

Abstract
In this mini-review we outline a model depicting the immunologic mechanisms by which psychological stress can exacerbate clinical symptoms in patients with asthma. This model highlights the importance of both social and physical exposures in the exacerbation of asthma symptoms. The basic premise of the model is that psychological stress operates by altering the magnitude of the airway inflammatory response that irritants, allergens, and infections bring about in persons with asthma. The biological pathways for how stress amplifies the immune response to asthma triggers include the hypothalamic-pituitary-adrenal (HPA) axis, the sympathetic-adrenal-medullary (SAM) axis, and the sympathetic (SNS) and parasympathetic (PNS) arms of the autonomic nervous system. Empirical evidence for this model is reviewed, and conclusions and future research directions are discussed.
Keywords: stress, asthma, cytokines, glucocorticoids, autonomic nervous system
1. Introduction
Physicians, scientists, and laypeople have long believed that stress contributes to exacerbations of asthma. However, it has only been in the past two decades that convincing scientific evidence has accumulated to substantiate this hypothesis. For example, in an 18-month prospective study of children with asthma, the experience of an acute negative life event (e.g., death of a close family member) increased the risk of a subsequent asthma attack by nearly 2-fold (Sandberg et al., 2000). The impact of an acute negative event was accentuated when it occurred in the context of chronic stress. Children exposed to high levels of acute and chronic stress showed a 3-fold increase in risk for an attack in the two weeks that followed the acute event. Despite the recent empirical evidence linking stress with the clinical profile of asthma, much remains to be learned about the biological mechanisms underlying this phenomenon. In this mini-review we outline a model highlighting inflammation as a central mediating pathway. It focuses on immunologic mechanisms that allow stress to “get inside the body” and exacerbate symptoms of patients with asthma. Because the role of stress in asthma onset has been thoughtfully discussed elsewhere (Wright, 2005; Wright, Cohen, & Cohen, 2005), and probably involves different behavioral and biological mediators, we do not attempt to cover it here.
2. What is stress?
Stress has been defined in many ways in the scientific literature. One of the most common psychological definitions has been that stress occurs when demands from the environment challenge an individual's adaptive capacity, or ability to cope (Cohen, Kessler, & Gordon, 1995). Frequently termed “stressors,” these demands include negative life events such as job loss, death of a loved one, and family conflict. Within this conceptualization of stress, researchers have typically distinguished between acute and chronic stressors. Acute stressors are time-limited in duration, typically with a clear onset and offset. In contrast, chronic stressors are ongoing, stable life difficulties that elicit prolonged responses and often have no clear endpoint in sight. Both have been linked to changes in various immune parameters in humans, although the directionality can vary depending on the type of stressor, and whether the person is healthy or suffers from illnesses like asthma (Segerstrom & Miller, 2004).
Because the same environmental demand can produce considerable variability in people's emotional, behavioral, or physiological responses, some theorists have argued that it is essential to consider an individual's subjective perception of the stressor. This view holds that the critical feature of stress is not the demand itself, but instead how an individual comes to interpret, or appraise, the situation. The important outcomes of this appraisal process are whether a person views the situation as threatening, and believes that s/he has the resources necessary to cope with it effectively. To the extent that the stressor is appraised as threatening and unmanageable, it elicits negative emotional responses, which in turn give rise to the behavioral and biological sequelae of stress (Lazarus & Folkman, 1984). Unfortunately, very little research in PNI has adopted this sort of conceptual framework (6). Because of its importance to understanding linkages between stress and asthma, we give special attention in the review to studies that feature appraisal-based conceptualizations.
3. How does stress affect asthma?
Figure 1 depicts our working model of stress and asthma. It highlights the importance of both social and physical exposures in the exacerbation of symptoms. The basic premise of the model is that psychological stress operates by altering the magnitude of the airway inflammatory response that irritants, allergens, and infections bring about in persons with asthma. It is important to note that the model suggests that stress on its own is NOT capable of modifying immune functions in a way that leads to asthmatic symptoms. Rather, stress is viewed as a process that accentuates the airway inflammatory response to environmental triggers and, in doing so, increases the frequency, duration, and severity of patients' symptoms.
Figure 1.
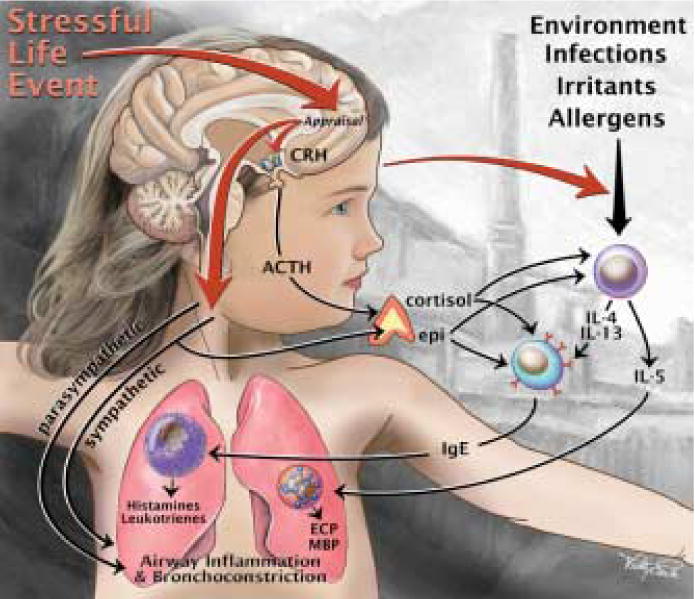
Model depicting the interaction of psychological stress with environmental triggers in influencing asthma exacerbations. The basic premise of the model is that stress operates by altering the magnitude of the airway inflammatory response that irritants, allergens, and infections bring about in persons with asthma. The figure provides an overview of the relevant biological pathways to airway inflammation and bronchoconstriction, including the hypothalamic-pituitary-adrenal (HPA) axis, the sympathetic-adrenal-medullary (SAM) axis, and the sympathetic (SNS) and parasympathetic (PNS) arms of the autonomic nervous system.
A variety of indoor and outdoor triggers have been found to contribute to exacerbations of asthma. For example, exposure to tobacco smoke can lead to wheezing and asthmatic symptoms in vulnerable individuals (Jaakkola, Nafstad, & Magnus, 2001), and so can infections in the upper and lower respiratory tracts (Sigurs et al., 2005). In sensitized individuals, exposure to allergens, such as cat dander and mould, can bring about bronchial hyperresponsiveness (Nelson et al., 1999). One of the common pathways these triggers operate through is airway inflammation. Upon entry into the body, for example, inhaled allergens are taken up by dendritic cells, and subsequently presented to T helper (Th) cells (Busse & Lemanske, 2001). Th cells have two phenotypes known as Th-1 and Th-2. Th-1 cells generally initiate and coordinate cellular immune responses by deploying cytokines such as IL-2 and IFN-g. By contrast, Th-2 cells promote B cell proliferation and differentiation, which leads to a humoral response involving antibody synthesis. They do so by releasing cytokines such as IL-4, IL-5, & IL-13. It is these humoral pathways that are generally implicated in flare-ups of asthma.
The Th-2 cytokines IL-4 and IL-13 operate by binding to B cells, and inducing them to synthesize and release IgE antibodies. IgE then binds to mast cells residing in the airways. When IgE molecules bind their cognate allergen, it causes the mast cell to degranulate, leading to the release of allergic mediators such as histamines and leukotrienes. Histamines and leukotrienes cause edema, smooth muscle constriction, and mucus, resulting in clinical symptoms of asthma including wheezing, chest tightness, and shortness of breath. This pathway constitutes the early response. A more prolonged late-phase response is generated when Th2 cells release IL-5. This cytokine recruits eosinophils into the airways, where they bring about inflammation and obstruction. Eosinophils also release mediators, such as eosinophil cationic protein and major basic protein, which can bring about damage to airway cells, and leukotrienes, which cause edema and further bronchial constriction. Hence, this late-phase response worsens clinical symptoms by promoting airway inflammation and obstruction.
Our model suggests that stress accentuates this immune response to environmental triggers. It views stress as an external demand from an individual's social environment. To have downstream influences on inflammatory processes in the airways, the external stressor must be appraised as threatening and unmanageable. This appraisal pattern brings about increases in negative emotions (e.g., anger, fear, shame) and decreases in positive emotions (e.g., vigor, joy, calmness) as well as changes in thoughts about the self and the future. These emotional and cognitive processes sensitize the Th-2 pathway, such that upon exposure to a trigger, there is a more pronounced inflammatory response, leading to increased frequency, duration, and severity of symptoms.
We note that our model differs from other depictions of stress and asthma in its explicit focus on the interaction between social and physical exposures and in its emphasis on the pathway of inflammation. Other models have also noted that stress has direct effects on other biological systems relevant to asthma (Wright, 2005; Wright, Rodriguez, & Cohen, 1998) – for example, through vagally mediated bronchoconstriction or heightened interoceptive sensitivity to symptoms - and we agree that these are likely to be important contributory mechanisms as well. We also acknowledge that there are numerous other factors that may interact with stress or independently operate to affect asthma morbidity, including genetics and access to health care (Andrulis, 1998; Ober & Hoffjan, 2006). Nonetheless, we view the model's central precept – that stress worsens asthma symptoms by accentuating inflammatory responses to environmental triggers – as a useful heuristic framework for thinking about the big-picture of asthma.
Before reviewing evidence for this model, we first acknowledge a paradox in the stress and asthma literature. It is well-known that upon exposure to many forms of acute stress there is activation of the hypothalamic-pituitary-adrenocortical (HPA) and sympathetic-adrenal-medullary (SAM) axes, leading to increased secretion of the hormones cortisol, epinephrine and norepinephrine (Baum & Greenberg, 1995). High levels of cortisol diminish inflammation in the airways and the periphery; similarly, β-adrenergic agonists such as epinephrine are potent bronchodilators. It is also well-known that certain stressors, especially those which are severe and chronic, can suppress cellular immune functions in humans (Segerstrom et al., 2004). Together, these observations suggest the paradoxical conclusion that stressful experiences should ameliorate rather than exacerbate symptoms of asthma. Of course, the findings from studies of asthma patients are inconsistent with this conclusion; stress has been shown repeatedly to worsen the course of illness. To resolve this paradox, we argue that after long periods of exposure to stress hormones, receptors for these molecules become down-regulated, leading to diminished regulation of inflammatory responses to asthma triggers. This is manifest in exaggerated production of Th2 cytokines and recruitment of eosinophils, both of which are known to accompany life stress in asthma patients (but not in healthy adults, see (Segerstrom et al., 2004). These ideas are discussed in greater detail in the ‘How does stress modify inflammation?’ section below.
4. What's the evidence for the model?
With the basic outline of the model sketched, we next describe a number of human studies that illustrate empirical approaches to assessing one or more aspects of this model.
Patients with Asthma
A handful of research projects have directly evaluated whether stress amplifies the immune response to asthma triggers. One team studied college students with asthma during periods of high stress (final exam period) and low stress (no major exam) (Liu et al., 2002). At each visit patients inhaled increasing dosages of allergens to which they were sensitized (ragweed, cat, or dust mite) until their pulmonary functioning had declined by ≥20%. There was evidence of a greater immune response to challenge under stressful conditions. During the session that occurred around final exams, the allergen challenge elicited greater numbers of eosinophils in both sputum and blood. A parallel finding emerged for in vitro production of IL-5 in sputum treated with phytohemagglutinin. These findings provide direct support for the basic premise of our model – that stress amplifies the immune response to environmental triggers of asthma. It does so in a particularly compelling fashion, by using an in vivo exposure paradigm, and collecting samples from both sputum and peripheral blood.
Other projects have used in vitro exposures to evaluate this hypothesis. These studies typically involve culturing peripheral blood mononuclear cells (PBMCs) with a mitogen cocktail that includes phorbol myristate acetate and ionomycin (PMA/INO). The volume of cytokine production is then used as an indicator of the magnitude of immune response. While this paradigm is limited because the allergen exposure occurs in vitro, it can be done easily and safely in community patients, so that high stress and low stress groups or time periods can be compared. In two studies our group has done of children with asthma, low socioeconomic status (SES) has been associated with greater PMA/INO stimulated production of IL-5 and IL-13, which are critical mediators of humoral immune response that brings about asthma. Further analyses indicated that low SES patients had been exposed to higher levels of chronic stress, and were inclined to appraise their life situations as being more threatening. When statistical analyses were conducted to evaluate relationships among these processes, they showed that low SES were associated with exaggerated Th-2 cytokine responses via chronic stress and threat appraisal (Chen et al., 2006; Chen, Fisher, Jr., Bacharier, & Strunk, 2003). In other words, data were consistent with the notion that low SES children may have exhibited amplified cytokine responses because they had more chronic stress at home, and tended to view their lives as threatening and unmanageable.
In a series of studies from another research group, high school students were studied before an exam (baseline period of low stress) and after exams (high stress period), and their peripheral blood cells were stimulated in vitro with various mitogen cocktails. In one study, students with asthma had greater production of IL-5 post-exam compared to students who were healthy. In contrast, there were no group differences in IL-5 production at baseline. This suggests that under conditions of low stress, individuals with asthma do not differ from healthy individuals in their responsiveness to mitogen, but that periods of stress heighten the responsiveness of Th-2 immune cells to mitogens specifically in individuals with asthma (Kang et al., 1997). A second study from this group documented that exam stress was associated with reduced production of the Th-1 cytokines IFN-g and IL-2, but increased production of the pro-inflammatory cytokine IL-6 (argued by this group to represent Th-2) across both a sample of students with asthma and healthy students (Kang & Fox, 2001).
Finally, in one study of 2-year old children with a family history of asthma or allergy, PBMC's were stimulated in vitro with allergens (dust mite, cockroach) as well as PHA. Because the children were too young to provide accurate reports, their caregivers were asked to what extent they found their lives threatening and unmanageable. Children whose caregivers appraised their lives as high in stress had greater stimulated production of the pro-inflammatory cytokine TNF-α, and reduced production of the Th-1 cytokine IFN-γ (Wright et al., 2004). Importantly, these effects were prospective, with stress temporally preceding immune response.
Collectively, these studies suggest that among patients suffering from asthma, stress can amplify the Th-2 cytokine response to asthma triggers and mitogen cocktails, and in some cases also blunt the release of Th-1 cytokines. The latter finding is intriguing because Th-1 and Th-2 cytokines often have opposing actions on their targets and can act in a mutually inhibitory fashion. Thus, a stress-evoked decline in Th-1 output could “permit” greater expression of Th-2 cytokines. Regardless, the studies indicate that an amplified humoral response to asthma triggers is found in the context of many kinds of stressors, including chronic situations like low SES and more acute ones like a school exam, and emerges both when stress is conceptualized as a demand from the environment or the result of a cognitive appraisal process. Over time, a pattern of exaggerated inflammatory responses may lead to more frequent or severe symptoms of the illness.
Healthy Adults
Because of the small number of studies in patients with asthma, we also review one compelling paradigm that has been used with healthy adults. It involves assessing the extent of psychological stress in healthy adults and, after they have been quarantined, exposing them to viruses that cause upper respiratory infections. The volunteers are then monitored for 3-5 days so that instances of infection (viral replication) and clinical illness (infection accompanied by symptoms) can be recorded. Because the dose and kind of pathogen exposure is identical across volunteers, one can evaluate the contribution of stressful experiences to rates of infection and illness, without concerns about the potential confounding explanation that people experiencing different degrees of stress will have differential exposure to pathogens in their daily lives. In an elegant series of projects using this paradigm, Cohen and his colleagues have shown that high levels of stress (both in the form of life events and appraisals of threat and manageability) are associated with heightened susceptibility to respiratory illness (both viral infection and clinical illness) (Cohen, Tyrrell, & Smith, 1991). These effects are attributable to chronic, rather than acute, forms of stressful experience (Cohen et al., 1998). In follow-up research projects designed to identify the responsible underlying mechanisms, this team found that stressed volunteers produced excessive amounts of IL-6 in the nasal cavity following inoculation. Nasal levels of IL-6 were related, in turn, to more severe illness symptoms. Statistical analyses of mediation indicated that, of the variance that stress accounts for in respiratory illness, 58% of this variance could be explained by excessive IL-6 production (Cohen, Doyle, & Skoner, 1999). Even though they were obtained in healthy adults, these findings suggest two conclusions relevant to our model – that stress is able to accentuate the inflammatory response to an in vivo pathogenic exposure, and that excessive cytokine production can serve as a primary mediating pathway linking stress and disease.
These findings are also important because among patients with asthma, viral infections are a potent trigger for the exacerbation of symptoms. Thus, enhanced susceptibility to respiratory infection may serve as an additional mechanism of action for stress. This logic may seem initially puzzling because viruses primarily activate Th-1 immune responses, and it is the opposing Th-2 cascade that is usually identified as the culprit in asthma flare-ups. However, asthma patients routinely display evidence of an antiviral immune response in the airways, and this has generated speculation that viruses or the immune signals they elicit activate airway epithelial cells in a fashion that contributes to asthma exacerbations (Holtzman et al., 2002). For example, activated Th-1 cells may facilitate recruitment of Th-2 cells to the airways, thus also exacerbating responses to allergen exposures (Holtzman et al., 2002). It is possible, then, that psychological stress in patients with asthma may operate by increasing the sensitivity of Th-1 cells to viral signals, thus increasing vulnerability to virally-induced exacerbations of asthma. Previous clinical research has found that patients with asthma who report more negative life events in combination with low social support experience a higher number of asthma exacerbations induced by upper respiratory infections (Smith & Nicholson, 2001).
5. How does stress modify inflammation?
Having provided support for the basic premises of our working model, we now turn to details of how stress amplifies the immune response to asthma triggers. Figure 1 provides an overview of the relevant pathways, including the hypothalamic-pituitary-adrenal (HPA) axis, the sympathetic-adrenal-medullary (SAM) axis, and the two major divisions of the autonomic nervous system, sympathetic (SNS) and parasympathetic (PNS).
5.1 Glucocorticoid pathways
The HPA axis becomes activated when neurons in the paraventricular nucleus of the hypothalamus secrete corticotropin-releasing hormone (CRH). This molecule travels to the anterior pituitary gland, which responds to its presence by secreting a pulse of adrenocorticotropin hormone (ACTH). The ACTH signal is carried through the peripheral circulation to the adrenal glands, which synthesize and release cortisol in a tissue layer called the zona fasciculate. In nervous, muscle, lymphoid, and other tissues, cortisol binds to an intracellular glucocorticoid receptor (GR), which is the starting point for a number of critical signaling pathways. On T-lymphocytes, GR plays key roles in regulating expression of interleukins-4, -5 and -13 following allergen exposure. Mast cells also constitutively express these receptors, and ligation can inhibit release of histamine and other allergic mediators, as well as diminish recruitment and activation of eosinophils.
Certain situations are known to be potent triggers of the HPA axis. Specifically, acute situations that involve high levels of social evaluation, and elicit self-conscious emotions like shame, reliably boost output of ACTH and cortisol in humans (Dickerson & Kemeny, 2004). In contrast, acute laboratory stressors that do not involve social evaluation, such as watching emotionally distressing films, do not activate the HPA axis (Dickerson et al., 2004). HPA activity is also reliably increased shortly after the onset of severe, chronic stressors, especially when they threaten physical integrity and are completely out of a person's control (Miller, Chen, & Zhou, 2007). We have argued that such situations are likely to increase vulnerability to asthma exacerbations. The idea here is that with prolonged exposure to high levels of cortisol, white blood cells mount a counterregulatory response- that is, they compensate for the high exposure to cortisol by downregulating the expression and function of the receptors that bind cortisol. This response diminishes the sensitivity of immune cells to glucocorticoid signaling, making them more resistant to these molecules' potent anti-inflammatory properties (Miller, Cohen, & Ritchey, 2002). While this may be an adaptive short-term response, it is likely to have long-term costs for patients with asthma. For example, the next time they are exposed to a trigger, it may be more difficult for them to regulate the magnitude and duration of airway inflammation. Another cost over the long-term might be diminished sensitivity to glucocorticoid medications, which are central to effective clinical management of asthmatic symptoms. This glucocorticoid resistance could help explain the paradox of why stress is not beneficial for asthma if it increases HPA activity.
Our first evaluation of this hypothesis involved healthy adults facing a severe, chronic stressor – having a child who was in the midst of being treated for pediatric cancer. Whole blood was collected from volunteers and incubated with LPS – which triggers the release of inflammatory cytokines – and varying dosages of the synthetic glucocorticoid dexamethasone. Parents of cancer patients exhibited diminished sensitivity to the anti-inflammatory properties of dexamethasone, as manifest by greater production of the cytokine IL-6 compared with a matched sample of parents with medically healthy children. These findings provide initial evidence that, in healthy humans, chronic stress diminishes sensitivity to glucorticoids. We recently conducted a follow-up project in children suffering from asthma (Miller & Chen, 2006). This work focused on expression of glucocorticoid receptor mRNA in leukocytes, rather than the indirect measures of its functional capacity used in the intitial project. We found that among children with asthma, chronic stress in the family was marginally associated with reduced expression of GR mRNA. We also assessed the occurrence of acute life events such as failing an exam or the death of a grandparent. Having an acute event in the six months before the study was not sufficient to influence glucocorticoid receptor mRNA. When such events occurred in the context of a chronic stressor, however, their association with patterns of gene expression was accentuated. Children with asthma who simultaneously experienced acute and chronic stress exhibited a 5.5-fold reduction in glucocorticoid receptor mRNA. These findings suggest that when children with asthma face this particular combination of experiences, the bioavailability of GR in leukocyte populations is likely to be significantly diminished. This could have important implications for regulating inflammation in the airways upon exposure to asthma triggers.
Stress-evoked changes in HPA activity could worsen asthma in ways other than glucocorticoid resistance. For instance, exposure to high doses of cortisol can bias the immune system towards an excessive Th-2 cytokine response (Marshall & Agarwal, 2000), which would increase the likelihood of an asthmatic patient experiencing severe or prolonged symptoms following exposure to a trigger. There is also mounting evidence that as time passes, chronic stressors become associated with reduced HPA axis output (Miller et al., 2007). These data indicate that when chronic stress first begins, there is an initial activation of the HPA axis, resulting in elevated concentrations of ACTH and cortisol. But as time passes this activity diminishes, and cortisol secretion rebounds to below normal levels. To the extent that this process results in deficient GR signaling in lymphoid tissues, it could have some of the same consequences for asthmatic patients as stress-related glucocorticoid resistance.
5.2 Autonomic pathways
Stressors also have the capacity to activate the sympathetic nervous system (SNS). SNS stimulation results in systemic release of epinephrine from the adrenal medulla, as well as activation of noradrenergic fibers that innervate pulmonary and lymphoid tissues. In the lungs sympathetic activation facilitates the dilation of bronchi, acting through smooth muscle in blood vessels and submucosal glands. In lymphoid organs such as the spleen, thymus, and lymph nodes, catecholamines released as part of SNS activation can bind to lymphocytes, and modify their functions in a fashion that may promote exaggerated humoral responses (Marshall et al., 2000). Adrenergic receptors are present on T- and B-cells, as well as other populations of leukocytes. They are able to regulate various features of the humoral response involved in asthma, including expression of IL-4, 5, and 13 following allergen exposure, the release of histamines by activated mast cells, and the recruitment and activation of eosinophils in the airways. Because they capitalize on these bronchodilatory and immunomodulatory properties, β-agonist medications are commonly prescribed to asthma patients.
With these important sympathetic mechanisms for asthma in mind, our team recently extended its reasoning on glucocorticoid resistance. We argued that long-term exposure to high levels of epinephrine and norepinephrine would lead to downregulation of β2-adrenergic receptors (β2AR) in pulmonary and lymphoid tissues. This might contribute to heightened expression of Th-2 cytokines, mast cell degranulation, and eosinophil activation, as well as diminish the bronchodilatory effects of beta-agonist therapies in patients with asthma. To begin evaluating this possibility we measured stressful experiences and mRNA for β2AR in leukocytes in a cohort of children with asthma (Miller et al., 2006). To the extent they had high levels of chronic stress in the family, children exhibited reduced quantities of mRNA for the β2AR. As was the case for the GR, there was no effect of acute events. However, when children experienced acute and chronic stressors at the same time, they showed a marked reduction of 9.5-fold in mRNA for β2AR. These findings provide initial evidence in support of our hypothesis that over extended periods of time, high levels of stress downregulate β2AR, which if true, could provide one explanation for why activation of stress systems is not more beneficial for asthma. Future work is needed, however, to determine the extent to which this downregulation translates into decrements in pulmonary and immunologic functions and clinical symptoms of asthma.
In contrast, parasympathetic fibers descend from the vagus nerve and innervate the airways. These fibers signal smooth muscle cells and submucosal glands in the airways. When activated, these fibers release the neurotransmitter acetylcholine, which facilitates bronchoconstriction and mucus secretion. While stress is typically thought to decrease parasympathetic activity in healthy individuals, some researchers have postulated that in patients with asthma, stress is associated with hyperresponsiveness of both the parasympathetic and alpha-sympathetic pathways (Lehrer, Isenberg, & Hochron, 1993). Previous research has documented that the experience of acute negative emotions is linked to autonomic reactivity in patients with asthma. In this research paradigm, children with asthma were shown a sad film. The more emotional children reported being over the film, the greater their airway reactivity, as measured by challenge with methacholine, a chemical that causes smooth muscle contraction in the airways via cholinergic pathways (Miller & Wood, 1994). In follow-up research this team has shown that sadness produces the greatest heart rate variability (indicating greater parasympathetic activation) and instability of oxygen saturation (indicating airway instability) as compared with happiness or neutral feelings. These findings suggest that the types of negative emotional responses that our model postulated would be associated with stressful life experiences may elicit cholinergic responses that contribute to bronchoconstriction and exacerbations of asthma.
6. What's next?
We have used this mini-review to outline the premises of a working model of stress and asthma. Although a number of the model's important predictions have been confirmed, more work needs to be done before its overall utility can be evaluated. Next steps for future research should include addressing questions such as: Do stressful experiences foster asthmatic symptoms by accentuating inflammatory responses to allergic, infectious, and chemical triggers? To what degree are diminished sensitivity to glucocorticoids and catecholamines responsible for the heightened inflammatory cascade? To more definitively answer these questions in human clinical research, researchers will need to conduct multi-wave prospective investigations, frequently assessing psychological, immunologic, and clinical variables. It will also be important to assess specific types of stress experiences and specific types of triggers, as there are likely to be more and less potent combinations of psychological experiences and environmental triggers. Finally, research is needed to determine whether other causal pathways in the model are operative, and to incorporate additional mechanisms linking the major constructs, since airway inflammation is unlikely to be the only critical mediator in these processes. To the extent that the next wave of studies can meet these challenges, they will allow us to develop convincing mechanistic explanations for how stress “gets inside the body” to worsen asthma.
Acknowledgments
This article was supported by NIH grant HL073975.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Andrulis DP. Access to care is the centerpiece in the elimination of socioeconomic disparities in health. Annals of Internal Medicine. 1998;129:412–416. doi: 10.7326/0003-4819-129-5-199809010-00012. [DOI] [PubMed] [Google Scholar]
- Busse WW, Lemanske RF. Asthma. New England Journal of Medicine. 2001;344:350–362. doi: 10.1056/NEJM200102013440507. [DOI] [PubMed] [Google Scholar]
- Chen E, Fisher EB, Jr, Bacharier LB, Strunk RC. Socioeconomic status, stress, and immune markers in adolescents with asthma. Psychosomatic Medicine. 2003;65:984–992. doi: 10.1097/01.psy.0000097340.54195.3c. [DOI] [PubMed] [Google Scholar]
- Chen E, Hanson MD, Paterson LQ, Griffin MJ, Walker HA, Miller GE. Socioeconomic status and inflammatory processes in childhood asthma: The role of psychological stress. Journal of Allergy and Clinical Immunology. 2006;117:1014–1020. doi: 10.1016/j.jaci.2006.01.036. [DOI] [PubMed] [Google Scholar]
- Cohen S, Doyle WJ, Skoner DP. Psychological stress, cytokine production, and severity of upper respiratory illness. Psychosomatic Medicine. 1999;61:175–180. doi: 10.1097/00006842-199903000-00009. [DOI] [PubMed] [Google Scholar]
- Cohen S, Frank E, Doyle WJ, Skoner DP, Rabin BS, Gwaltney JM., Jr Types of stressors that increase susceptibility to the common cold in healthy adults. Health Psychology. 1998;17:214–223. doi: 10.1037//0278-6133.17.3.214. [DOI] [PubMed] [Google Scholar]
- Cohen S, Kessler RC, Gordon LU. Strategies for measuring stress in studies of psychiatric and physical disorders. In: Cohen S, Kessler RC, Gordon LU, editors. Measuring stress: A guide for health and social scientists. New York, NY: Oxford University Press; 1995. pp. 3–26. [Google Scholar]
- Cohen S, Tyrrell DA, Smith AP. Psychological stress in humans and susceptibility to the common cold. New England Journal of Medicine. 1991;325:606–612. doi: 10.1056/NEJM199108293250903. [DOI] [PubMed] [Google Scholar]
- Dickerson SS, Kemeny ME. Acute stressors and cortisol responses: A theoretical integration and synthesis of laboratory research. Psychological Bulletin. 2004;130:355–391. doi: 10.1037/0033-2909.130.3.355. [DOI] [PubMed] [Google Scholar]
- Holtzman MJ, Morton JD, Shornick LP, Tyner JW, O'Sullivan MP, Antao A, et al. Immunity, inflammation, and remodeling in the airway epithelial barrier: Epithelial-viral-allergic paradigm. Physiological Reviews. 2002;82:19–46. doi: 10.1152/physrev.00020.2001. [DOI] [PubMed] [Google Scholar]
- Jaakkola JJ, Nafstad P, Magnus P. Environmental tobacco smoke, parental atopy, and childhood asthma. Environ Health Perspect. 2001;109:579–582. doi: 10.1289/ehp.01109579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang D, Coe C, McCarthy DO, Jarjour NN, Kelly EA, Rodriguez RR, et al. Cytokine profiles of stimulated blood lymphocytes in asthmatic and healthy adolescents across the school year. Journal of Interferon and Cytokine Research. 1997;17:481–487. doi: 10.1089/jir.1997.17.481. [DOI] [PubMed] [Google Scholar]
- Kang DH, Fox C. Th1 and Th2 cytokine responses to academic stress. Research in Nursing & Health. 2001;24:245–257. doi: 10.1002/nur.1027. [DOI] [PubMed] [Google Scholar]
- Lazarus RS, Folkman S. Stress, appraisal, and coping. New York: Springer Publishing Company; 1984. [Google Scholar]
- Lehrer PM, Isenberg S, Hochron SM. Asthma and emotion: A review. Journal of Asthma. 1993;30:5–21. doi: 10.3109/02770909309066375. [DOI] [PubMed] [Google Scholar]
- Liu LY, Coe CL, Swenson CA, Kelly EA, Kita H, Busse WW. School examinations enhance airway inflammation to antigen challenge. American Journal of Respiratory and Critical Care Medicine. 2002;165:1062–1067. doi: 10.1164/ajrccm.165.8.2109065. [DOI] [PubMed] [Google Scholar]
- Marshall GD, Agarwal SK. Stress, immune regulations, and immunity: Applications for asthma. Allergy and Asthma Proc. 2000;21:241–246. doi: 10.2500/108854100778248917. [DOI] [PubMed] [Google Scholar]
- Miller BD, Wood BL. Psychophysiologic reactivity in asthmatic children: A cholinergically mediated confluence of pathways. Journal of the American Academy of Child and Adolescent Psychiatry. 1994;33:1236–1244. doi: 10.1097/00004583-199411000-00004. [DOI] [PubMed] [Google Scholar]
- Miller GE, Chen E. Life stress and diminished expression of genes encoding glucocorticoid receptor and beta(2)-adrenergic receptor in children with asthma. Proceedings of the National Academy of Sciences. 2006;103:5496–5501. doi: 10.1073/pnas.0506312103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller GE, Chen E, Zhou E. If it goes up, must it come down? Chronic stress and the hypothalamic-pituitary-adrenocortical axis in humans. Psychological Bulletin. 2007;133:25–45. doi: 10.1037/0033-2909.133.1.25. [DOI] [PubMed] [Google Scholar]
- Miller GE, Cohen S, Ritchey AK. Chronic psychological stress and the regulation of pro-inflammatory cytokines: A glucocorticoid resistance model. Health Psychology. 2002;21:531–541. doi: 10.1037//0278-6133.21.6.531. [DOI] [PubMed] [Google Scholar]
- Nelson HS, Szefler SJ, Jacobs J, Huss K, Shapiro G, Sternberg AL. The relationships among environmental allergen sensitization, allergen exposure, pulmonary function, and bronchial hyperresponsiveness in the Childhood Asthma Management Program. Journal of Allergy and Clinical Immunology. 1999;104:775–785. doi: 10.1016/s0091-6749(99)70287-3. [DOI] [PubMed] [Google Scholar]
- Ober C, Hoffjan S. Asthma genetics 2006: the long and winding road to gene discovery. Genes Immun. 2006;7:95–100. doi: 10.1038/sj.gene.6364284. [DOI] [PubMed] [Google Scholar]
- Sandberg S, Paton JY, Ahola S, McCann DC, McGuinness D, Hillary CR. The role of acute and chronic stress in asthma attacks in children. Lancet. 2000;356:982–987. doi: 10.1016/S0140-6736(00)02715-X. [DOI] [PubMed] [Google Scholar]
- Segerstrom SC, Miller GE. Psychological stress and the human immune system: A meta-analytic study of 30 years of inquiry. Psychological Bulletin. 2004;130:601–630. doi: 10.1037/0033-2909.130.4.601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigurs N, Gustafsson PM, Bjarnason R, Lundberg F, Schmidt S, Sigurbergsson F, et al. Severe respiratory syncytial virus bronchiolitis in infancy and asthma and allergy at age 13. Am J Respir Crit Care Med. 2005;171:137–141. doi: 10.1164/rccm.200406-730OC. [DOI] [PubMed] [Google Scholar]
- Smith A, Nicholson K. Psychological factors, respiratory viruses and exacerbation of asthma. Psychoneuroendocrinology. 2001;26:411–420. doi: 10.1016/S0306-4530(00)00063-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright RJ. Stress and atopic disorders. Journal of Allergy and Clinical Immunology. 2005;116:1301–1306. doi: 10.1016/j.jaci.2005.09.050. [DOI] [PubMed] [Google Scholar]
- Wright RJ, Cohen RT, Cohen S. The impact of stress on the development and expression of atopy. Current Opinion in Allergy and Clinical Immunology. 2005;5:23–29. doi: 10.1097/00130832-200502000-00006. [DOI] [PubMed] [Google Scholar]
- Wright RJ, Finn P, Contreras JP, Cohen S, Wright RO, Staudenmayer J, et al. Chronic caregiver stress and IgE expression, allergen-induced proliferation, and cytokine profiles in a birth cohort predisposed to atopy. Journal of Allergy and Clinical Immunology. 2004;113:1051–1057. doi: 10.1016/j.jaci.2004.03.032. [DOI] [PubMed] [Google Scholar]
- Wright RJ, Rodriguez M, Cohen S. Review of psychosocial stress and asthma: An integrated biopsychosocial approach. Thorax. 1998;53:1066–1074. doi: 10.1136/thx.53.12.1066. [DOI] [PMC free article] [PubMed] [Google Scholar]