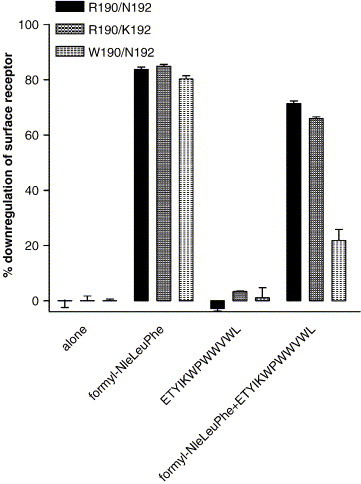
Down regulation of FPR surface expression in response to various ligands. Surface expression of FPR in W190/N192 FPR expressing cells, R190/K192 FPR expressing cells, or R190/N192 FPR expressing cells was determined after exposure for 1 h at 37° to either 10 μM formyl-Nle–Leu–Phe, 10 μM ETYIKWPWWVWL, or 10 μM formyl-Nle–Leu–Phe + 10 μM ETYIKWPWWVWL. ETYIKWPWWVWL inhibited receptor downregulation in response to formyl-Nle–Leu–Phe by 16 ± 2% in R190/N192 FPR expressing cells, 20 ± 2% in R190/K192 FPR expressing cells, and 64 ± 6% in W190/N192 FPR expressing cells. (P < 0.001 W190/N192 vs. either R190/N192 or R190/K192).
We also quantitated formyl-Nle–Leu–Phe–Nle–Tyr–Lys–Alexa Fluor internalization in R190/N192, R190/K192, and W190/N192, by fluorescence spectroscopy in the absence or presence of 0.1% SDS to correct for any quantum yield change in Alexa Fluor fluorescence which might occur upon binding to FPR. Interestingly, formyl-Nle–Leu–Phe–Nle–Tyr–Lys–Alexa Fluor fluorescence internalized by W190/N192 FPR was enhanced in the presence of 0.1% SDS by 95 ± 5%, but 0.1% SDS did not affect the fluorescence of formyl-Nle–Leu–Phe–Nle–Tyr–Lys–Alexa Fluor internalized by either R190/N192 FPR or R190/K192 FPR. This indicates that W190 quenches the fluorescence of formyl-Nle–Leu–Phe–Nle–Tyr–Lys–Alexa Fluor by ∼ 50% when it is bound to the receptor and thus must be sufficiently near the Alexa Fluor moiety of formyl-Nle–Leu–Phe–Nle–Tyr–Lys–Alexa Fluor when the peptide is bound to W190/N192 FPR so that W190 can contact AlexFluor during the excited state [38]. This indicates that W190 resides in or very close to the ligand binding pocket (∼1 to 1.5 nm average distance [38]). Since Alexa Fluor-ETYIKWPWWVWL exhibited twice the fluorescence of formyl-Nle–Leu–Phe–Nle–Tyr–Lys–Alexa Fluor when bound to W190/N192 (the expected value if it was not quenched), it is unlikely that W190 can come in contact with the Alexa Fluor of ETYIK-(Alexa Fluor)WPWWVW when bound to FPR. After correcting for the quenching of formyl-Nle–Leu–Phe–Nle–Tyr–Lys–Alexa Fluor on W190/N192, we estimate that ETYIK-(Alexa Fluor)WPWWVW is internalized at about 20–30% of the rate of formyl-Nle–Leu–Phe–Nle–Tyr–Lys–Alexa Fluor in R190/N192, R190/K192, and W190/N192, based on their relative internalization rates by FACScan. This level of internalization was too low to accurately quantitate by direct fluorescence readings so the degree of quenching of Alexa Fluor- ETYIKWPWWVWL could only be estimated by FACScan analysis.
We tested several peptides including formyl-Nle–Leu–Phe, ETYIKWPWWVWL, MYVKWPWYVWL and formyl-MYVKWPWYVWL for their ability to stimulate GTPγS binding. Both 300 nM formyl-Nle–Leu–Phe and 300 nM formyl-MYVKWPWYVWL stimulated GTPγS binding in all three FPR variants, but neither ETYIKWPWWVWL nor MYVKWPWYVWL had any effect on the three FPR variants (Fig. 5 ). This indicated that the formyl group on formyl- MYVKWPWYVWL was essential for agonist activity but not for binding. 2 μM MYVKWPWYVWL inhibited 300 nM formyl-MYVKWPWYVWL stimulation of GTPγS binding by W190/N192 FPR by 60% but did not have a significant effect on formyl-MYVKWPWYVWL stimulation of GTPγS binding in either R190/N192 FPR or R190/K192 FPR. This is consistent with the data in Table 2 where formylation of MYVKWPWYVWL enhanced the affinity for R190/N192 and R190/K192 by 6 fold but only 2 fold for W190/N192. We then tested ETYIKWPWWVWL for its ability to inhibit the stimulation of GTPγS by 300 nM formyl-Nle–Leu–Phe (Fig. 6 ). ETYIKWPWWVWL was more effective in inhibiting the stimulation of GTPγS binding in W190/N192 (EC50 = 300 ± 120 nM; (P = 0.003 vs. R190/K192; P = 0.005 vs. R190/N192)) than either R190/N192 (EC50 = 1600 ± 400 nM) or R190/K192 (EC50 = 1600 ± 440 nM). These results are consistent with the results in Table 2, which show that W190/N192 has a similar affinity for formyl-Nle–Leu–Phe and ETYIKWPWWVWL but that R190/N192 and R190/K192 have 6–8 fold higher affinities for formyl-Nle–Leu–Phe than ETYIKWPWWVWL. We also tested ETYIKWP and WWVWL for their ability to inhibit the stimulation of GTPγS binding by 300 nM formyl-Nle–Leu–Phe in W190/N192 FPR. The results are shown in Fig. 7 . WWVWL inhibited formyl-Nle–Leu–Phe stimulation of GTPγS binding with a slightly higher EC50 (EC50 = 1300 nM) than ETYIKWPWWVWL (EC50 = 300 ± 120 nM). ETYIKWP had no effect. Again this is consistent with the results shown in Table 2. We also tested the HIV-1 36 mer and the Ebola virus 12 mer for their effects on GTPγS binding. As observed with ETYIKWPWWVWL, neither peptide at 10 μM stimulated GTPγS binding but both peptides at 10 μM markedly inhibited the stimulation by 300 nM formyl-Nle–Leu–Phe (data not shown).
Fig. 5.
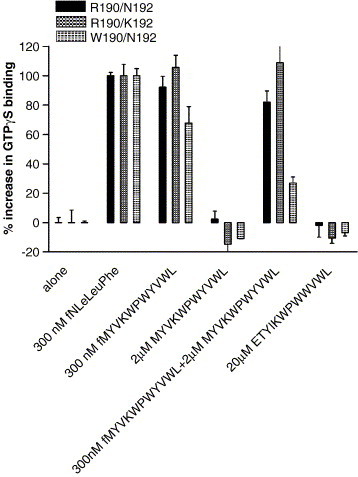
Stimulation of GTPγS binding by formyl-Nle–Leu–Phe, formyl-MYVKWPWYVWL, MYVKWPWYVWL or ETYIKWPWWVWL in cells expressing W190/N192, R190/K192, or R190/N192 FPR.
Fig. 6.
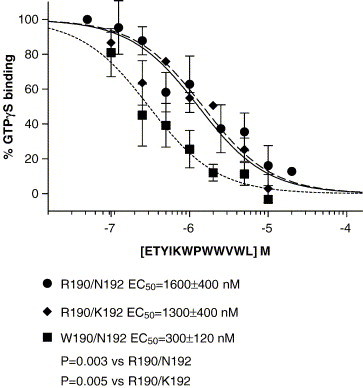
Inhibition of stimulation of GTPγS binding induced by formyl-Nle–Leu–Phe by ETYIKWPWWVWL in FPR variants R190/N192, R190/K192, or W190/N192. Inhibition of FPR activation by viral peptides. Inhibition of stimulation of GTPγS binding induced by formyl-Nle–Leu–Phe by ETYIKWPWWVWL. ETYIKWP, or WWVWL in W190/N192 FPR.
Fig. 7.
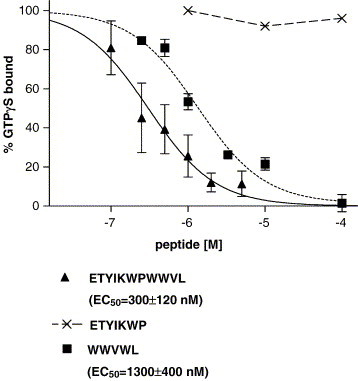
Inhibition of FPR activation by viral peptides. Inhibition of stimulation of GTPγS binding induced by formyl-Nle–Leu–Phe by ETYIKWPWWVWL. ETYIKWP, or WWVWL in W190/N192 FPR.
Previous studies with other G protein coupled receptors have indicated that some ligands that fail to activate G proteins are nonetheless able to promote β-arrestin translocation and activate β-arrestin dependent signaling pathways [39], [40], [41], [42]. Therefore, we also tested R190/N192, W190/N192, and R190/K192 expressing CHO S cells for the ability to undergo β-arrestin translocation in response to 10 μM formyl-Nle–Leu–Phe, 10 μM ETYIKWPWWVWL, or 10 μM each of both peptides (Fig. 8 ). Formyl-Nle–Leu–Phe caused β-arrestin translocation in CHO S cells expressing R190/N192, R190/K192, and W190/N192 but not in CHO S cells not expressing FPR. ETYIKWPWWVWL did not cause translocation in CHO S cells expressing R190/N192, W190/N192, or R190/K192 FPR. ETYIKWPWWVWL inhibited translocation in response to formyl-Nle–Leu–Phe by 18 ± 4% in R190/N192 FPR expressing cells, 20 ± 9% in R190/K192 FPR expressing cells, and 77 ± 10% in W190/N192 FPR expressing cells (P < 0.006; W190/N192 vs. either R190/N192 or R190/K192). Again ETYIKWPWWVWL was more effective against W190/N192 than with R190/N192 or R190/K192, but it exhibited only antagonist activity in this assay.
Fig. 8.
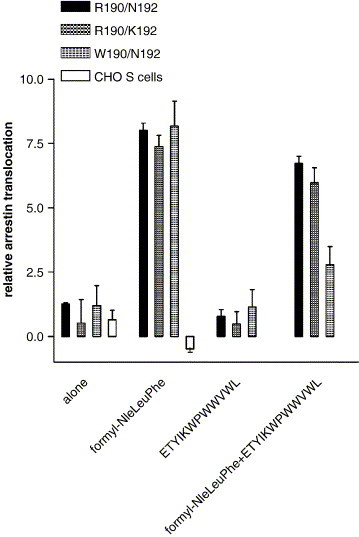
Arrestin translocation in the presence of various FPR ligands. Relative arrestin translocation was determined in non-expressing CHO cells, R190/N192 FPR expressing cells, R190/K192 FPR expressing cells, or W190/N192 FPR expressing cells in response to 10 μM formyl-Nle–Leu–Phe, 10 μM ETYIKWPWWVWL, or 10 μM formyl-Nle–Leu–Phe + 10 μM ETYIKWPWWVWL. 10 μM ETYIKWPWWVWL inhibited translocation in response to 10 μM formyl-Nle–Leu–Phe by 18 ± 4% in R190/N192 FPR expressing cells, 20 ± 9% in R190/K192 FPR expressing cells, and 77 ± 10% in W190/N192 expressing cells (P < 0.006 for W190/N192 FPR vs. either R190/N192 or R190/K192).
Finally, we tested R190N192, R190/K192, and W190/N192 expressing CHO S cells to undergo surface receptor downregulation in response to 10 μM formyl-Nle–Leu–Phe, 10 μM ETYIKWPWWVWL, or 10 μM each of both peptides (Fig. 9 ). Formyl-Nle–Leu–Phe caused receptor down regulation in CHO S cells expressing R190/N192, R190/K192, or W190/N192 FPR variants; 76 ± 1.2%, 85 ± 0.4%, and 76 ± 3%, respectively. ETYIKWPWWVWL did not cause surface receptor downregulation in CHO S cells expressing R190/N192, R190/K192, or W190/N192 FPR. ETYIKWPWWVWL inhibited receptor downregulation in response to formyl-Nle–Leu–Phe by 16 ± 2% in R190/N192 FPR expressing cells, 20 ± 2% in R190/K192 FPR expressing cells, and 64 ± 6% in W190/N192 FPR expressing cells (P < 0.001 for W190/N192 vs. either R190/N192 or R190/K192). As with the previous experiments, ETYIKWPWWVWL was more effective against W190/N192 than R190/N192 or R190/K192. The effects on downregulation closely correlated with β-arrestin translocation, and this was not unexpected given association between β-arrestin translocation and downregulation [43].
4. Discussion
R190/K192 exhibited the highest affinity for formyl-Nle–Leu–Phe, formyl-Nle–Leu–Phe–Nle–Tyr–Lys–FITC, and formyl-MYVKWPWYVWL, which indicates that it is not defective in binding formyl peptides. African American patients expressing the R190/K192 polymorphism were associated with increased risk of aggressive periodontitis whereas those expressing the W190/N192 polymorphism were associated with reduced risk [24]. Our data clearly show no evidence that R190/K192 is defective and we find that W190/N192 exhibited a 2.6 fold lower affinity for formyl peptides than R190/K192. Zhang et al. also reported that R190/K192 was not associated with an increased risk of aggressive periodontitis among either Brazilians or Turks nor was W190/N192 associated with reduced incidence of aggressive periodontal disease in these two populations [24] so it was unclear why this association was only true for African Americans. A recent study by Maney et al. [44] was unable to confirm the association of aggressive periodontitis with R190/K192 in African Americans, but did find an association with a silent polymorphism I116I. Our data finding no defect in R190/K192 are consistent with the study of Maney et al. [44] and also with the observation that R190/K192 was not associated with increased risk for aggressive periodontitis in either Turks or Brazilians.
In Fig. 1, we show the relative positions of R/W 190 and N/K 192 at the extracellular loop 2-TMH5 interface to other residues in FPR which we previously identified as important in ligand binding [25], [26], [35]. We previously determined that D106 and R201 likely form an ion pair between TMH3 and TMH5 and that this ion pair imparts formyl group selectivity to ligand binding since mutation of either residue changes the preference of formylated to non-formylated peptide from 4000/1 to ∼ 1/3 and 1/1, respectively, for D106N and R201A [25]. Here we observe that W190 is likely to be close to the lys residue of formyl-Nle–Leu–Phe–Nle–Tyr–Lys–Alexa Fluor since quenching of the Alexa Fluor fluorophore is observed only in W190/N192 and not in either R190/N192 or R190/K192. It is likely therefore that when bound, formyl-Nle–Leu–Phe–Nle–Tyr–Lys–Alexa Fluor is in a somewhat helical conformation (it contains residues with high helical propensity) with the formyl group near R201 and the AlexFluor moiety near W190 and presumably near R190 as well. This mode of binding is different from that proposed for fMLF where the C-terminus of fMLF interacts with R205 [25]. This is consistent with our previous observations that mutation of R205 to A results in a 1000 fold reduction of affinity to fMLF [25] but only an 8 fold reduction in affinity for formyl-Nle–Leu–Phe–Nle–Tyr–Lys–FITC [26]. Given that W190/N192 binds coronaviral peptides better than R190/N192 or R190/K192, it is likely that W190 interacts with some as yet unidentified residues in these peptides. One explanation of why R190/K192 binds formyl peptides better than W190/N192 is that the former contains two additional positive charges. Formylation removes the positive charge from the N-terminus of peptides. If R190 is near the entry site of the ligand, the positive charge would repel peptides with free N-termini but allow entry of formylated ones. Since formyl-Nle–Leu–Phe has a net negative charge, it would be attracted toward R190 but not W190. W190 would be expected to exhibit a relative preference for peptides with free N-termini and peptides with net positive charges compared to R190. This is consistent with our observation that MYVKWPWYVWL exhibits a 3 fold higher affinity for W190/N192 than for R190/N192 or R190/K192 and that formylation of MYVKWPWYVWL enhances its affinity 6 fold with R190/N192 and R190/K192 but only 2 fold with W190/N192. Formyl-MYVKWPWYVWL has a net charge of zero, and it binds equally well to all three variants.
Su et al. [4] originally observed that DP178/T-20, a peptide derived from the C-terminal HR and membrane proximal region of gp41 of human immunodeficiency virus type 1 (HIV-1), elicited chemotaxis in neutrophils and monocytes. They determined that this chemotaxis was a result of its ability to bind to and activate FPR. Su et al. [4] also observed that shorter versions of DP-178 acted as antagonists of FPR. We subsequently showed that DP178 binds to FPR expressed in CHO S cells and observed that two FPR mutants (D106N and R201A), which bind fMLF poorly, exhibited an enhanced ability (30 fold and 3 fold, respectively) to bind DP178/T20 [25]. Here we observed that R190/N192, W190/N192, and R190/N192 FPR variants all exhibited similar affinities for DP178/T20 and that DP178/T20 behaved as an antagonist based upon its inability to stimulate GTPγS binding and its ability to inhibit the stimulation of GTPγS by formyl-Nle–Leu–Phe.
HR regions appear to be a common motif in many viral fusion proteins [45]. They exist as sequential helical domains; one n terminal HR region (HR1) adjacent to the fusion peptide and a c-terminal HR region (HR2) close to the transmembrane anchor. Peptides derived from the HR2 and proximal membrane regions of HIV-1, SIV, HRSV, human parainfluenza virus type 3, measles virus, and a coronavirus have all been shown to be able to block virus infection [28], [46], [47], [48]. A twelve mer peptide derived from the proximal membrane region of FIV gp41, QLQKWEDWVRWI, was a potent (low nanomolar) inhibiter of FIV infection [49]. Here we observed that peptides derived from the proximal membrane region of HIV-1, HIV-2, Ebola virus, SARS coronavirus, and coronavirus 229E act as antagonists to the formyl peptide receptor. The coronavirus peptides are the most potent antagonists and these peptides exhibited an enhanced affinity for W190/N192 FPR. Our observation that tryptophan-rich peptides act as antagonists is consistent with the observation of Bae et al., who observed that WRWWWW was a potent antagonist for FPRL1 and FPRL2 [50], [51]. WRWWWW did not affect FPR [50], and it has been shown that DP178/T20 does not bind to FPRL1 [4], indicating that these receptors have slightly different ligand binding specificities but that both receptors may bind tryptophan rich peptides. A recently discovered FPRL2 agonist is also tryptophan rich [52] and the C-terminus of that peptide, WPWQVL, is remarkably similar to WPWWVWL which we found to be a potent antagonist for FPR. The FPRL2 agonist did not, however, bind to or activate FPR.
Coronaviruses are common infections among the human population [53], [54] and as such might affect the evolution of FPR if they were able to attach to FPR, become internalized, and spread through the body on the many cells that express FPR [55]. On the other hand, it is possible that FPR's ability to bind virally derived peptides allows it in some way to recognize the presence of a viral infection and inhibit its spread. A recent report showed that HIV-1 viruses are inactivated by transport to acidic endosomes and proteosomes and that blocking these processes can increase HIV-1 infectivity by up to 400 fold [56]. If FPR could bind viruses and transport them to endosomes and proteosomes, this might result in protection against viral infection. Another possibility is that FPR could serve as a “fly-trap” for viruses and prevent them from finding their appropriate receptor. Also, FPR might interact with the aromatic rich region subsequent to virus binding to a surface receptor and prevent membrane fusion. The scenarios are not of course mutually exclusive and the binding of viruses to FPR could have positive or negative consequences depending on the exact situation. Klestadt et al. [57] observed that activation of FPR in THP-1 cells markedly downregulated the expression of CD4 and CD13, the receptors for HIV-1/HIV-2 and coronavirus 229E, respectively [11], [58], [59]. In addition, activation of FPR downregulates the expression of CCR5 and CXCR4 [60], the co-receptors of HIV-1 and HIV-2 [9], [58]. Thus activation of FPR could serve as protection against viral infection by downregulation of viral receptors, and presumably FPR antagonists would prevent downregulation. Ueda et al. noted that the HIV-1 envelope gp41 was able to down regulate the expression of FPR and other chemokine receptors at low nanomolar concentrations, but that the downregulation of FPR and the chemokine receptors was dependent upon expression of CD4 [61]. Thus, there are a complicated set of interactions between FPR and the receptors and co-receptors of viruses, and FPR may play an important role in viral infections.
Acknowledgements
I thank Heini Miettinen for supplying PGBSA FPR expressing plasmid. I thank Heini Miettinen, Martin Teinze and John Walters for helpful discussions and critical reading of the manuscript.
These studies were supported by NIH grants 1R21DE016114-01 and P20 RR-16455-01* from the BRIN Program of the National Center for Research Resources.
References
- 1.Katanaev V.L. Signal transduction in neutrophil chemotaxis. Biochemistry (Moscow) 2001;66:351. doi: 10.1023/a:1010293809553. [DOI] [PubMed] [Google Scholar]
- 2.Springer T.A. Traffic signals on endothelium for lymphocyte recirculation and leukocyte emigration. Annu. Rev. Physiol. 1995;57:827. doi: 10.1146/annurev.ph.57.030195.004143. [DOI] [PubMed] [Google Scholar]
- 3.Schiffman E., Corcorin B., Wahl B. N-formyl methionine peptides as chemoattractants for leukocytes. Proc. Natl. Acad. Sci. U. S. A. 1975;72:1059. doi: 10.1073/pnas.72.3.1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Su S.B., Gong W.H., Gao J.L., Shen W.P., Grimm M.C., Deng X., Murphy P.M., Oppenheim J.J., Wang J.M. T20/DP178, an ectodomain peptide of human immunodeficiency virus type 1 gp41, is an activator of human phagocyte N-formyl peptide receptor. Blood. 1999;93:3885. [PubMed] [Google Scholar]
- 5.Su S.B., Gao J., Gong W., Dunlop N.M., Murphy P.M., Oppenheim J.J., Wang J.M. T21/DP107, a synthetic leucine zipper-like domain of the HIV-1 envelope gp41, attracts and activates human phagocytes by using G-protein-coupled formyl peptide receptors. J. Immunol. 1999;162:5924. [PubMed] [Google Scholar]
- 6.Postma B., Poppelier M.J., van Galen J.C., Prossnitz E.R., van Strijp J.A., de Haas C.J., van Kessel K.P. Chemotaxis inhibitory protein of Staphylococcus aureus binds specifically to the C5a and formylated peptide receptor. J. Immunol. 2004;172:6994. doi: 10.4049/jimmunol.172.11.6994. [DOI] [PubMed] [Google Scholar]
- 7.Haas P.J., de Haas C.J., Kleibeuker W., Poppelier M.J., van Kessel K.P., Kruijtzer J.A., Liskamp R.M., van Strijp J.A. N-terminal residues of the chemotaxis inhibitory protein of Staphylococcus aureus are essential for blocking formylated peptide receptor but not C5a receptor. J. Immunol. 2004;173:5704. doi: 10.4049/jimmunol.173.9.5704. [DOI] [PubMed] [Google Scholar]
- 8.Wenzel-Seifert K., Seifert R. Cyclosporin H is a potent and selective formyl peptide receptor antagonist. Comparison with N-t-butoxycarbonyl-l-phenylalanyl-l-leucyl-l-phenylalanyl-l-leucyl-l-phenylalanine and cyclosporins A B, C, D, and E. J. Immunol. 1993;150:4591. [PubMed] [Google Scholar]
- 9.Oostendorp R.A., Knol E.F., Verhoeven A.J., Scheper R.J. An immunosuppressive retrovirus-derived hexapeptide interferes with intracellular signaling in monocytes and granulocytes through N-formylpeptide receptors. J. Immunol. 1992;149:1010. [PubMed] [Google Scholar]
- 10.Bellner L., Thoren F., Nygren E., Liljeqvist J.A., Karlsson A., Eriksson K. A proinflammatory peptide from herpes simplex virus type 2 glycoprotein G affects neutrophil, monocyte, and NK cell functions. J. Immunol. 2005;174:2235. doi: 10.4049/jimmunol.174.4.2235. [DOI] [PubMed] [Google Scholar]
- 11.Overbaugh J., Luciw P.A., Hoover E.A. Models for AIDS pathogenesis: simian immunodeficiency virus, simian-human immunodeficiency virus and feline immunodeficiency virus infections. AIDS. 1997;11(Suppl. A):S47–S54. [PubMed] [Google Scholar]
- 12.Kubes P., Heit B., van M.G., Johnston J.B., Knight D., Khan A., Power C. In vivo impairment of neutrophil recruitment during lentivirus infection. J. Immunol. 2003;171:4801. doi: 10.4049/jimmunol.171.9.4801. [DOI] [PubMed] [Google Scholar]
- 13.Sahagun-Ruiz A., Colla J.S., Juhn J., Gao J.L., Murphy P.M., McDermott D.H. Contrasting evolution of the human leukocyte N-formylpeptide receptor subtypes FPR and FPRL1R. Genes Immun. 2001;2:335. doi: 10.1038/sj.gene.6363787. [DOI] [PubMed] [Google Scholar]
- 14.Liston A., McColl S. Subversion of the chemokine world by microbial pathogens. BioEssays. 2003;25:478. doi: 10.1002/bies.10273. [DOI] [PubMed] [Google Scholar]
- 15.Pease J.E., Murphy P.M. Microbial corruption of the chemokine system: an expanding paradigm. Semin. Immunol. 1998;10:169. doi: 10.1006/smim.1998.0129. [DOI] [PubMed] [Google Scholar]
- 16.Willett B.J., Picard L., Hosie M.J., Turner J.D., Adema K., Clapham P.R. Shared usage of the chemokine receptor CXCR4 by the feline and human immunodeficiency viruses. J. Virol. 1997;71:6407. doi: 10.1128/jvi.71.9.6407-6415.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kuhmann S.E., Madani N., Diop O.M., Platt E.J., Morvan J., Muller-Trutwin M.C., Barre-Sinoussi F., Kabat D. Frequent substitution polymorphisms in African green monkey CCR5 cluster at critical sites for infections by simian immunodeficiency virus SIVagm, implying ancient virus–host coevolution. J. Virol. 2001;75:8449. doi: 10.1128/JVI.75.18.8449-8460.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Daniel M.A., McDonald G., Offenbacher S., Van Dyke T.E. Defective chemotaxis and calcium response in localized juvenile periodontitis neutrophils. J. Periodontol. 1993;64:617. doi: 10.1902/jop.1993.64.7.617. [DOI] [PubMed] [Google Scholar]
- 19.Lavine W.S., Maderazo E.G., Stolman J., Ward P.A., Cogen R.B., Greenblatt I., Robertson P.B. Impaired neutrophil chemotaxis in patients with juvenile and rapidly progressing periodontitis. J. Periodontal Res. 1979;14:10. doi: 10.1111/j.1600-0765.1979.tb00213.x. [DOI] [PubMed] [Google Scholar]
- 20.Perez H.D., Kelly E., Elfman F., Armitage G., Winkler J. Defective polymorphonuclear leukocyte formyl peptide receptor(s) in juvenile periodontitis. J. Clin. Invest. 1991;87:971. doi: 10.1172/JCI115105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shibata K., Warbington M.L., Gordon B.J., Kurihara H., Van Dyke T.E. Nitric oxide synthase activity in neutrophils from patients with localized aggressive periodontitis. J. Periodontol. 2001;72:1052. doi: 10.1902/jop.2001.72.8.1052. [DOI] [PubMed] [Google Scholar]
- 22.Sigusch B., Eick S., Pfister W., Klinger G., Glockmann E. Altered chemotactic behavior of crevicular PMNs in different forms of periodontitis. J. Clin. Periodontol. 2001;28:162. doi: 10.1034/j.1600-051x.2001.028002162.x. [DOI] [PubMed] [Google Scholar]
- 23.Van Dyke T.E., Levine M.J., Tabak L.A., Genco R.J. Reduced chemotactic peptide binding in juvenile periodontitis: a model for neutrophil function. Biochem. Biophys. Res. Commun. 1981;100:1278. doi: 10.1016/0006-291x(81)91962-8. [DOI] [PubMed] [Google Scholar]
- 24.Zhang Y., Syed R., Uygar C., Pallos D., Gorry M.C., Firatli E., Cortelli J.R., VanDyke T.E., Hart P.S., Feingold E., Hart T.C. Evaluation of human leukocyte N-formylpeptide receptor (FPR1) SNPs in aggressive periodontitis patients. Genes Immun. 2003;4:22. doi: 10.1038/sj.gene.6363900. [DOI] [PubMed] [Google Scholar]
- 25.Mills J.S., Miettinen H.M., Cummings D., Jesaitis A.J. Characterization of the binding site on the formyl peptide receptor using three receptor mutants and analogs of Met–Leu–Phe and Met–Met–Trp–Leu–Leu. J. Biol. Chem. 2000;275:39012. doi: 10.1074/jbc.M003081200. [DOI] [PubMed] [Google Scholar]
- 26.Miettinen H.M., Mills J.S., Gripentrog J.M., Dratz E.A., Granger B.L., Jesaitis A.J. The ligand binding site of the formyl peptide receptor maps in the transmembrane region. J. Immunol. 1997;159:4045. [PubMed] [Google Scholar]
- 27.Jones B.E., Miettinen H.M., Jesaitis A.J., Mills J.S. Mutations of F110 and C126 of the formyl peptide receptor interfere with G-protein coupling and chemotaxis. J. Periodontol. 2003;74:475. doi: 10.1902/jop.2003.74.4.475. [DOI] [PubMed] [Google Scholar]
- 28.Wild C.T., Shugars D.C., Greenwell T.K., McDanal C.B., Matthews T.J. Peptides corresponding to a predictive alpha-helical domain of human immunodeficiency virus type 1 gp41 are potent inhibitors of virus infection. Proc. Natl. Acad. Sci. U. S. A. 1994;91:9770. doi: 10.1073/pnas.91.21.9770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sainz B., Jr., Rausch J.M., Gallaher W.R., Garry R.F., Wimley W.C. The aromatic domain of the coronavirus class I viral fusion protein induces membrane permeabilization: putative role during viral entry. Biochemistry. 2005;44:947. doi: 10.1021/bi048515g. [DOI] [PubMed] [Google Scholar]
- 30.Fouchier R.A., Hartwig N.G., Bestebroer T.M., Niemeyer B., de Jong J.C., Simon J.H., Osterhaus A.D. A previously undescribed coronavirus associated with respiratory disease in humans. Proc. Natl. Acad. Sci. U. S. A. 2004;101:6212. doi: 10.1073/pnas.0400762101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.van der H.L., Pyrc K., Jebbink M.F., Vermeulen-Oost W., Berkhout R.J., Wolthers K.C., Wertheim-van Dillen P.M., Kaandorp J., Spaargaren J., Berkhout B. Identification of a new human coronavirus. Nat. Med. 2004;10:368. doi: 10.1038/nm1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Woo P.C., Lau S.K., Chu C.M., Chan K.H., Tsoi H.W., Huang Y., Wong B.H., Poon R.W., Cai J.J., Luk W.K., Poon L.L., Wong S.S., Guan Y., Peiris J.S., Yuen K.Y. Characterization and complete genome sequence of a novel coronavirus, coronavirus HKU1, from patients with pneumonia. J. Virol. 2005;79:884. doi: 10.1128/JVI.79.2.884-895.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cardoso R.M., Zwick M.B., Stanfield R.L., Kunert R., Binley J.M., Katinger H., Burton D.R., Wilson I.A. Broadly neutralizing anti-HIV antibody 4E10 recognizes a helical conformation of a highly conserved fusion-associated motif in gp41. Immunity. 2005;22:163. doi: 10.1016/j.immuni.2004.12.011. [DOI] [PubMed] [Google Scholar]
- 34.Zwick M.B., Jensen R., Church S., Wang M., Stiegler G., Kunert R., Katinger H., Burton D.R. Anti-human immunodeficiency virus type 1 (HIV-1) antibodies 2F5 and 4E10 require surprisingly few crucial residues in the membrane-proximal external region of glycoprotein gp41 to neutralize HIV-1. J. Virol. 2005;79:1252. doi: 10.1128/JVI.79.2.1252-1261.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mills J.S., Miettinen H.M., Barnidge D., Vlases M.J., Wimer-Mackin S., Dratz E.A., Sunner J., Jesaitis A.J. Identification of a ligand binding site in the human neutrophil formyl peptide receptor using a site-specific fluorescent photoaffinity label and mass spectrometry. J. Biol. Chem. 1998;273:10428. doi: 10.1074/jbc.273.17.10428. [DOI] [PubMed] [Google Scholar]
- 36.Kyte J., Doolittle R.F. A simple method for displaying the hydropathic character of a protein. J. Mol. Biol. 1982;157:105. doi: 10.1016/0022-2836(82)90515-0. [DOI] [PubMed] [Google Scholar]
- 37.Chen J., Bernstein H.S., Chen M., Wang L., Ishii M., Turck C.W., Coughlin S.R. Tethered ligand library for discovery of peptide agonists. J. Biol. Chem. 1995;270:23398. doi: 10.1074/jbc.270.40.23398. [DOI] [PubMed] [Google Scholar]
- 38.Mansoor S.E., McHaourab H.S., Farrens D.L. Mapping proximity within proteins using fluorescence spectroscopy. A study of T4 lysozyme showing that tryptophan residues quench bimane fluorescence. Biochemistry. 2002;41:2475. doi: 10.1021/bi011198i. [DOI] [PubMed] [Google Scholar]
- 39.Azzi M., Charest P.G., Angers S., Rousseau G., Kohout T., Bouvier M., Pineyro G. Beta-arrestin-mediated activation of MAPK by inverse agonists reveals distinct active conformations for G protein-coupled receptors. Proc. Natl. Acad. Sci. U. S. A. 2003;100:11406. doi: 10.1073/pnas.1936664100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hunton D.L., Barnes W.G., Kim J., Ren X.R., Violin J.D., Reiter E., Milligan G., Patel D.D., Lefkowitz R.J. Beta-arrestin 2-dependent angiotensin II type 1A receptor-mediated pathway of chemotaxis. Mol. Pharmacol. 2005;67:1229. doi: 10.1124/mol.104.006270. [DOI] [PubMed] [Google Scholar]
- 41.Lefkowitz R.J., Shenoy S.K. Transduction of receptor signals by beta-arrestins. Science. 2005;308:512. doi: 10.1126/science.1109237. [DOI] [PubMed] [Google Scholar]
- 42.Wei H., Ahn S., Shenoy S.K., Karnik S.S., Hunyady L., Luttrell L.M., Lefkowitz R.J. Independent beta-arrestin 2 and G protein-mediated pathways for angiotensin II activation of extracellular signal-regulated kinases 1 and 2. Proc. Natl. Acad. Sci. U. S. A. 2003;100:10782. doi: 10.1073/pnas.1834556100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lefkowitz R.J., Whalen E.J. beta-arrestins: traffic cops of cell signaling. Curr. Opin. Cell Biol. 2004;16:162. doi: 10.1016/j.ceb.2004.01.001. [DOI] [PubMed] [Google Scholar]
- 44.P. Maney, J.S. Mills, and J.D. Walters, Formylpeptide Receptor Polymorphisms in African-Americans With Aggressive Periodontitis., 2006, p. 2335.
- 45.Skehel J.J., Wiley D.C. Coiled coils in both intracellular vesicle and viral membrane fusion. Cell. 1998;95:871. doi: 10.1016/s0092-8674(00)81710-9. [DOI] [PubMed] [Google Scholar]
- 46.Bosch B.J., van der Z.R., de Haan C.A., Rottier P.J. The coronavirus spike protein is a class I virus fusion protein: structural and functional characterization of the fusion core complex. J. Virol. 2003;77:8801. doi: 10.1128/JVI.77.16.8801-8811.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lambert D.M., Barney S., Lambert A.L., Guthrie K., Medinas R., Davis D.E., Bucy T., Erickson J., Merutka G., Petteway S.R., Jr. Peptides from conserved regions of paramyxovirus fusion (F) proteins are potent inhibitors of viral fusion. Proc. Natl. Acad. Sci. U. S. A. 1996;93:2186. doi: 10.1073/pnas.93.5.2186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Malashkevich V.N., Chan D.C., Chutkowski C.T., Kim P.S. Crystal structure of the simian immunodeficiency virus (SIV) gp41 core: conserved helical interactions underlie the broad inhibitory activity of gp41 peptides. Proc. Natl. Acad. Sci. U. S. A. 1998;95:9134. doi: 10.1073/pnas.95.16.9134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Giannecchini S., Di F.A., D'Ursi A.M., Matteucci D., Rovero P., Bendinelli M. Antiviral activity and conformational features of an octapeptide derived from the membrane-proximal ectodomain of the feline immunodeficiency virus transmembrane glycoprotein. J. Virol. 2003;77:3724. doi: 10.1128/JVI.77.6.3724-3733.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bae Y.S., Lee H.Y., Jo E.J., Kim J.I., Kang H.K., Ye R.D., Kwak J.Y., Ryu S.H. Identification of peptides that antagonize formyl peptide receptor-like 1-mediated signaling. J. Immunol. 2004;173:607. doi: 10.4049/jimmunol.173.1.607. [DOI] [PubMed] [Google Scholar]
- 51.Shin E.H., Lee H.Y., Kim S.D., Jo S.H., Kim M.K., Park K.S., Lee H., Bae Y.S. Trp–Arg–Trp–Trp–Trp–Trp antagonizes formyl peptide receptor like 2-mediated signaling. Biochem. Biophys. Res. Commun. 2006;341:1317. doi: 10.1016/j.bbrc.2006.01.098. [DOI] [PubMed] [Google Scholar]
- 52.Migeotte I., Riboldi E., Franssen J.D., Gregoire F., Loison C., Wittamer V., Detheux M., Robberecht P., Costagliola S., Vassart G., Sozzani S., Parmentier M., Communi D. Identification and characterization of an endogenous chemotactic ligand specific for FPRL2. J. Exp. Med. 2005;201:83. doi: 10.1084/jem.20041277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Baker S.C. Coronaviruses: from common colds to severe acute respiratory syndrome. Pediatr. Infect. Dis. J. 2004;23:1049. doi: 10.1097/01.inf.0000145815.70485.f7. [DOI] [PubMed] [Google Scholar]
- 54.Holmes K.V. Coronaviruses. In: Knipe D., Howley P., editors. Fields Virology. Lippincott, Williams and Wilkins; Philadelphia: 2001. pp. 1187–1203. [Google Scholar]
- 55.Becker E.L., Forouhar F.A., Grunnet M.L., Boulay F., Tardif M., Bormann B.J., Sodja D., Ye R.D., Woska J.R., Jr., Murphy P.M. Broad immunocytochemical localization of the formylpeptide receptor in human organs, tissues, and cells. Cell Tissue Res. 1998;292:129. doi: 10.1007/s004410051042. [DOI] [PubMed] [Google Scholar]
- 56.Wei B.L., Denton P.W., O'Neill E., Luo T., Foster J.L., Garcia J.V. Inhibition of lysosome and proteasome function enhances human immunodeficiency virus type 1 infection. J. Virol. 2005;79:5705. doi: 10.1128/JVI.79.9.5705-5712.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Klestadt D., Laval-Gilly P., Foucaud L., Falla J. Modification of membrane markers on THP-1 cells after ozone exposure in the presence or absence of fMLP. Toxicol. In Vitro. 2004;18:279. doi: 10.1016/j.tiv.2003.10.008. [DOI] [PubMed] [Google Scholar]
- 58.Philpott S.M. HIV-1 coreceptor usage, transmission, and disease progression. Curr. HIV Resour. 2003;1:217. doi: 10.2174/1570162033485357. [DOI] [PubMed] [Google Scholar]
- 59.Nomura R., Kiyota A., Suzaki E., Kataoka K., Ohe Y., Miyamoto K., Senda T., Fujimoto T. Human coronavirus 229E binds to CD13 in rafts and enters the cell through caveolae. J. Virol. 2004;78:8701. doi: 10.1128/JVI.78.16.8701-8708.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Shen W., Li B., Wetzel M.A., Rogers T.J., Henderson E.E., Su S.B., Gong W., Le Y., Sargeant R., Dimitrov D.S., Oppenheim J.J., Wang J.M. Down-regulation of the chemokine receptor CCR5 by activation of chemotactic formyl peptide receptor in human monocytes. Blood. 2000;96:2887. [PubMed] [Google Scholar]
- 61.Ueda H., Howard O.M., Grimm M.C., Su S.B., Gong W., Evans G., Ruscetti F.W., Oppenheim J.J., Wang J.M. HIV-1 envelope gp41 is a potent inhibitor of chemoattractant receptor expression and function in monocytes. J. Clin. Invest. 1998;102:804. doi: 10.1172/JCI3273. [DOI] [PMC free article] [PubMed] [Google Scholar]