

Abstract
Hepatitis C virus (HCV) core protein (core) plays a significant role in the development of chronic liver diseases caused by HCV infection. We have discovered that the core sensitized all-trans-retinoic acid (ATRA)-induced cell death in MCF-7 cells. Activation of retinoic acid receptor alpha (RARα)-mediated transcription by the core was also seen in all the cell lines tested. By use of a yeast two-hybrid system, we identified Sp110b as a candidate for a core-interacting cellular factor. Although the function of Sp110b has remained unknown, we observed that Sp110b interacts with RARα and suppresses RARα-mediated transcription. These data suggest that Sp110b is a transcriptional cofactor negatively regulating RARα-mediated transcription. RNA interference-mediated reduction of endogenous Sp110b levels depressed the ability of the core to activate RARα-mediated transcription, suggesting an essential role for Sp110b in this pathway. The normal nuclear subcellular localization of Sp110b was altered by molecular interaction with the core to the cytoplasmic surface of the endoplasmic reticulum. This evidence suggests a model in which the core sequesters Sp110b from the nucleus and inactivates its corepressor function to activate RARα-mediated transcription. These findings likely describe a novel system in which a cytoplasmic viral protein regulates host cell transcription.
Hepatitis C virus (HCV) is a causative agent of liver diseases such as chronic hepatitis, liver cirrhosis, and hepatocellular carcinoma (HCC) (1, 8, 20). HCV core protein (core), one of the viral structural proteins (18), is a multifunctional protein regulating several defined cellular events. Core gene transgenic mice develop hepatic steatosis and HCC (23, 31, 32). Also, expression of the core protein has the potential to transform several cultured rodent cells (40, 51) and has been observed to modulate apoptotic responses of the cells (28, 41, 43). From these observations, core protein function is thought to be closely related to the molecular basis of HCV-related diseases. Despite the efforts of many researchers, the precise mechanisms governing the phenomena caused by core expression have remained unknown.
Other viral proteins, such as adenovirus E1A and E1B (44), human T-cell leukemia virus type 1 (HTLV-1) Tax (29), and human papillomavirus (HPV) E6 and E7 (33), also mediate a variety of functions in the cells. A variety of novel cellular mechanisms and proteins have been disclosed through the investigation of these viral proteins. Therefore, understanding the molecular mechanisms governing the phenomena induced by the core, such as tumor development, cellular transformation, and modulation of apoptotic responses, may also disclose a novel cellular signaling pathway. In this study, we investigated the molecular mechanism underlying modulation of apoptosis by core expression.
Of the various apoptotic stimuli tested, we determined that all-trans-retinoic acid (ATRA)-induced cell death was sensitized by core expression. This sensitization correlated with the activation of ATRA-induced transcription and the enhancement of downstream proapoptotic gene expression. To investigate the mechanism by which the core activates ATRA-induced transcription, we screened for cellular factors interacting with the core by the yeast two-hybrid system. This search identified Sp110b, whose function is unknown, as a core-interacting protein. Here we demonstrate that Sp110b is a potent transcriptional corepressor of retinoic acid receptor alpha (RARα), playing a critical role in core-mediated activation of RAR signaling.
Until now, several viral proteins, such as adenovirus E1A (2, 26) and E1B (24), human immunodeficiency virus type 1 (HIV-1) Tat (3), HTLV-1 Tax (4, 46, 53), and HPV E6 (38) and E7 (37), have been reported to regulate cellular transcriptional activity via interactions with transcriptional cofactors such as CBP/p300, PCAF, histone deacetylase complex, and SRC-1. These viral proteins are primarily located in the nucleus, directly modulating the function of transcriptional cofactors. In this study, however, we determined that HCV core interacted with Sp110b on the cytoplasmic surface of the endoplasmic reticulum (ER) to modulate retinoid-dependent nuclear receptor signaling. It is suggested that the core sequesters Sp110b away from the nucleus and inactivates the corepressor function of Sp110b, resulting in the activation of ATRA-induced transcription and sensitization to ATRA-mediated cell death. This is the first model explaining the mechanism of transcriptional regulation by the core. Furthermore, this seems to be a novel system of host transcriptional regulation by a cytoplasmic viral protein through the altered localization of a cellular transcriptional cofactor from the nucleus.
MATERIALS AND METHODS
Plasmid constructs.
The pcDNA-myc, pCMV-FLAG, and pCA-FLAG vectors were obtained by inserting the Myc, FLAG, and FLAG tag coding sequences into the BamHI-XhoI sites of pcDNA3 (Invitrogen), the EcoRI-HindIII sites of pKS(+)/CMV (28), and the EcoRI site of pCAGGS (a gift from J. Miyazaki, Osaka University Medical School) (34), respectively. Sp110 and Sp110b cDNAs covering the entire open reading frame regions were obtained by reverse transcriptase PCR (RT-PCR) from a human liver total-RNA (Clontech) template by using primers 5′-GTTGAATTCATGTTCACCATGACAAGAG-3′ and 5′-GTTCTCGAGTCAAGGAAGAGTCCAG-3′ for Sp110 and primers 5′-GTTGAATTCATGTTCACCATGACAAGAG-3′ and 5′-GTTCTCGAGTTATTCTTGGAGGACAG-3′ for Sp110b. To obtain pcDNA-Sp110, pCA-Sp110, pcDNA-Sp110b, and pCMV-Sp110b, the Sp110 or Sp110b PCR product was subcloned into the EcoRI-XhoI site of pcDNA-myc, pCA-FLAG, or pCMV-FLAG. The series of plasmids expressing deletion mutants of Sp110 and Sp110b, pcDNA-Sp110(1-276), pcDNA-Sp110(277-453), pcDNA-Sp110(454-689), pcDNA-Sp110(277-388), pcDNA-Sp110(389-689), pcDNA-Sp110b(389-453) (CBR fragment), pcDNA-Sp110b(389-539), and pcDNA-Sp110b(1-276 + 454-539), was constructed by inserting PCR fragments generated by using appropriate synthetic oligonucleotides as primers and either pcDNA-Sp110 or pcDNA-Sp110b as the template.
The generation of pCMV-core has been described previously (28). pcDNA-core was obtained by inserting a BamHI-EcoRI pCMV-core fragment encoding the core protein into the BamHI-EcoRI site of pcDNA3. pCMV-core(6162M) was made by oligonucleotide-directed mutagenesis using oligonucleotides 5′-CCTCGTGGAGGGCTACAACCTATCCC-3′ and 5′-GGGATAGGTTGTAGCCCTCCACGAGG-3′ as primers. pGEX-Sp110 and pGEX-Sp110b, encoding a fusion of glutathione S-transferase (GST) and either Sp110 or Sp110b, respectively, were created by insertion of the EcoRI-XhoI Sp110 or Sp110b fragment, respectively, into the EcoRI-XhoI site of the pGEX-6P1 vector (Clontech). To obtain pGEX-core(1-80), the DNA fragment generated by PCR using oligonucleotides 5′-GTTGGATCCATGAGCACAAATCC-3′ and 5′-GTTCTCGAGTCACCCGGGCTGAGCCC-3′ as primers and pcDNA-core as a template was inserted into the BamHI-XhoI site of pGEX-6P1. All expression plasmids encoding other core derivatives were made as described previously (54).
pRARE-Luc, a reporter plasmid containing three copies of the RARα binding element (RARE), was obtained by inserting the MluI-XhoI PCR fragment prepared with primers 5′-GTTACGCGTGTTCACCGAAAGTTCACCGAGAGTTCACCGAAAGTTCACAGCCA-3′ and 5′-GTTCTCGAGGTGAACTTTCGGTGAACTGGCTGTGAACTTTCGGTGAACTCTCG-3′ into the MluI-XhoI site of p-55BLuc (kindly provided by T. Kiyono, National Cancer Center Research Institute, Tokyo, Japan) (21). pSG5-RARα, encoding RARα, was kindly provided by P. Chambon of the Institut de Genetique et de Biologie Moleculaire et Cellulaire, CNRS/INSERM/ULP/College de France, and N. Kato at the Institute of Molecular and Cellular Biosciences, The University of Tokyo.
Cell culture and transfection.
MCF-7, COS-7, 293T, and HeLa cells were cultured in Dulbecco's modified Eagle's medium (Nissui) supplemented with 10% fetal bovine serum and l-glutamine. Plasmid transfection of cells was performed as described previously (54).
Establishment of HCV core-producing cell lines.
After transfection with pLXSH-core or pLXSH (51), MCF-7 cells were selected in the culture medium containing 250 μg of hygromycin B (Wako)/ml to establish several clones of stable transfectants containing these plasmids.
Immunoblot analysis.
Immunoblot analysis was performed essentially as described previously (54). The antibodies used in these experiments were specific for HCV core (515S; a gift from M. Kohara, Tokyo Metropolitan Institute of Medical Science), α-tubulin (Ab-1; Calbiochem), FLAG (M2; Sigma), and SC-35 (Sigma).
Reporter assay.
The pRARE-Luc reporter plasmid, containing three copies of RARE upstream of a basal transcription promoter region, was used for the reporter assay as described previously (28).
Yeast two-hybrid screening.
The region of the core containing amino acids (aa) 1 to 80, subcloned into the pHybLex/Zeo vector (Invitrogen), was used to screen a human prostate cDNA library (Clontech) according to the Hybrid Hunter protocol (Invitrogen). A total of 5 × 106 transformants were selected based on histidine prototrophy and β-galactosidase activity.
GST pulldown assay.
GST and the GST-Sp110, GST-Sp110b, and GST-core(1-80) fusion proteins, encoded by pGEX-6P1, pGEX-Sp110, pGEX-Sp110b, and pGEX-core(1-80), respectively, were produced in BL21 cells (Amersham Biosciences) following treatment with 1 mM isopropyl-β-d-thiogalactopyranoside(IPTG). After preparation of proteins by using glutathione-Sepharose resin (Amersham Biosciences), proteins bound to the resin were incubated with in vitro-translated, radiolabeled proteins at 4°C for 1 h. 35S-radiolabeled proteins were prepared with the TNT T7 quick coupled transcription-translation system (Promega) using 0.25 μg of expression plasmid and l-[35S]methionine (Amersham Biosciences), according to the manufacturer's protocol. After five washes in binding buffer (containing 50 mM Tris-HCl [pH 8.0], 150 mM NaCl, 1 mM EDTA, 0.5% NP-40, 1 mM dithiothreitol, and 1 mM phenylmethylsulfonyl fluoride for the experiment for which results are shown in Fig. 3B and 5D, and containing those reagents supplemented with 0.5% NP-40, 0.5% sodium deoxycholate, and 0.05% sodium dodecyl sulfate (SDS) for the experiment for which results are shown in Fig. 3A), resin-bound radiolabeled proteins were fractionated by SDS-polyacrylamide gel electrophoresis and detected by autoradiography.
FIG. 3.

Interaction of the core with Sp110b. (A) The 35S-labeled in vitro transcription-translation product of full-length core (top panel) or the core(6162M) mutant (center panel) was incubated with recombinant Sp110 or Sp110b fused to GST (GST-Sp110 or GST-Sp110b) or with GST as a negative control. The GST pulldown assay was performed as described in Materials and Methods. 1/10 input, signal for 1/10 the amount of 35S-labeled product used in the pulldown assay. Coomassie brilliant blue (CBB) staining patterns of pulled-down proteins are shown in the bottom panel. Arrowhead, circle, and square indicate the bands corresponding to GST, GST-Sp110, and GST-Sp110b, respectively. (B) Mapping of the region interacting with the core in Sp110b by deletion analysis. (Left) Schematic representations of the full-length and truncated mutants of Sp110 and Sp110b are shown. Numbers above the diagrams indicate the amino acid positions from the amino terminus of Sp110 or Sp110b. The Sp100-like domain (Sp), SAND domain (S), PHD (P), and bromodomain (B) are indicated. (Right) Designations of the GST fusion protein and 35S-labeled derivatives of Sp110 and Sp110b are given above and to the left of the autoradiograms (a through k), respectively. GST-core80, GST fused with the region of the core from aa 1 to 80. CBB staining patterns of pulled-down proteins are shown in the bottom panel. Arrow and arrowhead indicate bands corresponding to GST and GST-core80, respectively. Two dots indicate apparent degradation products of GST-core80. (C) Interaction between the core and Sp110b produced in the cells. Lysates from COS-7 cells transfected with 1 μg of pCMV-Sp110b and/or pCMV-core were used for coimmunoprecipitation, followed by immunoblot analysis. The combinations of plasmids used for the transfection are indicated at the top. “IP” designates the antibodies used for immunoprecipitation, either the anti-FLAG antibody (FL) or normal mouse IgG (used as a negative control). Coimmunoprecipitated core with FLAG-tagged Sp110b was detected with an anti-core antibody (top panel). Center and bottom panels show results of experiments in which the core and FLAG-tagged Sp110b in total-cell lysates from transfectants were detected by anti-core and anti-FLAG antibodies, respectively.
FIG. 5.

Sp110b is a potent transcriptional corepressor of RARα. (A through C) Activation and suppression of RARα-mediated transcription by Sp110 and Sp110b, respectively. COS-7 cells were transfected with 25 ng of pRARE-Luc with pCA-Sp110 (A), pCMV-Sp110b (B), or pCMV-Sp110b with pSG5-RARα (C) at the indicated doses for a total of 400 ng of plasmids by adjusting with the empty vector. Additional conditions were as described in the legend to Fig. 2A. RARα protein production levels for each condition were examined by immunoblot analysis (C, lower panel). (D) Interaction of Sp110 and Sp110b with RARα. A GST pulldown assay was performed as described in Materials and Methods. 35S-labeled RARα was incubated with GST-Sp110, GST-Sp110b, or GST in the absence [ATRA(−)] or presence [ATRA (+)] of 1 μM ATRA. The Coomassie brilliant blue (CBB) staining pattern of pulled-down products is shown in the bottom panel. Positions of molecular size markers are given on the left. Arrowhead, circle, and square indicate bands corresponding to GST, GST-Sp110, and GST-Sp110b, respectively. (E) Coimmunoprecipitation assay detecting the interaction between Sp110 and RARα. Lysates from ATRA-treated 293T cells overproducing FLAG-tagged Sp110b (FL-Sp110b) and/or RXRα with HA-tagged RARα (HA-RARα) were used for coimmunoprecipitation followed by immunoblot analysis. Data are presented essentially as described in the legend to Fig. 3C. IgG, normal mouse IgG; HA, anti-HA antibody. FL-Sp110b coimmunoprecipitated (co-IP) with HA-RARα was detected by using an anti-FLAG antibody (upper panel). Center and lower panels show HA-RARα and FL-Sp110b in total-cell lysates detected by anti-HA and anti-FLAG antibodies, respectively. (F) DNA-protein complex immunoprecipitation assay detecting the RARα-Sp110b-RARE complex. 293T cells overproducing either RXRα with HA-RARα or FL-Sp110b, or both, and carrying either pRARE-Luc (RARE +) or the p-55BLuc reporter plasmid lacking RARE (RARE −), were treated with ATRA. Formaldehyde-cross-linked DNA-protein complexes were then immunoprecipitated with anti-HA (HA), anti-FLAG (FL), or normal mouse IgG (IgG). The DNA extracted from the respective immunoprecipitates was amplified by PCR as described in Materials and Methods.
Immunoprecipitation.
Cells were lysed in immunoprecipitation buffer, containing 50 mM Tris-HCl (pH 8.0), 150 mM NaCl, and 0.5% NP-40 for the experiment for which results are shown in Fig. 3C. For Fig. 5E, the buffer was composed of 50 mM Tris-HCl (pH 8.0), 150 mM NaCl, 0.1% NP-40, 0.1% sodium deoxycholate, and 0.01% SDS. After centrifugation, the supernatant was incubated with an anti-FLAG antibody (M2; Sigma), an anti-hemagglutinin (HA) antibody (3F10; Roche), or normal mouse immunoglobulin G (IgG; Zymed Laboratories) for at least 1 h. Immune complexes were recovered by adsorption to protein G-Sepharose resin (Amersham Biosciences). After four washes in immunoprecipitation buffer, the immunoprecipitates were analyzed by immunoblot analysis.
Northern blot analysis.
Total RNA, isolated from human tissue (Clontech), was analyzed by Northern blot hybridization using ULTRAhyb (Ambion) according to the manufacturer's protocol. Probes were prepared by PCR in the presence of [32P]dCTP (Amersham Biosciences) by using HindIII-XbaI-digested pCMV-Sp110b as a template and 5′-TGAGGCTGTCCCTCTTGG-3′ as a primer.
RT-PCR.
The relative expression of Sp110 and Sp110b mRNAs was evaluated by semiquantitative RT-PCR with a one-step RNA PCR kit (Takara) by using one sense primer (5′-GTTGAATTCATGCTCCAAGTGGTGGATAAG-3′) and two antisense primers (5′-GTTCTCGAGTCAAGGAAGAGTCCAG-3′ and 5′-GTTCTCGAGTTATTCTTGGAGGACAG-3′). Sp110 and Sp110b RNAs synthesized from pcDNA-Sp110 and pcDNA-Sp110b in vitro by using the MEGAscript T7 kit (Ambion) were used as standards. The total RNA of cultured cells was isolated with Sepasol-RNA I Super (Nacalai Tesque, Kyoto, Japan) according to the manufacturer's protocol. The primers, 5′-GCATGGTCAACTGCAACGATG-3′ and 5′-GGGCGCATCGTACTTGGTG-3′, were used for detection of tissue transglutaminase (tTGase) mRNA.
DNA-protein complex immunoprecipitation assay.
293T cells treated with 1 μM ATRA were transfected with expression plasmids together with either the reporter plasmid pRARE-Luc, containing RARE, or p-55BLuc, which lacks RARE, as a negative control. After cross-linking with formaldehyde for 15 min, cells were lysed, sonicated, and subjected to immunoprecipitation using anti-FLAG, anti-HA, or normal mouse IgG. Recovered immunocomplexes were incubated at 65°C for 16 h and then digested with proteinase K for 2 h. DNA was extracted from the immunocomplexes with phenol and precipitated with ethanol. The primers used for detection of DNA were 5′-CACTGCATTCTAGTTGTGG-3′ and 5′-ACCAACAGTACCGGAATG-3′.
Production of an antibody against Sp110 and Sp110b.
A rabbit polyclonal antiserum against Sp110 and Sp110b was generated by immunization with a synthesized peptide (STPSDKKGKKRKRC, corresponding to aa 270 to 283 of Sp110 and Sp110b) conjugated to keyhole limpet hemocyanin (MBL, Nagoya, Japan).
RNA interference (RNAi) technique.
A 21-nucleotide small interfering RNA (siRNA) duplex (5′-AAGCUUCAAACGUGUUGGUGC-3′) containing 3′ dTdT overhanging sequences was synthesized (Dharmacon). siRNA transfection was performed using the Oligofectamine reagent (Invitrogen) according to the manufacturer's protocol.
Indirect immunofluorescence analysis.
Indirect immunofluorescence analysis was performed as described previously (54). Cells were permeabilized with 0.05% Triton X-100 and treated with the primary anti-FLAG (M2; Sigma), anti-Myc (9E10; Santa Cruz), anti-core (515S), or anti-RARα (C-20; Santa Cruz) antibody and the rabbit antiserum against Sp110 and Sp110b. The secondary antibodies conjugated to Alexa 488 and 568 series (Molecular Probes) were used to visualize primary antibody staining. Nuclei were stained with 4′,6′-diamidino-2-phenylindole (DAPI).
Fractionation of cell extracts.
Cells transfected with various plasmids were harvested and homogenized in a buffer containing 50 mM Tris-HCl (pH 8.0), 150 mM NaCl, and 1 mM EDTA. After centrifugation (MX-150; Tomy) at 3,900 rpm for 7 min, postnuclear supernatants were further centrifuged at 7,000 rpm for 5 min. The resulting supernatant was subjected to ultracentrifugation (himac CS120FX; Hitachi) at 50,000 rpm for 1 h. The pellet and supernatant fractions were collected as microsomal membranes and cytosolic fractions, respectively.
Evaluation of cell death.
Dead cell numbers were evaluated as described previously (28). The cells were stained with trypan blue dye; the percentage of cell death was measured by scoring at least 500 cells per experiment.
Concentration of cells transiently transfected with the expression plasmids.
The specific concentration of transiently transfected cells from heterogeneous cell populations was determined with the MACSelect system (Miltenyi Biotec) as described previously (28).
RESULTS
HCV core sensitizes ATRA-induced cell death.
To investigate the effect of the core on sensitivity to apoptosis induced by a variety of stimuli, we established MCF-7 cells constitutively producing the core. Examination of the response of these cells to a variety of apoptotic stimuli determined that ATRA-induced cell death was enhanced in core-producing cells (Fig. 1A). Similar results were obtained for MCF-7 cells transiently producing the core protein (Fig. 1B, bars 1 to 6). This enhancement of apoptotic induction was accompanied by an enhancement of the expression of tTGase (Fig. 1C, lanes 4 and 5), which is downstream of ATRA and involved in ATRA-mediated apoptosis (19, 35, 36). It was confirmed that suppression of tTGase function by a tTGase inhibitor, monodansylcadaverine (36), reduced ATRA-induced cell death in core-producing MCF-7 cells (Fig. 1B, bars 3, 4, 11, and 12). Thus, the core appears to sensitize cells to ATRA-mediated cell death, accompanied by an enhancement of proapoptotic tTGase gene induction.
FIG. 1.

HCV core sensitized ATRA-induced cell death. (A) Production levels of the core in each clone of MCF-7 cells stably transfected with pLXSH-core (MCF-7-core 6 and -core 21) or the empty vector pLXSH (MCF-7-vec) were analyzed by immunoblot analysis with an anti-core (right, upper panel) and an anti-α-tubulin (right, lower panel) antibody as an internal control. MCF-7-core 6, MCF-7-core 21, and MCF-7-vec cells (5 × 104 each) were incubated in the absence (no treatment) or presence (ATRA) of 1 μM ATRA for 96 h. Living and dead cells were quantitated by staining with trypan blue. The average percentage of cell death from three independent experiments is presented. Open (bars 1 and 4), solid (bars 2 and 5), and hatched (bars 3 and 6) bars, MCF-7-vec, -core 6, and -core 21, respectively. (B) MCF-7 cells (5 × 104) transfected with 2.5 μg of pMACS-Kk and each expression plasmid given below were treated with (solid bars) or without (open bars) 1 μM ATRA for 96 h after the concentration of transfected cells by using the MACSelect system (see Materials and Methods). For bars 11 and 12, 50 μM monodansylcadaverine (MDC) was added simultaneously with ATRA. The percentage of cell death was estimated as described for panel A. Bars 1 and 2, empty vector; bars 3, 4, 11, and 12, pCMV-core (3 μg); bars 5 and 6, pCMV-core (6 μg); bars 7 and 8, pCMV-core (3 μg) and pCMV-Sp110b(389-453) (CBR fragment; 4.5 μg); bars 9 and 10, pCMV-core(6162M) (3 μg). (C) Enhancement of ATRA-induced tTGase expression by the core. MCF-7 cells (2 × 105) transfected with pMACS-Kk and each expression plasmid given below were treated for 48 h with (lanes 4 to 6) or without (lanes 1 to 3) 1 μM ATRA after cell concentration as described for panel B. Following the extraction of total RNA from these cells, mRNA levels of tTGase (upper panel) and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) as an internal control (lower panel) were semiquantified by RT-PCR as described in Materials and Methods. RTase(−), experimental control treated like other samples, but without reverse transcriptase. Lanes: 1 and 4, empty vector; 2 and 5, pCMV-core; 3 and 6, pCMV-core and pCMV-Sp110b(389-453) (CBR fragment).
HCV core protein activates RAR-mediated transcription.
ATRA-induced apoptosis and tTGase expression are reportedly mediated by the transcriptional activity of RAR, the nuclear receptor for ATRA (11, 19, 36). Therefore, we next examined the effect of core expression on the activity of RAR-mediated transcription by a reporter assay. We used the RAR-responsive reporter plasmid pRARE-Luc, which includes a basal promoter with three copies of RARE upstream of the luciferase gene, to assess the RARα-mediated transcriptional activity of the cells producing the core either transiently (Fig. 2A) or constitutively (Fig. 2B). Luciferase activity increased in a manner dependent on core concentration. Similar results were obtained with additional cell lines, including HeLa, Huh-7, and MCF-7 cells, producing the core transiently (data not shown). Both the whole HCV polyprotein and the core alone activated luciferase activity (data not shown). In Huh-7 cells carrying an HCV full-genome replicon, established in our laboratory (unpublished data), RARα activity increased approximately threefold over the levels observed in parental Huh-7 cells (data not shown). Since the luciferase activity driven by a reporter plasmid lacking RARE was not affected by core expression (data not shown), these results suggested that the core protein activates RARα-mediated transcription independently of cell type.
FIG. 2.

Activation of RARα-mediated transcription in cells expressing the core. (A) COS-7 cells were transfected with 25 ng of pRARE-Luc, pCMV-core at the doses indicated, and the empty vector for a total amount of 400 ng of plasmids. Cells were then treated with (solid bars) or without (open bars) 1 μM ATRA. At 24 h posttransfection, luciferase activities of whole-cell lysates were measured. Data are means of the relative luciferase activities in three independent experiments. Error bars, standard deviations. (B) MCF-7-core 6, MCF-7-core 21, and MCF-7-vec cells were transfected with 25 ng of the pRARE-Luc reporter plasmid. (C) Identification of the region within the core essential for activation of RARα-mediated transcription. COS-7 cells were transfected with 25 ng of pRARE-Luc and 350 ng of a series of truncated core expression plasmids [core full, pCMV-core; coreΔ(101-191), pCMV-coreΔ(101-191); coreΔ(1-20 + 81-191), pCMV-coreΔ(1-20 + 81-191); coreΔ(21-80)-p7, pCMV-coreΔ(21-80)-p7] or control plasmids (control, pKS+/CMV; p7, pCMV-p7). Experimental conditions and presentation of data for panels B and C were the same as those described for panel A.
To identify the region of the core protein responsible for RARα activation, we performed a detailed deletion analysis as described previously (54). We prepared carboxy-terminal deletion constructs fused with HCV p7 containing a signal sequence to allow localization to the ER. We confirmed that these fused proteins reside on ER membranes, as seen with the wild-type core (27, 54) (data not shown). Cells producing the core constructs lacking the amino-terminal 100 aa [coreΔ(101-191)] or aa 21 to 80 [coreΔ(1-20 + 81-191)] demonstrated reporter activities similar to those of negative-control cells transfected with the empty vector (Fig. 2C). Production of the region from aa 21 to aa 80 [coreΔ(21-80)-p7] upregulated reporter activity significantly from the levels observed in negative-control cells and cells producing p7 alone. These data indicated that the region from aa 21 to aa 80 of the core [coreΔ(21-80)] is responsible for the activation of RARα-mediated transcription.
Identification of cellular factors interacting with HCV core by yeast two-hybrid screening.
To clarify the molecular mechanisms governing transcriptional activation of RARα by the core protein, we tried to identify cellular factors interacting with coreΔ(21-80) by yeast two-hybrid screening. Using the amino-terminal 80 aa of the core (core80) fused to the LexA DNA binding region as bait, we screened a human prostate cDNA library for interacting proteins. Six independent candidate clones for binding with the bait were selected from 5 × 106 transformants. Sequencing analysis revealed that one of the candidate clones encoded Sp110b. A splicing variant of this protein, Sp110, reportedly activates RARα-mediated transcription. A schematic diagram of the molecular structures of Sp110 and Sp110b is shown in Fig. 3B (left).
Sp110b interacts with HCV core through its central region.
To confirm an interaction between the core and Sp110b, we performed a GST pulldown assay in vitro. Recombinant Sp110 and Sp110b fused to GST (GST-Sp110 and GST-Sp110b) were incubated with in vitro-translated wild-type 35S-labeled core. The 35S-labeled core was copurified with Sp110b but not with Sp110 (Fig. 3A, top panel). In vitro-translated Sp110b was efficiently pulled down with GST-core80, but only a small amount of Sp110 could be observed in the pulled-down fraction (Fig. 3Ba and b). These results suggested that the core interacts more efficiently with Sp110b than with Sp110. The efficient interaction of the core with Sp110b appeared to be specific, as no association was observed with Sp100, which exhibits significant homology to Sp110 and Sp110b (Fig. 3Bk).
We used deletion analysis to determine the region of Sp110b interacting with the core. The in vitro-synthesized fragment of Sp110b from aa 277 to 453, but not fragments of Sp110 from aa 1 to 276 or aa 454 to 689, was copurified with GST-core80 (Fig. 3Bc to e). Sp110b lacking the region from aa 277 to 453 could not be copurified with GST-core80 (Fig. 3Bj), suggesting that the region of Sp110b from aa 277 to 453 is both necessary and sufficient for the interaction with core80. Dissection of this region determined that the region of Sp110b from aa 389 to 453, but not that from aa 277 to 388, was essential for this interaction (Fig. 3Bf and g). Therefore, we have named this region the core-binding region (CBR). Although the CBR is shared by Sp110b and Sp110, the affinity of Sp110 to the core was low in vitro, as described above. Similar results were obtained when the C-terminal fragments of Sp110b and Sp110, which include the CBR, were examined in this assay system (Fig. 3Bh and i). From these results, it appears to be possible that the C-terminal region of Sp110, containing the plant homeobox domain (PHD) and the bromodomain, has an inhibitory effect on core association. Analysis of additional deletion mutants showed that the entire C-terminal domain harboring both the PHD and the bromodomain, rather than either domain alone, was likely to be responsible for blocking the interaction with the core (data not shown).
To further confirm the interaction of Sp110b with the core, we performed a coimmunoprecipitation assay. The core was detected in immune complexes pulled down with an anti-FLAG antibody, but not with normal mouse IgG, from the lysates of COS-7 cells producing both FLAG-tagged Sp110b and the core simultaneously (Fig. 3C). Cumulatively, these results indicate that Sp110b interacts specifically with the core.
Sp110b is expressed ubiquitously in human tissues and more abundantly than Sp110.
Sp110b expression in human tissues was examined by Northern blot analysis using the 32P-labeled 5′-terminal cDNA region of Sp110b as a probe. A 1.9-kb band was detected in all the human tissues investigated (Fig. 4A). From the information that the Sp110 and Sp110b mRNAs are approximately 2.3 and 1.9 kb, respectively, this band seemed to correspond to Sp110b mRNA. The 2.3-kb band of Sp110 mRNA was not observed in this experiment. To compare the expression levels of Sp110 and Sp110b mRNAs in these specimens, a semiquantitative analysis was conducted by RT-PCR. The band for Sp110 mRNA was detected in total RNAs from human spleens and Jurkat cells, a human T-lymphoma cell line (Fig. 4B, lanes 11 and 14). Little or no signal was observed in other tissues and cell lines (Fig. 4B, lanes 10 to 15). We observed, however, that Sp110b mRNA was more abundant than Sp110 mRNA in all the tissues and cell lines investigated. In vitro-synthesized Sp110 and Sp110b mRNAs were mixed at variable ratios as templates in this system (Fig. 4B, lanes 1 to 9); the expression of Sp110b mRNA was estimated to be about 10- and 3- to 5-fold higher than that of Sp110 mRNA in nonleukocytes and leukocytes, respectively. These results were consistent with data in which Sp110b protein but not Sp110 protein was detected in lysates of HeLa (Fig. 6A) and Huh-7 (data not shown) cells by using an antibody recognizing both Sp110 and Sp110b protein. Thus, Sp110b mRNA is expressed ubiquitously in all the human tissues examined, more abundantly than Sp110.
FIG. 4.

Expression of Sp110 and Sp110b mRNAs in human tissues and cell lines. (A) Northern blot analysis detecting Sp110 and Sp110b mRNAs in human tissues. (Upper panel) Ten-microgram portions of total RNAs from human brain, heart, liver, lung, kidney, bone marrow, placenta, thymus, testis, and spleen were analyzed by using a 32P-labeled Sp110b fragment as a probe. Positions of molecular size markers are indicated on the left. (Lower panel). Ethidium bromide staining of 28S rRNA served as a loading control. (B) Semiquantitative RT-PCR analysis of Sp110 and Sp110b mRNAs in human tissues and cell lines. (Upper panel) cDNA fragments of Sp110 and Sp110b mRNAs were simultaneously amplified by RT-PCR using a common sense primer and two antisense primers specific to Sp110 and Sp110b mRNAs, respectively. To evaluate the quantifying ability of this system, mixtures of in vitro-synthesized Sp110 and Sp110b RNAs at various concentrations (given above the gel, in femtograms) were used as templates (lanes 1 to 9). Total RNAs from human kidneys (lane 10) and spleens (lane 11) and from MCF-7 (lane 12), Huh-7 (lane 13), Jurkat (lane 14), and HeLa (lane 15) cells were examined. (Lower panel) As an internal control, glyceraldehyde-3-phosphate dehydrogenase (GAPDH) mRNA was detected by a similar protocol (lanes 10 to 15). RTase(−), experimental control treated like other samples, but without reverse transcriptase.
FIG. 6.

The RARα activation capacity of HCV core was reduced by RNAi elimination of endogenous Sp110b. (A) RNAi elimination of endogenous Sp110b protein in HeLa cells. Extracts from cells treated with 300 U of gamma interferon/ml after transfection with either an siRNA specific for Sp110 and Sp110b [Sp110(b)-siRNA] or a randomized siRNA (control-siRNA) was analyzed with an antibody recognizing both Sp110 and Sp110b (upper panel) or anti-α-tubulin (lower panel). As a positive control, small amounts of extracts from cells overproducing FLAG-tagged Sp110 or Sp110b were also analyzed. Positions of molecular size markers (in kilodaltons) are given on the right. (B) HeLa cells were transfected with control-siRNA (bars 1 and 2) or Sp110(b)-siRNA (bars 3 and 4). At 24 h posttransfection, plasmid transfection with 25 ng of pRARE-Luc and 300 ng of either pKS+/CMV (bars 1 and 3) or pCMV-core (bars 2 and 4) was carried out. An additional 24 h later, the cells were harvested; luciferase activity was measured as described in the legend to Fig. 2A. Data are means of the relative luciferase activities in three independent experiments. Comparisons of luciferase activities (bars 1 versus 2 [×2.69] and bars 3 versus 4 [×1.30]) are shown above the graph. The combinations of siRNAs and plasmids used are indicated below the graph.
Sp110b is a potent transcriptional corepressor of RAR.
Although Sp110 is reported to activate RARα-mediated transcription (6), the molecular function of Sp110b remains unknown. To characterize the function of Sp110b, we examined the effect of this protein on RARα-mediated transcription by a reporter assay. As reported previously (6), ectopic expression of Sp110 augmented reporter activity in a dose-dependent manner (Fig. 5A). In contrast, luciferase activity was drastically reduced in cells producing Sp110b in a dose-dependent manner, irrespective of ectopic RARα production (Fig. 5B and C). Reporter activity from the reporter plasmid lacking RARE was not affected by ectopically produced Sp110 or Sp110b (data not shown). Similar results were obtained in all the cell lines examined, including Huh-7, MCF-7, and HeLa cells (data not shown). Moreover, we observed the downregulation of RAR-responsive genes, including tTGase and transforming growth factor β2 (TGF-β2), in cells producing Sp110b (data not shown). These data suggest that Sp110b suppresses RARα function. To investigate the function of endogenous Sp110b, we introduced an siRNA, Sp110(b)-siRNA, to specifically knock down the expression of Sp110 and Sp110b in HeLa cells in which Sp110b was expressed but Sp110 protein was not detected (Fig. 6A). Endogenous Sp110b levels were dramatically reduced, while α-tubulin levels remained the same (Fig. 6A). Under these conditions, RARα-mediated transcription was activated two- to threefold over that in cells transfected with a randomized siRNA (Fig. 6B, bars 1 and 3). These findings indicated that Sp110b suppresses the transcriptional activity of RARα without altering RARα production levels (Fig. 5C, lower panel).
The existence of a liganded nuclear receptor binding motif (LXXLL) (17) in Sp110 and Sp110b (aa 525 to 529; LGELL) raised the possibility that Sp110 and Sp110b may interact with the nuclear receptor RARα in the presence of RARα ligands. We investigated this possibility using a GST pulldown assay. RARα was copurified with both GST-Sp110 and GST-Sp110b in the presence, but not in the absence, of ATRA (Fig. 5D, top and center panels), indicating that RARα interacts with Sp110 and Sp110b in vitro in a ligand-dependent manner. The interaction between Sp110b and RARα in the presence of ATRA was confirmed by a coimmunoprecipitation assay with 293T cells (Fig. 5E). Further analysis on whether Sp110b, together with RARα, associates with the target promoter was performed by a DNA-protein complex immunoprecipitation assay. A RARE-containing promoter was immunoprecipitated with either Sp110b or RARα in the presence of ATRA (Fig. 5F). These data suggest that, in conjunction with RARα, Sp110b associates with the target promoter containing RARE in the presence of ATRA. In addition, Sp110 and Sp110b were observed in the nuclei of cells in a dense granular staining pattern (Fig. 7A, panels 2, 9, and 30), as previously reported for Sp110 (6). RARα was also located primarily in the nucleus (Fig. 7A, panels 3 to 5 and 10 to 12), as reported previously (7).
FIG. 7.

The subcellular localization of Sp110b was altered from the nucleus to an area around the cytoplasmic surface of ER membranes by core expression. (A) Indirect immunofluorescence analysis was performed on COS-7 cells transfected with pCMV-core (panel 1), pCA-Sp110 (panel 2), pCA-Sp110 with pSG5-RARα (panels 3 to 5), pCA-Sp110 with pCMV-core (panels 6 to 8), pCMV-Sp110b (panel 9), pCMV-Sp110b with pSG5-RARα (panels 10 to 12), pCMV-Sp110b with pCMV-core (panels 13 to 15), pCMV-core(6162 M) (panel 16), pCMV-Sp110b with pCMV-core(6162 M) (panels 17 to 19), pCMV-Sp110b(1-276 + 454-539) (panel 20), pCMV-Sp110b(1-276 + 454-539) with pCMV-core (panels 21 to 23), or pCMV-Sp110b and pCMV-core with pcDNA-Sp110b(389-453) (myc-CBR fragment) (panels 24 to 29) and on HeLa cells transfected with pKS+/CMV (panel 30) or pCMV-core (panels 31 to 33) following treatment with 300 U of gamma interferon/ml to allow the detection of endogenous Sp110b. The primary antibodies used were anti-FLAG (panels 2, 3, 6, 9, 10, 13, 17, 20, 21, and 27) (green), anti-Myc (panel 24) (green), anti-Sp110(b) (panels 30 and 31) (green), anti-core (panels 1, 7, 14, 16, 18, 22, 25, 28, and 32) (red), and anti-RARα (panels 4 and 11) (red). Merged images of green and red signals are shown in panels 5, 8, 12, 15, 19, 23, 26, 29, and 33. DAPI was used to visualize nuclear staining (right panels). (B) Subcellular fractionation was performed on cells transfected with 1 μg of pCMV-Sp110b (lanes 1 to 3), 1 μg of pCMV-core (lanes 4 to 6), and 1 μg each of pCMV-Sp110b and pCMV-core (lanes 7 to 9). The transfectants were homogenized and separated into nuclear (N), microsomal-membrane (MM), and cytosolic (C) fractions by centrifugation as described in Materials and Methods. The FLAG-tagged Sp110b, core, α-tubulin, and SC-35 proteins in those fractions were detected by immunoblot analysis.
These results suggest that Sp110b is a transcriptional corepressor downregulating RARα activity in the nucleus.
The ability of HCV core to activate RARα was reduced by RNAi elimination of endogenous Sp110b.
The accumulated evidence suggests that Sp110b, a potent transcriptional corepressor of RARα, may play a role in the activation of RARα-mediated transcription by the core. We therefore examined the capacity of the core to activate RARα upon elimination of endogenous Sp110b protein in HeLa cells by the RNAi technique. When endogenous Sp110b protein was knocked down by siRNA treatment (Fig. 6A), the effect of core expression on RARα-mediated transcription (about 1.30-fold activation) was significantly reduced from levels observed in cells treated with a control siRNA (about 2.69-fold activation) (Fig. 6B). This result suggests that endogenous Sp110b plays an important role in the activation of RARα-mediated transcription by the core protein.
HCV core changes the subcellular localization of Sp110b from the nucleus to the cytoplasmic surface of the ER.
Our finding that Sp110b interacts with the core and plays an important role in RARα activation by the core presented a paradoxical problem in that Sp110b was likely to be a nuclear factor but the core was located mainly around the perinuclear region in the cytoplasm (Fig. 7A, panel 1). To investigate this question, we examined the subcellular localization of each protein by indirect immunofluorescence analysis. Upon coproduction of the core protein, the localization of Sp110b changed dramatically from the nucleus to the perinuclear region of the cytoplasm, where the core was located (Fig. 7A, panels 9 and 13 to 15). In contrast, Sp110 was observed in the nucleus, irrespective of core production (Fig. 7A, panels 2 and 6 to 8). Similar results were obtained in 293T cells (data not shown). The subcellular localization of other well-known nuclear proteins—p53 (12), poly(ADP-ribose) polymerase (45), and RARα (7)—was not influenced by core expression, in a manner similar to that observed for Sp110 (data not shown). These results indicate that the core specifically alters the subcellular localization of Sp110b. We further confirmed this phenomenon in HeLa cells by observation of the alteration of the location of endogenous Sp110b by the core. In the absence of core expression, a nuclear staining pattern was seen (Fig. 7A, panel 30) as described above. However, perinuclear localization was observed in certain cells producing the core, similar to the pattern seen for exogenous Sp110b (Fig. 7A, panels 31 to 33). Since Sp110b but not Sp110 was detected in HeLa cells by using this antibody (as shown in Fig. 6A), the endogenous protein detected around the perinuclear region in the presence of the core appears to be primarily Sp110b. Thus, core expression altered the subcellular localization of Sp110b.
To investigate the colocalization of Sp110b with the core, we fractionated cell extracts into nuclear, microsomal-membrane, and cytosolic fractions for detection of Sp110b (Fig. 7B). Fraction identities were confirmed by detection of SC-35 (49), the core (27, 54), and α-tubulin for the nuclear, microsomal-membrane, and cytosolic fractions, respectively (Fig. 7B, lower panels). Following coproduction of the core, Sp110b shifted into the microsomal-membrane fraction (Fig. 7B, top panel). Core expression thus altered the subcellular localization of Sp110b from the nucleus to the cytoplasmic surface of the ER, where the core was originally located.
To investigate the molecular mechanisms governing the change in Sp110b subcellular localization by the core, we utilized a core point mutant, core(6162M). This protein, containing substitutions of arginines at positions 61 and 62 for glycine and leucine, respectively, has little affinity for Sp110b (Fig. 3A, center panel). Production of the core(6162M) mutant had no effect on the subcellular localization of Sp110b (Fig. 7A, panels 16 to 19). In addition, Sp110b(1-276 + 454-539), which lacks the CBR and does not associate with the core in vitro (Fig. 3Bj), remained in the nucleus irrespective of core production (Fig. 7A, panels 20 to 23). We also performed an interaction-competition analysis using a peptide fragment containing the CBR of Sp110b (myc-CBR fragment), a fragment capable of colocalizing with the core in the cytoplasm (Fig. 7A, panels 24 to 26). Excess myc-CBR fragment inhibited the colocalization of Sp110b with the core (Fig. 7A, panels 27 to 29), suggesting that molecular interaction of Sp110b with the core is essential for the alteration of Sp110b subcellular localization by core production.
Transcriptional activation of RARα by HCV core was exerted via sequestration of the transcriptional corepressor Sp110b away from the nucleus.
The above results raised the possibility that Sp110b, a potent nuclear transcriptional corepressor of RARα, was functionally modulated by the core through sequestration from the nucleus. To investigate this possibility, we examined by a reporter assay whether the core modulates the suppressive effect of Sp110b on RARα activity (Fig. 8A). Sp110b-induced transcriptional suppression was overcome by production of the core in a dose-dependent manner (Fig. 8A). As the expression levels of both ectopically produced Sp110b and RARα were not affected by core production (data not shown), the core likely inactivates the function of Sp110b suppressing RARα-mediated transcription.
FIG. 8.

Sequestration of Sp110b from the nucleus plays a significant role in the activation of RARα-mediated transcription by core expression. (A) A reporter assay was performed using COS-7 cells transfected with a total of 400 ng of plasmids including 25 ng of pRARE-Luc, the effector plasmids pCMV-Sp110b and pCMV-core, and empty vector. Other conditions were the same as those described in the legend to Fig. 2A. The amounts of the effector plasmids (in nanograms) in each experiment are given below the graph. (B) A reporter assay was performed using COS-7 cells transfected with a total of 400 ng of plasmids including 25 ng of pRARE-Luc, the effector plasmids pCMV-core, pCMV-Sp110b(389-453) (CBR fragment), and pCMV-core(6162M), and empty vector. Data are means of the relative luciferase activities from three independent experiments.
To determine if the sequestration of Sp110b from the nucleus is essential for the activation of RARα-mediated transcription by the core, we performed an interaction-competition analysis. The CBR fragment, which itself did not affect the transcriptional activity of RARα in the absence of the core (Fig. 8B, bars 7 and 8), served as a competitor for Sp110b sequestration by the core (Fig. 7A, panels 24 to 29). Coproduction of this CBR fragment with the core reversed the core-induced transcriptional activation in a dose-dependent manner (Fig. 8B, bars 3, 4, and 9 to 14). Moreover, we observed that the core mutant core(6162M), which could not alter the localization of Sp110b, also lacked the ability to augment transcriptional activity (Fig. 8B, bars 5 and 6). From the above results, we concluded that the activation of RARα-mediated transcription by the core results from the sequestration from the nucleus and subsequent inactivation of the transcriptional corepressor Sp110b.
Sequestration of Sp110b from the nucleus by HCV core caused sensitization to ATRA-mediated cell death.
We analyzed the relationship of Sp110b sequestration by the core to sensitization to ATRA-induced cell death and enhancement of tTGase gene induction (Fig. 1) by an interaction-competition analysis using the CBR fragment. tTGase mRNA levels, increased by core expression in the presence of ATRA, decreased following coproduction of the CBR fragment (Fig. 1C, upper panel, lanes 4 to 6). When the core and the CBR fragment were simultaneously produced in transiently transfected cells, the observed levels of ATRA-induced cell death were similar to those found in cells with no ectopic protein production (Fig. 1B, bars 7 and 8). This result suggested that the CBR fragment reverses the promotion of ATRA-induced cell death mediated by the core. Moreover, the core(6162M) mutant did not enhance cell death induced by ATRA (Fig. 1B, bars 9 and 10).
The data cumulatively suggest that the interaction of HCV core with Sp110b and the subsequent sequestration of Sp110b from the nucleus play a significant role in the sensitization to ATRA-mediated cell death induced by HCV core protein.
DISCUSSION
Sp110b as a transcriptional corepressor of RARα.
In this study, we have identified Sp110b as a cellular factor interacting with HCV core. Although the function of Sp110b has remained unknown, we found in this study that Sp110b had the potential to suppress RARα-mediated transcription and that it associated with RARα and the target promoter containing RARE in the presence of ATRA. Moreover, Sp110b mRNA was expressed both ubiquitously and abundantly in human tissues at levels much higher than those of Sp110, a splicing variant of Sp110b. These results suggest that Sp110b is a transcriptional corepressor of RARα, contrasting with the transcriptional coactivating function of Sp110.
The only structural difference between Sp110b and Sp110 is the absence in Sp110b of a C-terminal extension of Sp110, which contains the PHD and the bromodomain (Fig. 3B). Although the function of the PHD is still unknown, the bromodomain is known as a structure for interaction with an acetylated lysine residue (10, 57). This domain has been identified in many proteins involved in transcriptional regulation, likely associating with histone acetyltransferase activity, which is involved in the activation of gene expression (9). From these facts, it seemed reasonable that the bromodomain of Sp110 may be essential for its transcriptional coactivating activity and that Sp110b, lacking this region, would have no potential for transcriptional activation. The suppressive effect of Sp110b on transcription may result from competitive exclusion of other coactivators, such as SRC-1, TIF2/GRIP1, RAC3/ACTR/pCIP/AIB-1, and CBP/p300, via the LXXLL motif from the RARα-containing transcription complex. Further analysis will be necessary to clarify the molecular mechanisms of transcriptional regulation in which Sp110b participates.
Role of Sp110b in the activation of RARα-mediated transcription by HCV core.
The present study suggested that the core activates RARα-mediated transcription by sequestering Sp110b from the nucleus, where Sp110b could exert its suppressive function on RARα-mediated transcription. Sp110b is instead relocalized to the cytoplasm and concentrated at ER membranes (Fig. 9). This phenomenon appears to be independent of cell type, as Sp110b is expressed ubiquitously and can suppress RARα activity in all cell lines examined. In fact, despite the restriction of HCV infection to hepatocytes, the core activated RARα-mediated transcription in all cell lines tested. We also confirmed the physiological relevance of RARα activation by the core in hepatocytes through observations that the core increased mRNA levels of TGF-β2, a gene downstream of RAR, in hepatoma Huh-7 cells (data not shown).
FIG. 9.
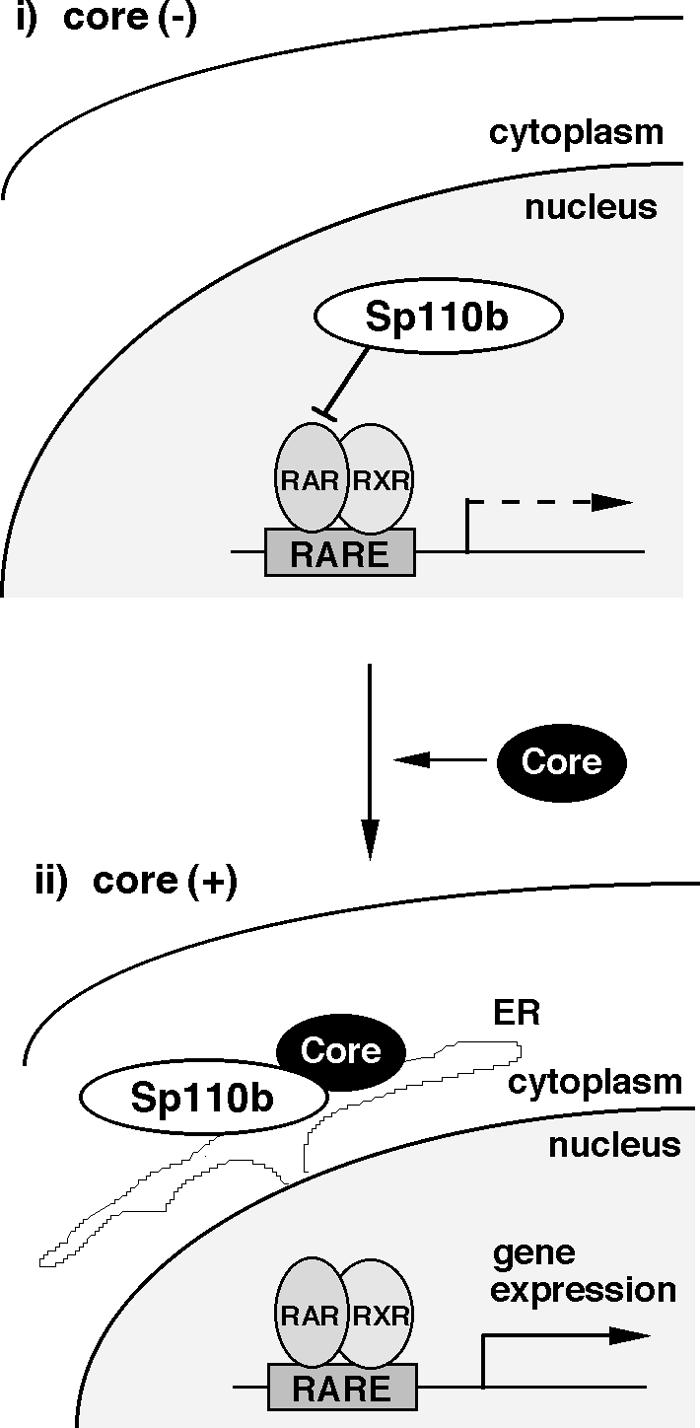
Schematic representation of the mechanistic model of activation of RARα-mediated transcription by the core. (i) In the absence of the core, Sp110b is located in the nucleus, playing a suppressive role in RARα-mediated transcription. (ii) In the presence of the core, Sp110b is sequestered to ER membranes through interaction with the core, which is located on the cytoplasmic surfaces of ER membranes. This sequestration results in a reduction of the transcriptional suppressive effect of Sp110b in the nucleus. Consequently, ATRA-induced transcription and expression of RARα-responsive genes, such as tTGase, are enhanced.
Although the mechanism of Sp110b relocalization by the core remains unknown, some speculations can be put forward. Nascent Sp110b protein, newly synthesized in ribosomes around the ER, is trapped by the core, which resides around the ER, preventing its translocation into the nucleus. Sp110b may also shuttle between the nucleus and the cytoplasm; when Sp110b is in the cytoplasm, Sp110b may be trapped by the core through molecular interaction.
There have been numerous reports that the core modulates the activities of a variety of transcriptional promoters for various genes, including a wide range of transcription factors such as NF-κB (28, 48, 56), AP-1 (48), Elk (14, 16), STAT (55), NF-AT (5), RXR (52), and p53 (25, 42). In some reports, it was speculated that the core, which is mainly located in the cytoplasm, modulates the transcriptional activities of nuclear factors via direct interaction. Previously, however, there been no evidence demonstrating the precise location of this interaction in the cells. In addition, the relationship between the interaction and the transcriptional modulation has remained obscure. To our knowledge, this study is the first to demonstrate the molecular mechanism of transcription factor activity modulation by the core; sequestration of a transcriptional cofactor is essential. Recently, the core was reported to interact with RXRα and activate its transcriptional activity, although the mechanism of this activation was unclear (52). While the authors speculated that the core functions as an RXRα cofactor, we demonstrate that the core interacts with a transcriptional cofactor, Sp110b, in the cytoplasm to inhibit its corepressor function. Although experiments analyzing the interaction between Sp110b and RXRα are under way, a similar mechanism of sequestration of Sp110b may be involved in RXRα activation by the core.
Transcriptional cofactors, such as CBP/p300, SRC-1, TIF2/GRIP1, and RAC3/ACTR/pCIP/AIB-1, participate in the regulation of multiple transcription factors (22). These transcriptional cofactors play an important role in the cross talk between different signal transduction pathways; activation of NF-κB represses the transcriptional activation of glucocorticoid receptors, and vice versa, by competition for interaction with CBP and SRC-1, which are required for their activities and are limiting in cells (47). Similar cross talk mechanisms have been reported between AP-1 and androgen receptor (13), NF-κB and p53 (39), c-Myb and GATA-1 (50), and estrogen receptor alpha and NF-κB (15). Constitutive androstane receptor inhibited the activity of estrogen receptor through sequestration of a transcriptional coactivator, GRIP1 (30). Although it remains unknown if Sp110b is involved in the regulation of additional transcription pathways, some of the effects of the core on a variety of transcriptional events may result from the depletion of Sp110b from the nucleus by the core. Further biochemical analysis of Sp110b may shed more light on this point.
Acknowledgments
We are grateful to P. Chambon, S. Kato, T. Kiyono, and J. Miyazaki for providing plasmids, M. Kohara for the anti-core antibody, and M. Hosaka for Huh-7 cells carrying an HCV full-genome replicon.
This work was supported by grants-in-aid for cancer research and for the second-term comprehensive 10-year strategy for cancer control from the Ministry of Health, Labor, and Welfare of Japan; by grants-in-aid for scientific research from the Ministry of Education, Culture, Sports, Science and Technology of Japan; by grants-in-aid for research for the future from the Japanese Society for the Promotion of Science; and by the Program for Promotion of Fundamental Studies in Health Science of the Organization for Pharmaceutical Safety and Research (OPSR) of Japan.
REFERENCES
- 1.Alter, H. J., R. H. Purcell, J. W. Shih, J. C. Melpolder, M. Houghton, Q. L. Choo, and G. Kuo. 1989. Detection of antibody to hepatitis C virus in prospectively followed transfusion recipients with acute and chronic non-A, non-B hepatitis. N. Engl. J. Med. 321:1494-1500. [DOI] [PubMed] [Google Scholar]
- 2.Arany, Z., D. Newsome, E. Oldread, D. M. Livingston, and R. Eckner. 1995. A family of transcriptional adaptor proteins targeted by the E1A oncoprotein. Nature 374:81-84. [DOI] [PubMed] [Google Scholar]
- 3.Ariumi, Y., A. Kaida, M. Hatanaka, and K. Shimotohno. 2001. Functional cross-talk of HIV-1 Tat with p53 through its C-terminal domain. Biochem. Biophys. Res. Commun. 287:556-561. [DOI] [PubMed] [Google Scholar]
- 4.Ariumi, Y., A. Kaida, J. Y. Lin, M. Hirota, O. Masui, S. Yamaoka, Y. Taya, and K. Shimotohno. 2000. HTLV-1 tax oncoprotein represses the p53-mediated trans-activation function through coactivator CBP sequestration. Oncogene 19:1491-1499. [DOI] [PubMed] [Google Scholar]
- 5.Bergqvist, A., and C. M. Rice. 2001. Transcriptional activation of the interleukin-2 promoter by hepatitis C virus core protein. J. Virol. 75:772-781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bloch, D. B., A. Nakajima, T. Gulick, J. D. Chiche, D. Orth, S. M. de La Monte, and K. D. Bloch. 2000. Sp110 localizes to the PML-Sp100 nuclear body and may function as a nuclear hormone receptor transcriptional coactivator. Mol. Cell. Biol. 20:6138-6146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chambon, P. 1996. A decade of molecular biology of retinoic acid receptors. FASEB J. 10:940-954. [PubMed] [Google Scholar]
- 8.Choo, Q. L., G. Kuo, A. J. Weiner, L. R. Overby, D. W. Bradley, and M. Houghton. 1989. Isolation of a cDNA clone derived from a blood-borne non-A, non-B viral hepatitis genome. Science 244:359-362. [DOI] [PubMed] [Google Scholar]
- 9.Denis, G. V. 2001. Duality in bromodomain-containing protein complexes. Front. Biosci. 6:D849-D852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dyson, M. H., S. Rose, and L. C. Mahadevan. 2001. Acetyllysine-binding and function of bromodomain-containing proteins in chromatin. Front. Biosci. 6:D853-D865. [DOI] [PubMed] [Google Scholar]
- 11.Eck-Enriquez, K., T. L. Kiefer, L. L. Spriggs, and S. M. Hill. 2000. Pathways through which a regimen of melatonin and retinoic acid induces apoptosis in MCF-7 human breast cancer cells. Breast Cancer Res. Treat. 61:229-239. [DOI] [PubMed] [Google Scholar]
- 12.Ferrigno, P., and P. A. Silver. 1999. Regulated nuclear localization of stress-responsive factors: how the nuclear trafficking of protein kinases and transcription factors contributes to cell survival. Oncogene 18:6129-6134. [DOI] [PubMed] [Google Scholar]
- 13.Fronsdal, K., N. Engedal, T. Slagsvold, and F. Saatcioglu. 1998. CREB binding protein is a coactivator for the androgen receptor and mediates cross-talk with AP-1. J. Biol. Chem. 273:31853-31859. [DOI] [PubMed] [Google Scholar]
- 14.Fukuda, K., K. Tsuchihara, M. Hijikata, S. Nishiguchi, T. Kuroki, and K. Shimotohno. 2001. Hepatitis C virus core protein enhances the activation of the transcription factor, Elk1, in response to mitogenic stimuli. Hepatology 33:159-165. [DOI] [PubMed] [Google Scholar]
- 15.Harnish, D. C., M. S. Scicchitano, S. J. Adelman, C. R. Lyttle, and S. K. Karathanasis. 2000. The role of CBP in estrogen receptor cross-talk with nuclear factor-κB in HepG2 cells. Endocrinology 141:3403-3411. [DOI] [PubMed] [Google Scholar]
- 16.Hayashi, J., H. Aoki, K. Kajino, M. Moriyama, Y. Arakawa, and O. Hino. 2000. Hepatitis C virus core protein activates the MAPK/ERK cascade synergistically with tumor promoter TPA, but not with epidermal growth factor or transforming growth factor alpha. Hepatology 32:958-961. [DOI] [PubMed] [Google Scholar]
- 17.Heery, D. M., E. Kalkhoven, S. Hoare, and M. G. Parker. 1997. A signature motif in transcriptional co-activators mediates binding to nuclear receptors. Nature 387:733-736. [DOI] [PubMed] [Google Scholar]
- 18.Hijikata, M., N. Kato, Y. Ootsuyama, M. Nakagawa, and K. Shimotohno. 1991. Gene mapping of the putative structural region of the hepatitis C virus genome by in vitro processing analysis. Proc. Natl. Acad. Sci. USA 88:5547-5551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Joseph, B., O. Lefebvre, C. Mereau-Richard, P. M. Danze, M. T. Belin-Plancot, and P. Formstecher. 1998. Evidence for the involvement of both retinoic acid receptor- and retinoic X receptor-dependent signaling pathways in the induction of tissue transglutaminase and apoptosis in the human myeloma cell line RPMI 8226. Blood 91:2423-2432. [PubMed] [Google Scholar]
- 20.Kato, N., M. Hijikata, Y. Ootsuyama, M. Nakagawa, S. Ohkoshi, T. Sugimura, and K. Shimotohno. 1990. Molecular cloning of the human hepatitis C virus genome from Japanese patients with non-A, non-B hepatitis. Proc. Natl. Acad. Sci. USA 87:9524-9528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kiyono, T., A. Hiraiwa, S. Ishii, T. Takahashi, and M. Ishibashi. 1994. Inhibition of p53-mediated transactivation by E6 of type 1, but not type 5, 8, or 47, human papillomavirus of cutaneous origin. J. Virol. 68:4656-4661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Leo, C., and J. D. Chen. 2000. The SRC family of nuclear receptor coactivators. Gene 245:1-11. [DOI] [PubMed] [Google Scholar]
- 23.Lerat, H., M. Honda, M. R. Beard, K. Loesch, J. Sun, Y. Yang, M. Okuda, R. Gosert, S. Y. Xiao, S. A. Weinman, and S. M. Lemon. 2002. Steatosis and liver cancer in transgenic mice expressing the structural and nonstructural proteins of hepatitis C virus. Gastroenterology 122:352-365. [DOI] [PubMed] [Google Scholar]
- 24.Liu, Y., A. L. Colosimo, X. J. Yang, and D. Liao. 2000. Adenovirus E1B 55-kilodalton oncoprotein inhibits p53 acetylation by PCAF. Mol. Cell. Biol. 20:5540-5553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lu, W., S. Y. Lo, M. Chen, K. Wu, Y. K. Fung, and J. H. Ou. 1999. Activation of p53 tumor suppressor by hepatitis C virus core protein. Virology 264:134-141. [DOI] [PubMed] [Google Scholar]
- 26.Lundblad, J. R., R. P. Kwok, M. E. Laurance, M. L. Harter, and R. H. Goodman. 1995. Adenoviral E1A-associated protein p300 as a functional homologue of the transcriptional co-activator CBP. Nature 374:85-88. [DOI] [PubMed] [Google Scholar]
- 27.Martire, G., A. Viola, L. Iodice, L. V. Lotti, R. Gradini, and S. Bonatti. 2001. Hepatitis C virus structural proteins reside in the endoplasmic reticulum as well as in the intermediate compartment/cis-Golgi complex region of stably transfected cells. Virology 280:176-182. [DOI] [PubMed] [Google Scholar]
- 28.Marusawa, H., M. Hijikata, T. Chiba, and K. Shimotohno. 1999. Hepatitis C virus core protein inhibits Fas- and tumor necrosis factor alpha-mediated apoptosis via NF-κB activation. J. Virol. 73:4713-4720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mesnard, J. M., and C. Devaux. 1999. Multiple control levels of cell proliferation by human T-cell leukemia virus type 1 Tax protein. Virology 257:277-284. [DOI] [PubMed] [Google Scholar]
- 30.Min, G., H. Kim, Y. Bae, L. Petz, and J. K. Kemper. 2002. Inhibitory cross-talk between estrogen receptor (ER) and constitutively activated androstane receptor (CAR). CAR inhibits ER-mediated signaling pathway by squelching p160 coactivators. J. Biol. Chem. 277:34626-34633. [DOI] [PubMed] [Google Scholar]
- 31.Moriya, K., H. Fujie, Y. Shintani, H. Yotsuyanagi, T. Tsutsumi, K. Ishibashi, Y. Matsuura, S. Kimura, T. Miyamura, and K. Koike. 1998. The core protein of hepatitis C virus induces hepatocellular carcinoma in transgenic mice. Nat. Med. 4:1065-1067. [DOI] [PubMed] [Google Scholar]
- 32.Moriya, K., H. Yotsuyanagi, Y. Shintani, H. Fujie, K. Ishibashi, Y. Matsuura, T. Miyamura, and K. Koike. 1997. Hepatitis C virus core protein induces hepatic steatosis in transgenic mice. J. Gen. Virol. 78:1527-1531. [DOI] [PubMed] [Google Scholar]
- 33.Munger, K. 2002. The role of human papillomaviruses in human cancers. Front. Biosci. 7:d641-d649. [DOI] [PubMed] [Google Scholar]
- 34.Niwa, H., K. Yamamura, and J. Miyazaki. 1991. Efficient selection for high-expression transfectants with a novel eukaryotic vector. Gene 108:193-199. [DOI] [PubMed] [Google Scholar]
- 35.Oliverio, S., A. Amendola, C. Rodolfo, A. Spinedi, and M. Piacentini. 1999. Inhibition of “tissue” transglutaminase increases cell survival by preventing apoptosis. J. Biol. Chem. 274:34123-34128. [DOI] [PubMed] [Google Scholar]
- 36.Ou, H., J. Haendeler, M. R. Aebly, L. A. Kelly, B. C. Cholewa, G. Koike, A. Kwitek-Black, H. J. Jacob, B. C. Berk, and J. M. Miano. 2000. Retinoic acid-induced tissue transglutaminase and apoptosis in vascular smooth muscle cells. Circ. Res. 87:881-887. [DOI] [PubMed] [Google Scholar]
- 37.Park, J. S., E. J. Kim, H. J. Kwon, E. S. Hwang, S. E. Namkoong, and S. J. Um. 2000. Inactivation of interferon regulatory factor-1 tumor suppressor protein by HPV E7 oncoprotein. Implication for the E7-mediated immune evasion mechanism in cervical carcinogenesis. J. Biol. Chem. 275:6764-6769. [DOI] [PubMed] [Google Scholar]
- 38.Patel, D., S. M. Huang, L. A. Baglia, and D. J. McCance. 1999. The E6 protein of human papillomavirus type 16 binds to and inhibits co-activation by CBP and p300. EMBO J. 18:5061-5072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ravi, R., B. Mookerjee, Y. van Hensbergen, G. C. Bedi, A. Giordano, W. S. El-Deiry, E. J. Fuchs, and A. Bedi. 1998. p53-mediated repression of nuclear factor-κB RelA via the transcriptional integrator p300. Cancer Res. 58:4531-4536. [PubMed] [Google Scholar]
- 40.Ray, R. B., L. M. Lagging, K. Meyer, and R. Ray. 1996. Hepatitis C virus core protein cooperates with ras and transforms primary rat embryo fibroblasts to tumorigenic phenotype. J. Virol. 70:4438-4443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ray, R. B., K. Meyer, and R. Ray. 1996. Suppression of apoptotic cell death by hepatitis C virus core protein. Virology 226:176-182. [DOI] [PubMed] [Google Scholar]
- 42.Ray, R. B., R. Steele, K. Meyer, and R. Ray. 1997. Transcriptional repression of p53 promoter by hepatitis C virus core protein. J. Biol. Chem. 272:10983-10986. [DOI] [PubMed] [Google Scholar]
- 43.Ruggieri, A., T. Harada, Y. Matsuura, and T. Miyamura. 1997. Sensitization to Fas-mediated apoptosis by hepatitis C virus core protein. Virology 229:68-76. [DOI] [PubMed] [Google Scholar]
- 44.Sang, N., J. Caro, and A. Giordano. 2002. Adenoviral E1A: everlasting tool, versatile applications, continuous contributions and new hypotheses. Front. Biosci. 7:d407-d413. [DOI] [PubMed] [Google Scholar]
- 45.Schreiber, V., M. Molinete, H. Boeuf, G. de Murcia, and J. Menissier-de Murcia. 1992. The human poly(ADP-ribose) polymerase nuclear localization signal is a bipartite element functionally separate from DNA binding and catalytic activity. EMBO J. 11:3263-3269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Scoggin, K. E., A. Ulloa, and J. K. Nyborg. 2001. The oncoprotein Tax binds the SRC-1-interacting domain of CBP/p300 to mediate transcriptional activation. Mol. Cell. Biol. 21:5520-5530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sheppard, K. A., K. M. Phelps, A. J. Williams, D. Thanos, C. K. Glass, M. G. Rosenfeld, M. E. Gerritsen, and T. Collins. 1998. Nuclear integration of glucocorticoid receptor and nuclear factor-κB signaling by CREB-binding protein and steroid receptor coactivator-1. J. Biol. Chem. 273:29291-29294. [DOI] [PubMed] [Google Scholar]
- 48.Shrivastava, A., S. K. Manna, R. Ray, and B. B. Aggarwal. 1998. Ectopic expression of hepatitis C virus core protein differentially regulates nuclear transcription factors. J. Virol. 72:9722-9728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Stein, G. S., A. J. van Wijnen, J. L. Stein, J. B. Lian, M. Montecino, J. Choi, K. Zaidi, and A. Javed. 2000. Intranuclear trafficking of transcription factors: implications for biological control. J. Cell Sci. 113:2527-2533. [DOI] [PubMed] [Google Scholar]
- 50.Takahashi, T., N. Suwabe, P. Dai, M. Yamamoto, S. Ishii, and T. Nakano. 2000. Inhibitory interaction of c-Myb and GATA-1 via transcriptional co-activator CBP. Oncogene 19:134-140. [DOI] [PubMed] [Google Scholar]
- 51.Tsuchihara, K., M. Hijikata, K. Fukuda, T. Kuroki, N. Yamamoto, and K. Shimotohno. 1999. Hepatitis C virus core protein regulates cell growth and signal transduction pathway transmitting growth stimuli. Virology 258:100-107. [DOI] [PubMed] [Google Scholar]
- 52.Tsutsumi, T., T. Suzuki, T. Shimoike, R. Suzuki, K. Moriya, Y. Shintani, H. Fujie, Y. Matsuura, K. Koike, and T. Miyamura. 2002. Interaction of hepatitis C virus core protein with retinoid X receptor alpha modulates its transcriptional activity. Hepatology 35:937-946. [DOI] [PubMed] [Google Scholar]
- 53.Van Orden, K., J. P. Yan, A. Ulloa, and J. K. Nyborg. 1999. Binding of the human T-cell leukemia virus Tax protein to the coactivator CBP interferes with CBP-mediated transcriptional control. Oncogene 18:3766-3772. [DOI] [PubMed] [Google Scholar]
- 54.Watashi, K., M. Hijikata, H. Marusawa, T. Doi, and K. Shimotohno. 2001. Cytoplasmic localization is important for transcription factor nuclear factor-κB activation by hepatitis C virus core protein through its amino terminal region. Virology 286:391-402. [DOI] [PubMed] [Google Scholar]
- 55.Yoshida, T., T. Hanada, T. Tokuhisa, K. Kosai, M. Sata, M. Kohara, and A. Yoshimura. 2002. Activation of STAT3 by the hepatitis C virus core protein leads to cellular transformation. J. Exp. Med. 196:641-653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.You, L. R., C. M. Chen, and Y. H. Lee. 1999. Hepatitis C virus core protein enhances NF-κB signal pathway triggering by lymphotoxin-beta receptor ligand and tumor necrosis factor alpha. J. Virol. 73:1672-1681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zeng, L., and M. M. Zhou. 2002. Bromodomain: an acetyl-lysine binding domain. FEBS Lett. 513:124-128. [DOI] [PubMed] [Google Scholar]