

Abstract
To investigate the role of the gene products encoded from the open reading frames 101, 142, and 144 of Autographa californica multiple nucleopolyhedrovirus (AcMNPV), a set of bacmid knockout and repair constructs were generated. The repair genes were engineered to contain an HA epitope tag at their C-termini. The results of transfection-infection assays and growth curve analyses showed that the Ac 101, 142, and 144 genes were required for infectious virus production. To better characterize the role of these genes in the baculovirus replication cycle, quantitative DNA replication assays were performed and demonstrated that in cells transfected with the Ac 101, 142, or 144 knockouts, DNA replicated with similar kinetics as a control virus. Western blot analyses of budded virus from cells infected with the repair viruses showed that these proteins are associated with the viral nucleocapsid. Furthermore, immunoelectron microscopy of cells transfected with the knockout bacmids revealed defects in nucleocapsid production for all three constructs. From these results we concluded that the gene products encoded from these open reading frames are essential for virus production and may be involved in DNA processing, packaging, or nucleocapsid morphogenesis.
Introduction
Baculoviruses have a complex structure and life cycle that is reflected in their large genome sizes that range from 82 to 178 kb depending on the virus strain. The recent accumulation of complete genome sequence data from over 30 baculoviruses has revealed patterns of gene content and diversity that further illuminates the evolutionary patterns characteristic of this family of viruses. Homologs of a number of genes are found in the genomes of most members of the family thereby implicating them as playing fundamental roles in the biology of these viruses. Three of these open reading frames were originally identified in the genome of the Autographa californica multiple nucleopolyhedrovirus (AcMNPV) and named ORFs 101, 142, and 144 and were predicted to encode gene products of 41.5, 55.4, and 33.5 kDa, respectively (Ayres et al., 1994). Homologs of these genes have been identified in all sequenced baculovirus genomes with the exception of the virus pathogenic for the dipteran, Culex nigripalpus (CuNiNPV) (Afonso et al., 2001). This could reflect either fundamental differences between CuNiNPV and the other baculoviruses, or simply that the homologous sequences can no longer be identified because of differences incorporated over time.
In addition to their conservation, the products of all three of these genes were identified as being associated with occlusion derived virions (ODV) (Braunagel et al., 2003) suggesting that they might be involved in capsid structure. Although data on Ac142 is limited, several studies have provided information regarding Ac101 and Ac144. The Ac101 ORF was reported to be transcribed from a single early and three late promoters, and to encode a product of about 42 kDa that is present in both ODV nucleocapsids and budded virions (BV) (Braunagel et al., 2001). Therefore, it was named BV/ODV-C42 indicating it was a protein of about 42 kDa that localized to the BV and ODV capsid (C) structures. In addition, it was reported to interact in a yeast two hybrid assay and by native gel electrophoresis (Braunagel et al., 2001) with phosphoprotein 78/83 (pp78/83) (also called AcMNPV orf8, or orf1629) which has been shown to localize to the basal end region of nucleocapsids (Russell et al., 1997; Vialard and Richardson, 1993). PP78/83 has also been implicated in the polymerization of actin that may be involved in capsid morphogenesis (Goley et al., 2006).
A variety of investigations have been conducted on Ac ORF 144. Initially it was confirmed that its transcript initiates at a late promoter element, and immunoelectron microscopy and fractionation studies suggested that it was a component of occlusion-derived virion capsids and envelopes, but was not associated with budded virions. In addition, Western blot analysis suggested that its molecular mass is 27 kDa which is less than the 33.5 kDa predicted from sequence analysis. Consequently it was named ODV-EC27 (Braunagel et al., 1996a) indicating it is a protein of 27 kDa that localized to the ODV envelope (E) and capsid (C) structures. Later it was suggested that it is a multi-functional cyclin and may be involved in regulating the cell cycle during virus infection, in particular causing arrest at the G2/M phase. It was found to be present in a complex with CDC2 or CDK6 capable of phosphorylating histone H1 and retinoblastoma protein in vitro (Belyavskyi et al., 1998). It was also reported to interact in a yeast two-hybrid assay with Ac ORF 101 described above (also named C42) and both Ac ORF 101 and p78/83 in native gel electrophoresis assays (Braunagel et al., 2001).
In this report we provide additional information on the role these genes play in baculovirus biology. To accomplish this we generated Ac ORF 101, 142, and 144 knockout bacmids as well as repair bacmid constructs that expressed an HA-tagged version of each gene. These constructs were then characterized in a tissue culture system to determine the role of the genes in virus infectivity and DNA replication. In addition, their localization in infected cells and the effect of deletion of each gene on capsid structure and on the infection cycle was investigated by electron microscopy.
Results
AcMNPV ORFs 101, 142, and 144 are essential genes
To determine whether the Ac 101, 142, and 144 ORFs are essential for viral replication, deletion and repair constructs of each gene were prepared. The deletion constructs were selected by their resistance to chloramphenicol that indicated that site-specific deletion of the target gene had occurred. The repair constructs were designed such that the HA-tagged target gene along with its native promoter region were inserted into the polyhedrin locus along with an egfp gene under the IE-1 promoter. The structure of all the deletion and repair constructs was confirmed by PCR (data not shown). To investigate the function of each gene, Sf-9 cells were transfected with either the knockout or repair bacmid construct and monitored for EGFP expression by fluorescence microscopy. By 120 hours post-transfection, widespread EGFP expression could be seen in cell monolayers that were transfected with the three repair constructs indicating that these bacmids were able to produce levels of budded virus sufficient to initiate secondary infection (Fig. 1A). In contrast, in cells transfected with either of the three knockout bacmids, EGFP expression was only observed in isolated cells that were initially transfected indicating that these bacmid constructs were unable to produce infectious BV that could initiate secondary infection (Fig. 1B). To confirm these results, supernatants from cells transfected with the repair or knockout bacmids were transferred to a new cell monolayer and monitored for infection. As expected, cells incubated with supernatants from the repair construct transfections showed numerous EGFP expressing cells. However, in cells incubated with supernatants from either of the three knockout transfections, no EGFP expression was detected at any time-point analyzed up to 120 h (Fig. 1 A and B). Therefore these results indicate that the Ac 101, 142, and 144 ORFs are essential for infectious BV production.
Fig. 1.
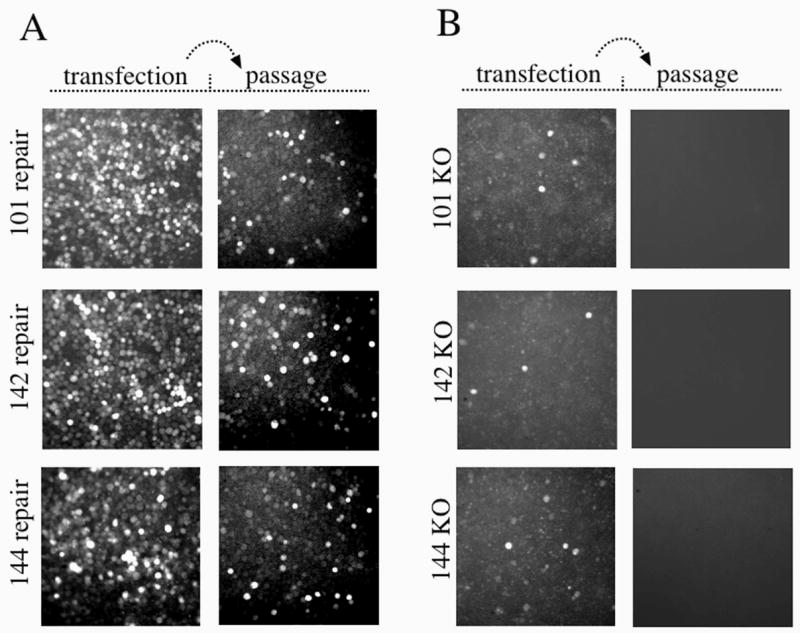
Analysis of viral replication in transfected Sf-9cells. (A) The left set of panels represent Sf-9 cells transfected with the Ac 101, Ac 142, and Ac 144 repair constructs. Cells were incubated for 120 h at 27° C, and 50 μl of the supernatant was passed on to a new monolayer of cells (right panel) and allowed to incubate for 120 h (passage). (B) The left set of panels represent Sf-9 cells transfected with the Ac 101, Ac 142, and Ac 144 knockout constructs. Cells were incubated for 120 h at 27° C, and 1ml of the supernatant was passed on to a new monolayer of cells and allowed to incubate for 120 h (passage). Infectivity was monitored by expression of GFP under the control of the baculovirus IE-1 promoter.
To further assess whether the Ac 101, 142, or 144 knockout bacmids were deficient in budded virus production and were restored to wild-type levels in the repair bacmids, viral growth curve analyses were performed. For these experiments, Sf-9 cells were transfected with bacmid DNA and at selected time-points the budded virus titers from the cell supernatants were determined by a TCID50 end-point dilution assay (see Materials and Methods). The results of these analyses showed that with Sf-9 cells transfected with the knockout bacmids, no budded virus titers could be detected in the supernatant at any of the time-points analyzed up to 120 hr post transfection (h.p.t.) (Fig. 2). However, Sf-9 cells transfected with either of the repair bacmids, BV production was similar to that produced by the infectious control bacmid indicating that re-inserting the deleted open reading frame under the control of their native promoters into the polyhedrin locus of the respective knockout bacmid genome was sufficient to restore normal replication. Therefore, these data are in accordance with the results of the transfection-infection assay and confirm that Ac 101, 142, and 144 are essential genes required for budded virus production.
Fig. 2.

Growth curves of knockout and repair constructs generated from a transfection time-course in Sf-9 cells. Shown are the results from three independent growth curve analyses. For these experiments, Sf-9 cells were transfected in triplicate with either the Ac 101, 142, or 144 knockout or repair bacmid or the infectious control bacmid (AcGFP), and at the indicated time-points the supernatants were removed and the titers determined by TCID50 end-point dilution assays (see Materials and Methods). Infectivity was determined by monitoring GFP expression. The points indicate the averages of triplicate transfections and the error bars represent standard deviation.
Quantitative analysis of viral DNA replication
To analyze DNA replication in cells transfected with the Ac 101, 142, or 144 knockout bacmids, quantitative DNA replication assays were performed with DNA extracts from cells transfected with the knockout bacmids along with DNA extracts from cells transfected with a gp64 knockout bacmid that serves as a non-infectious control virus (Vanarsdall, 2006). Because the viral fusion protein GP64 is required for egress of nucleocapsids from infected cells (Oomens and Blissard, 1999), this mutant lacks the ability to initiate cell-to-cell infection, but all other replication processes should remain normal, including DNA replication. When the DNA replication levels were analyzed over a 96 hour time-course, cells transfected with either the Ac 101, 142 or 144 knockout bacmid or with the gp64 knockout control showed similar levels of viral DNA replication by 24 to 72 hours post-transfection at which point DNA synthesis appeared to plateau (Fig. 3). In addition, the levels of viral DNA detected in all samples at the final time-point of 96 hours post-transfection were similar (Fig. 3). Therefore based on these analyses, the rate and level of viral DNA synthesis in cells transfected with the bacmids lacking either the Ac 101, Ac 142 or Ac 144 open reading frames was similar to the gp64 knockout control suggesting that these genes are not involved in viral DNA synthesis.
Fig. 3.

Real-time PCR analysis of viral DNA replication in transfected Sf-9 cells. Shown are the results of three independent DNA replication assays. For these analyses, total DNA was isolated from Sf-9 cells transfected with either the Ac 101 (panel A), 142 (panel B), or the 144 (panel C) knockout bacmid or a bacmid lacking the gp64 envelope fusion protein which served as the non-infectious control. At the designated time-point, total DNA was extracted and digested with the restriction enzyme DpnI to eliminate input bacmid DNA, and analyzed by real-time PCR. The values displayed represent the averages from transfections performed in triplicate with error bars indicating standard deviations.
Western blot analysis of purified budded virus
To gain insight into whether the proteins encoded by the Ac 101, 142 or 144 ORFs are associated with extra-cellular virions, Western blot analyses was performed with budded virus envelope and nucleocapsid fractions prepared from cells infected with the repair constructs expressing either the Ac 101, 142 or 144 HA-tagged proteins. To confirm the separation of BV preparations into nucleocapsid and envelope fractions, a set of control blots were probed with either a monoclonal antibody to the vp39 capsid protein or a monoclonal antibody to the GP64 envelope fusion protein. The results of these control experiments showed that a protein of 39 kDa was present in both virus infected cell extracts and the budded virus nucleocapsid fraction, but not in the BV fraction containing the envelope (Fig. 4A, lane IC, C, and EN). However, a 64 kDa protein was detected in the envelope fraction when probed with the anti-GP64 antibody (Fig. 4 B). Some residual GP64 contamination of the capsid preparation was also observed (data not shown). Western blot analyses of cell lysates infected with the Ac 101 repair construct using a monoclonal antibody to the HA epitope tag detected a single immunoreactive protein identical to the predicted molecular weight of ~41.5 kDa (Fig. 4 C, lane IC). This protein was also detected in the nucleocapsid fraction from budded virus (Fig. 4C, lane C), but no signal was detected in the fraction containing the budded virus envelope (Fig. 4 C, lane EN). Similarly, Western analysis of lysates from cells infected with either the Ac 142 or the Ac144-repair viruses using the monoclonal antibody to the HA epitope also detected proteins of the expected sizes of 48 kDa and 33.5 kDa, respectively (Fig. 4 D and C, lanes IC). Immunoreactive proteins of this size were also detected in the nucleocapsid fractions of budded virus, but not in the budded virus envelope fractions (Fig. 4 D and E, lanes C, and EN). Therefore the results of the Western blot analyses indicate that the proteins encoded by the ORFs Ac 101, 142, and 144 are associated with the nucleocapsid, but not the envelope of budded virus.
Fig. 4.

Western blot analysis of cell lysates and BV fractions from infected Sf-9 cells. Shown are the results of three independent infection experiments to determine if the proteins encoded by ORFs Ac 101, 142, or 144 are associated with BV. For these experiments, 100 mls of Sf-9 cells at a density of 1.5 × 106 were infected at an MOI of 2 with either the HA-tagged 101, 142, or 144 repair virus stocks. At 96 hours post-infection, the cells were harvested and the BV collected and fractionated according to the Material and Methods. Samples representing uninfected cell lysates (Sf-9), virus infected cell lysates (IC), or nucleocapsid (C) or envelope fractions (EN) of BV were analyzed by 10 % SDS-PAGE. Samples analyzed in panels A and B were derived from cells infected with the control virus Ac-GUS (Vanarsdall et al, 2004) and the samples analyzed in panels C, D, and E were derived from cells infected with the 101, 142, and 144 repair virus, respectively. Samples were probed with a monoclonal antibody to vp39 at a dilution of 1:100 (panel A), a monoclonal antibody to GP64 (AcV5) at a dilution of 1:5000 (panel B), or a monoclonal antibody to the HA-epitope at a dilution of 1:2500 (panels C-E). A goat- anti-mouse-HRP secondary antibody (Promega) was used at a dilution of 1:2500. The molecular weights in kilodaltons from protein standards are indicated on the left of the panels, protein samples applied to each lane are indicated on the top of the panels, and the antibody used is indicated on the bottom of the panels.
Immunoelectron microscopy of bacmid transfected cells
Because the results of the Western blot analyses described above indicated that the proteins encoded by Ac ORFs 101, 142, and 144 are associated with nucleocapsids of budded virions, immunoelectron microscopy was performed with cells transfected with the knockout constructs to determine if the lack of these proteins results in defects in nucleocapsid production. To facilitate these analyses, thin sections generated from transfected Sf-9 cells were immunostained with a monoclonal antibody to the major capsid protein vp39. Control experiments were performed with cells transfected with the GP64 knockout bacmid and showed typical characteristics of a baculovirus infection, including a well-defined virogenic stroma inundated with electron-dense rod-shaped nucleocapsids that reacted with the vp39 monoclonal antibody (Fig. 5A). In contrast, in cells transfected with the 101-knockout (KO) bacmid, no well-defined nucleocapsid structures could be discerned. However, concentrations of gold label could be seen in distinct regions adjacent to the inner-nuclear membrane (Fig. 5B). The gold label in these regions, although it did not interact with structures resembling nucleocapsids, did appear to localize to amorphous electron dense matter. Similarly, visual observation of immuno-stained thin-sections from cells transfected with the 142-KO bacmid construct also revealed striking differences compared to control samples. In these cells, although there was limited immuno-staining, some aberrant looking capsid structures could be seen randomly dispersed throughout the nucleus that reacted with the vp39 antibody. These capsid-like structures displayed a unique morphology and in some cases appeared as hollow tubes apparently lacking an electron dense core (Fig. 5C). Finally, analysis of cells transfected with the 144-KO bacmid revealed electron-dense structures within the nucleus that reacted with vp39 antibody. However, these structures were indistinct and lacked the typical well-defined nucleocapsid morphology (Fig. 5D). Therefore these results indicate that the protein products of ORFs Ac 101, 142, and 144 are required for normal nucleocapsid production.
Fig. 5.

Electron microscopic analysis of Sf-9 cells transfected with knockout bacmids. Sf-9 cells were transfected with either the Ac 101, 142, or 144 knockout bacmid and at 72 hours post-infection the cells were processed and the thin sections stained with monoclonal antibody to vp39. Controls experiments were performed with Sf-9 cells transfected with the gp64 knockout bacmid. (A) Nucleus of a cell transfected with the gp64 KO bacmid serving as the control. (B) Nucleus of a cell transfected with the Ac 101 knockout virus showing a region in the nucleus that lacks viral nucleocapsids, but is enriched in gold label for vp39. (C) Nucleus of cell transfected with the Ac 142 knockout virus showing amorphous electron dense matter staining with gold label for vp39. (D) Image from the nucleus of a cell transfected with the Ac 144 knockout virus also showing amorphous electron dense matter staining with gold label for vp39. For all samples, sections were stained with primary antibody to AcMNPV vp39 as undiluted tissue culture supernatant. The goat anti-mouse 10 nm gold-conjugated secondary antibody was used at 1:50 dilution. For images A, B, C and D the bar represents 0.25 mm.
Discussion
This report describes the construction and characterization of bacmids lacking either AcORF 101, 142, or 144, and shows that in cultured S. frugiperda cells, all three of these genes are essential for budded virus production. However, they do not appear to be required for DNA replication, and therefore are likely involved in other replicative processes. In order to analyze expression and localization of these gene products, repair constructs that were generated to confirm the phenotype of the original knockout bacmids were also designed to contain an HA-epitope fused to the C-terminal end of the repair genes. Therefore these viruses expressed single copies of Ac ORF 101, 142, or 144 whose products could be monitored using a commercially available monoclonal antibody to the HA epitope. The repaired viruses appeared to replicate normally. In addition, Western blot analyses of lysates from cells infected with either the Ac 101, 142, or 144 repair viruses detected proteins with molecular weights of ~42, ~48, and 33.5 kDa, respectively. These molecular masses are consistent with those predicted from the sequences of the open reading frames. Using a strategy in which purified budded virus was separated into envelope and nucleocapsid fractions, the proteins encoded by ORFs Ac 101, 142, and 144 were shown to be present exclusively in fractions containing the viral nucleocapsid.
A previous report investigating Ac ORF 144 using a polyclonal rabbit antibody concluded from Western blot analysis of cell extracts that the major protein encoded by this open reading frame had a molecular mass of 27 kDa (Braunagel et al., 1996b), in contrast to the predicted molecular weight of 33.5 kDa (Ayres et al., 1994). In addition, the ~27 kDa protein identified in the previous study was shown to be present only in occlusion derived and not in budded virus preparations. Therefore, it was suggested that this protein was selectively incorporated into occluded virions and it was designated ODV-EC27. In contrast, our Western blot analyses, using a highly specific monoclonal antibody to the HA epitope, detected a protein from both cell extracts and budded virus preparations that conforms to the predicted molecular weight of 33.5 kDa. In addition, there was no indication of a 27 kDa protein from either infected cell lysates or budded virus preparations. It is unclear why the previous report identified a major protein of 27 kDa. However, it could represent a non-specific cross-reaction of their polyclonal antibody preparation to another protein present in the extract.
The results of the Western blot analysis with budded virus indicating that the proteins encoded by Ac ORF 101, 142, or 144 are associated with viral nucleocapsids supports the finding that they are associated with ODV as determined by mass spectrometric analysis (Braunagel et al, 2003). Electron microscopic investigations of Ac101 (Braunagel et al, 2001) and 144 (Braunagel et al, 1996) also showed that they are nucleocapsid-associated. To investigate the significance of these observations further, we used electron microscopy to assess whether the knockout constructs were defective in nucleocapsid production. The results of these analyses showed that in cells transfected with the Ac ORF 101 knockout, no mature nucleocapsids could be seen, although significant amounts of anti-vp39-capsid immuno-gold label were concentrated in regions adjacent to the inner nuclear membrane. It is possible that these regions represent specific domains in the nucleus where nucleocapsid assembly occurs, but in the absence of Ac 101, this process is stalled. Similarly, in cells transfected with the Ac ORF142 or Ac ORF 144 knockout bacmids, mature nucleocapsids were not present. However, in contrast to the Ac101 knockout, gold label did not accumulate in distinct regions within the nucleus, but stained undefined structures that were dispersed more randomly throughout the nucleus. In general, these structures were not present in large quantities.
In a previous analysis, the protein encoded by Ac ORF 101 was shown to associate with the AcMNPV pp78/83 and Ac orf 144 (Braunagel et al., 2001). Pp78/83 localizes to the basal end region of nucleocapsids (Russell et al., 1997; Vialard and Richardson, 1993) and a recent report has implicated it in nuclear actin dynamics by activating the host Arp2/3 complex (Goley et al., 2006). Therefore, Ac101 and 144 may also play a role the dynamics of actin that may be involved in nucleocapsid assembly.
Whether the lack of normal nucleocapsid production was caused by the failure of Ac 101, 142 or 144 to fulfill a virion structural requirement, or indirectly due to a defect of other processes is unclear. Very late expression factor 1 (VLF-1) is also a nucleocapsid associated protein and the phenotype of the Ac 101, 142, and 144 deletion mutants is reminiscent of the deletion of VLF-1. The VLF-1 deletion mutant produces defective tube-like capsid structures and repair of the VLF-1 deletion with a point mutation in a conserved domain that is thought to be involved in a final step in DNA processing results in normal appearing, but non-infectious nucleocapsids. This suggests that VLF-1 is an important virion structural element in addition to having other functions (Vanarsdall et al, 2006). Similar to Ac 101, 142, 144 and VLF-1 knockouts, deletion of the single stranded DNA binding protein (DBP) gene also produces defective nucleocapsids. However DBP is not nucleocapsid-associated (Vanarsdall et al, 2007). The DBP deletion mutant caused a reduced level of DNA synthesis and appeared to produce defective genomes. It also did not produce a recognizable virogenic stroma. The virogenic stroma is a structure unique to baculovirus infected nuclei and likely serves as the scaffold for viral DNA replication and nucleocapsid assembly. Therefore the deletion of dbp may have resulted in defective nucleocapsid production by causing either a defect in genome structure or by the lack of a virogenic stroma to serve as a matrix for nucleocapsid assembly.
In summary, the characterization of the different types of mutations that are associated with defective nucleocapsids indicates that they may be caused by the absence of a virion structural protein, a defect in DNA processing, or by a defective scaffold or packaging protein that would be required for the assembly of virion DNA with nucleocapsid proteins. The association of Ac ORFs 101, 142, and 144 with the nucleocapsid, but not the envelope fraction of BV, suggests that they are integral structural elements of nucleocapsids and it is likely that their absence directly impairs the formation of nucleocapsids. However, if similar to VLF-1, other critical requirements for baculovirus replication can not be ruled out.
Material and Methods
Cells, virus, bacterial strains, and antibiotics
Spodoptera frugiperda (Sf-9) cells were cultured in Sf-900 II serum-free medium (Invitrogen), penicillin G (50 units/ml), streptomycin (50 units/ml, Whittaker Bioproducts), and fungizone (amphotericin, 375 ng/ml, Invitrogen) as previously described (Harwood et al., 1998). The Ac-GUS virus control was previously described (Vanarsdall et al, 2004). The E. coli strains BW25113 harboring plasmid pKD46 encoding the λ Red recombination system (Datsenko and Wanner, 2000), BW25141 harboring plamid pKD3 encoding the CAT gene (Datsenko and Wanner, 2000) and DH10B were kindly provided by G.W. Blissard (Boyce Thompson Institute, Cornell University, Ithaca, NY). The cell line DH10Bac (Invitrogen) was used to isolate bacmid bMON14272 that contains the AcMNPV genome and the helper plasmid pMON7124 encoding a transposase (Luckow et al., 1993). The concentration of antibiotics used in the various steps in manipulating the bacmid and associated plasmids was 100 μg/ml ampicillin, 50 μg/ml kanamycin, 20 μg/ml chloramphenicol, 10 μg/ml tetracycline, and 7 μg/ml gentamycin.
Knockout and repair bacmid construction
Bacmids containing deletions of either Ac ORF 101, 142, or 144, were made using the λ Red homologous recombination system in Escherichia coli as described previously (Bideshi and Federici, 2000; Datsenko and Wanner, 2000; Vanarsdall et al., 2004). The primer sequences and their genomic coordinates that were used for PCR amplification of a linear DNA fragment containing the chloramphenicol resistance gene are listed in Table 1. Parental knockout bacmid constructs were transposed with the pFBIE-GFP transfer vector (Vanarsdall, 2006) according to the methods described previously (Vanarsdall et al., 2004) to introduce the GFP reporter gene under control of the AcMNPV IE-1 promoter. To construct repair bacmids, gene fragments were PCR amplified from bacmid DNA using 5′ primers that annealed ~500 bp upstream of the ATG to include the native promoter region along with 3′ primers that annealed just upstream of the native stop codon and also contained additional DNA encoding an in-frame HA-epitope sequence. The 5′ and 3′ primers also included EcoRI and HindIII restriction sites (Table 1). The repair fragments containing the HA epitope tag were cloned into the pFBIE-GFP plasmid described above and used to transpose parental knockout bacmids. The control virus, Ac-GFP, was constructed by transposing bacmid pMON14272 with the pFBIE-GFP plasmid that has the EGFP gene under the control of the AcMNPV ie-1 promoter (Vanarsdall et al, 2006).
Oligonucleotides used to generate knockout and repair bacmid constructs
| primer | locus | sequence |
|---|---|---|
| orf101-5′cat primer1 | 87778-87827 | 5′-GTG GCT CGC AAA TCG CAA AAC ACA ATT TTA AAT AAC AAC GCT GCC ATA GCC ATA TGA ATA TCC TCC TTA G-3′ |
| orf101-3′cat primer1 | 86977-87026 | 5′-CGT CTT GGT CGT TTG AAT TTT TGT TGC TGT GTT TCC TAA TAT TTT CCA TCG TGT AGG CTG GAG CTG CTT C-3′ |
| orf142-5′cat primer1 | 123753-123802 | 5′-GCA ATT ACT TGA ATT GCA CGT TTG ATT TGC TAG ACG ATG CCG TGC TCA TGC ATA TGA ATA TCC TCC TTA G-3′ |
| orf142-3′cat primer1 | 124656-124705 | 5′-CAC AAA AAT GTA CGA CGA TTC ATG ATC GTA CAT GAC AGG AAA CTG GTT GGG TGT AGG CTG GAG CTG CTT C-3′ |
| orf144-5′cat primer1 | 125588-125637 | 5′-CGG CAG ATT GCC GCC GTG GTG TTT AGC ACA TTA GCT TTT ATA CAC AAT AGC ATA TGA ATA TCC TCC TTA G-3′ |
| orf144-3′cat primer1 | 126148-126198 | 5′-GAT GTT ACA GAA TTT GAG TTT ATT TTG TCA AAA TCT TCA ATG GTA AAT TTG GTG TAG GCT GGA GCT GCT TC-3′ |
| orf101-5′repair-EcoRI primer2 | 88472-88494 | 5′-GAA TTC GTG CTA ATG ACA GAA ATG ATT G-3′ |
| orf101-3′repair-HindIII primer2 | 86924-86946 | 5′-AAG CTT TTA GGC GTA ATC TGG GAC GTC GTA TGG GTA ATA TTT TTT ACG CTT TGC ATT CG-3′ |
| orf142-5′repair-EcoRI primer2 | 123111-123122 | 5′-GAA TTC CAA TGT GCC GAC ATT GCC AAA C-3′ |
| orf142-3′repair-HindIII primer2 | 125042-125062 | 5′-AAG CTT TTA GGC GTA ATC TGG GAC GTC GTA TGG GTA TTG TAC CGA GTC GGG GAT TA-3′ |
| orf144-5′repair-EcoRI primer2 | 124852-124872 | 5′-GAA TTC TTC GCC GCC GGT CCG TTT GAC-3′ |
| orf144-3′repair-HindIII primer2 | 126181-126226 | 5′-AAG CTT TTA GGC GTA ATC TGG GAC GTC GTA TGG GTA TTT ATT AAA ATT ATA TAT ATT AAA CCC TGA TGT TAC AGA ATT TGA G-3′ |
The underlined regions indicate sequence homology with the chloramphenicol resistance gene from plasmid pKD3.
The underlined regions indicate DNA sequences that were used to generate a HA epitope fusion tag, EcoRI, or HindIII restriction sites.
Bacmid purification and transfection
Bacmid DNA was purified from 0.5 L cultures using the plasmid Maxi plasmid DNA purification kit (Qiagen). Equimolar amounts (~2 μg) of purified bacmid DNA were transfected into Sf-9 cells (0.9 × 106/well) seeded in a six-well plate using a cationic liposome method (Campbell, 1995). Bacmid DNA was mixed with 200 μl of Sf-900 II medium containing 10 μl of liposomes and incubated at 27°C for 30-45 minutes. After incubation, the DNA solution was increased to 1 ml with SF 900 II medium, overlaid onto freshly plated Sf-9 cells, and transferred to 27°C for 4 h. After incubation, the transfection media was removed and the cells were replenished with 2 ml of fresh SF-900 II medium and returned to 27°C.
Quantitative DNA replication assay
To assess viral DNA replication, a quantitative real-time PCR assay was performed. Sf-9 cells were transfected as described above and at the indicated time-point the cells were harvested in 1 ml PBS, collected by centrifugation at 5000 rpm for 3 min, lysed in 200 μl cell lysis buffer (10 mM Tris-pH 8.0, 100 mM EDTA, 20 μg/ml RNAase A, 0.5% SDS) and incubated for 30 min at 37° C before adding 80 μg/ml of proteinase K and continuing incubation overnight at 65°C. Total DNA was extracted with 200 μl phenol-chloroform followed by 200 μl chloroform, and the aqueous layer was removed and diluted to 500 μl total volume. Prior to PCR, 5 μl of total DNA from each time-point was digested with 2 units of DpnI restriction enzyme (Fermentas) overnight in 20 μl total reaction volume. Quantitative PCR was performed with 2 μl of the digested DNA added to Platinum SYBR Green qPCR SuperMix UDG (Invitrogen) according to the manufacturer’s instructions and with primers and conditions described previously (Vanarsdall, 2006)
Virus growth curve
To determine the titers of virus supernatants, Sf-9 cells were transfected with the appropriate bacmid DNA (~2 μg/well), the cell culture supernatants from various time-points were collected, and the budded virus titers were determined using a TCID50 end-point dilution assay (O’Reilly et al., 1992). Virus infection was determined by cytopathology and by monitoring EGFP expression by fluorescence microscopy.
Budded virus purification and Western blotting
For purification of BV encoding the HA-tagged orf of interest, 100 mls of Sf-9 cells at a density of 1.5 × 106/ml were infected with a repair virus at an MOI of about 2. At 96 hours post-infection, the cells were harvested and collected by centrifugation at 5000 × g for 10 min. The clarified supernatants were then loaded onto a 30% sucrose cushion and ultracentrifuged at 24K RPM at 4°C for 75 min with a SW28.1 rotor. The BV pellet was resuspended in 500 ml of PBS supplemented with 1% NP-40 and rocked gently at room temperature to remove the budded virus envelope. The samples were then loaded onto a 25%–60% sucrose gradient and centrifuged at 32K RPM @ 4°C for 2 hours with a SW 50.1 rotor. After the final centrifugation, the envelope and capsid fractions were collected from the gradients. For Western blot analysis, BV fractions were analyzed by 10 % SDS-PAGE and the proteins transferred to PVDF membranes with a Bio-Rad semi-dry electrotransfer blotting apparatus according to the manufacture’s instructions. Blots were analyzed using a chemiluminescent detection method according to the manufacture’s instructions (Boehringer Mannheim). A monoclonal antibody to the HA-epitope (Covance) was used at a dilution of 1:2500, a monoclonal antibody to vp39 (a generous gift from Dr. Loy Volkman) was used at a dilution of 1:100, and a monoclonal antibody to the AcV5 epitope (Hohmann and Faulkner, 1983) was used at a dilution of 1:5000. A goat-anti-mouse-HRP secondary antibody (Promega) was used at a dilution of 1:2500.
Immunoelectron microscopy
Sf-9 cells were transfected as stated above, harvested at 72 h.p.t, and prepared for immunoelectron microscopy as described previously (Russell and Rohrmann, 1990). A mouse monoclonal antibody to AcMNPV vp39 was used as undiluted tissue culture supernatant. A goat anti-mouse IgG 10 nm gold secondary antibody was used at a dilution of 1:50. Images were obtained with a Phillips EM 300 electron microscope.
Acknowledgments
This research was supported by National Institutes of Health Grant GM060404 (to G. F. R.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Afonso CL, Tulman ER, Lu Z, Balinsky CA, Moser BA, Becnel JJ, Rock DL, Kutish GF. Genome Sequence of a Baculovirus Pathogenic for Culex nigripalpus. J Virol. 2001;75:11157–65. doi: 10.1128/JVI.75.22.11157-11165.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayres MD, Howard SC, Kuzio J, Lopez-Ferber M, Possee RD. The complete DNA sequence of Autographa californica nuclear polyhedrosis virus. Virology. 1994;202:586–605. doi: 10.1006/viro.1994.1380. [DOI] [PubMed] [Google Scholar]
- Belyavskyi M, Braunagel SC, Summers MD. The structural protein ODV-EC27 of Autographa californica nucleopolyhedrovirus is a multifunctional viral cyclin. Proc Natl Acad Sci. 1998;95:11205–10. doi: 10.1073/pnas.95.19.11205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bideshi DK, Federici BA. The Trichoplusia ni granulovirus helicase is unable to support replication of Autographa californica multicapsid nucleopolyhedrovirus in cells and larvae of T. ni. J Gen Virol. 2000;81:1593–9. doi: 10.1099/0022-1317-81-6-1593. [DOI] [PubMed] [Google Scholar]
- Braunagel SC, Guidry PA, Rosas-Acosta G, Engelking L, Summers MD. Identification of BV/ODV-C42, an Autographa californica nucleopolyhedrovirus orf101-encoded structural protein detected in infected-cell complexes with ODV-EC27 and p78/83. J Virol. 2001;75:12331–8. doi: 10.1128/JVI.75.24.12331-12338.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braunagel SC, He H, Ramamurthy P, Summers MD. Transcription, translation, and cellular localization of three Autographa californica nuclear polyhedrosis virus structural proteins: ODV-E18, ODV-E35 and ODV-EC27. Virology. 1996a;222:100–114. doi: 10.1006/viro.1996.0401. [DOI] [PubMed] [Google Scholar]
- Braunagel SC, He H, Ramamurthy P, Summers MD. Transcription, translation, and cellular localization of three Autographa californica nuclear polyhedrosis virus structural proteins: ODV-E18, ODV-E35, and ODV-EC27. Virology. 1996b;222:100–14. doi: 10.1006/viro.1996.0401. [DOI] [PubMed] [Google Scholar]
- Braunagel SC, Russell WK, Rosas-Acosta G, Russell DH, Summers MD. Determination of the protein composition of the occlusion-derived virus of Autographa californica nucleopolyhedrovirus. Proc Natl Acad Sci. 2003;100:9797–802. doi: 10.1073/pnas.1733972100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell MJ. Lipofection reagents prepared by a simple ethanol injection technique. Biotechniques. 1995;18:1027–32. [PubMed] [Google Scholar]
- Datsenko KA, Wanner BL. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci. 2000;97:6640–5. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goley ED, Ohkawa T, Mancuso J, Woodruff JB, D’Alessio JA, Cande WZ, Volkman LE, Welch MD. Dynamic nuclear actin assembly by Arp2/3 complex and a baculovirus WASP-like protein. Science. 2006;314:464–7. doi: 10.1126/science.1133348. [DOI] [PubMed] [Google Scholar]
- Harwood SH, Li L, Ho PS, Preston AK, Rohrmann GF. AcMNPV late expression factor-5 interacts with itself and contains a zinc ribbon domain that is required for maximal late transcription activity and is homologous to elongation factor TFIIS. Virology. 1998;250:118–34. doi: 10.1006/viro.1998.9334. [DOI] [PubMed] [Google Scholar]
- Hohmann AW, Faulkner P. Monoclonal antibodies to baculovirus structural proteins: determination of specificities by western blot analysis. Virology. 1983;125:432–444. doi: 10.1016/0042-6822(83)90214-3. [DOI] [PubMed] [Google Scholar]
- Luckow VA, Lee SC, Barry GF, Olins PO. Efficient generation of infectious recombinant baculoviruses by site-specific transposon-mediated insertion of foreign genes into a baculovirus genome propagated in Escherichia coli. J Virol. 1993;67:4566–79. doi: 10.1128/jvi.67.8.4566-4579.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Reilly DR, Miller L, Luckow VA. Baculovirus expression vectors : a laboratory manual. xiii. W.H. Freeman; New York: 1992. p. 347. [Google Scholar]
- Oomens AG, Blissard GW. Requirement for GP64 to drive efficient budding of Autographa californica multicapsid nucleopolyhedrovirus. Virology. 1999;254:297–314. doi: 10.1006/viro.1998.9523. [DOI] [PubMed] [Google Scholar]
- Russell RL, Rohrmann GF. A baculovirus polyhedron envelope protein: immunogold localization in infected cells and mature polyhedra. Virology. 1990;174:177–84. doi: 10.1016/0042-6822(90)90066-z. [DOI] [PubMed] [Google Scholar]
- Russell RLQ, Funk CJ, Rohrmann GF. Association of a baculovirus encoded protein with the capsid basal region. Virology. 1997;227:142–152. doi: 10.1006/viro.1996.8304. [DOI] [PubMed] [Google Scholar]
- Vanarsdall AL, Okano K, Rohrmann GF. Characterization of a baculovirus with a deletion of vlf-1. Virology. 2004;326:191–201. doi: 10.1016/j.virol.2004.06.003. [DOI] [PubMed] [Google Scholar]
- Vanarsdall AL, Okano K, George F, Rohrmann Characterization of the role of VLF-1 in baculovirus capsid structure and DNA processing. J Virol. 2006;80:1724–33. doi: 10.1128/JVI.80.4.1724-1733.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanarsdall AL, Mikhailov VS, Rohrmann GF. Characterization of a baculovirus lacking the DBP (DNA-binding protein) gene. Virology. 2007 doi: 10.1016/j.virol.2007.03.024. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vialard JE, Richardson CD. The 1,629-nucleotide open reading frame located downstream of the Autographa californica nuclear polyhedrosis virus polyhedrin gene encodes a nucleocapsid-associated phosphoprotein. J Virol. 1993;67:5859–5866. doi: 10.1128/jvi.67.10.5859-5866.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volkman LE. Autographa californica MNPV nucleocapsid assembly: inhibition by cytochalasin D. Virology. 1988;163:547–53. doi: 10.1016/0042-6822(88)90295-4. [DOI] [PubMed] [Google Scholar]