

Abstract
The relative gas-phase stabilities of seven quadruplex DNA structures, [d(TG4T)]4, [d(T2G3T)]4, [d(G4T4G4)]2, [d(T2AG3)2]2, d(T2AG3)4, d(T2G4)4, and d(G2T4)4, were investigated using molecular dynamics simulations and electrospray ionization mass spectrometry (ESI-MS). MD simulations revealed that the G-quadruplexes maintained their structures in the gas phase although the G-quartets were distorted to some degree and ammonium ions, retained by [d(TG4T)]4 and [d(T2G3T)]4, played a key role in stabilizing the tetrad structure. Energy-variable collisional activated dissociation was used to assess the relative stabilities of each quadruplex based on E1/2 values, and the resulting order of relative stabilities was found to be [d(TG4T)]4 ≫ d(T2AG3)4 ∼ d(T2G4)4 > [d(T2G3T)]4 > [d(T2AG3)2]2 ∼ d(G2T4)4 ∼ [d(G4T4G4)]2. The stabilities from the E1/2 values generally paralleled the RMSD and relative free energies of the quadruplexes based on the MD energy analysis. One exception to the general agreement is [d(G4T4G4)]2 which had the lowest E1/2 value, but was determined to be the most stable quadruplex according to the free energy analysis and ranked fourth based on the RMSD comparison. This discrepancy is attributed to differences in the fragmentation pathway of the quadruplex.
Keywords: quadruplex DNA, mass spectrometry, molecular dynamics
Introduction
The basis of many anticancer and antitumor therapies is the interaction between small molecule drugs and nucleic acid structures. While most current DNA-interactive therapies target duplex DNA, G-quadruplex DNA has attracted recent interest as a potential anti-cancer drug target because of its role in telomere maintenance (1-3). Telomeres are the regions of non-coding DNA found on the extremities of chromosomes (4). In addition to protecting the chromosomes from fusion and degradation, telomeres allow for the complete replication of the chromosomal DNA. With each subsequent cell division process, the length of the telomeres is shortened until a critical length is reached, leading to cell senescence and death (5). Telomerase is a reverse transcriptase enzyme that is responsible for maintaining the length of telomeric DNA. While this enzyme is inactive in most human somatic cells, high levels of telomerase activity is found in 80-90% of human cancer cells (6,7).
Telomeric DNA is composed of tandem repeats of G-rich sequences, such as the d(T2AG3)n sequence in mammals (8), d(T2G4)n in Tetrahymena (9) and d(T4G4)n in Oxytricha (10). These and other G-rich sequences have been shown to form G-quadruplex structures in vitro. G-quadruplex DNA forms via Hoogsteen hydrogen bonding between a planar arrangement of four guanine nucleobases, termed a G-quartet. In the quadruplex structure, the G-quartets stack on top of one another and the overall structure is stabilized by monovalent cations such as Na+, K+, or NH4+ coordinated in the central cavity of the tetrad (11). Structural polymorphism resulting from different quadruplex sequences and strand orientations has been demonstrated in vitro (12). A DNA sequence containing a single G-rich repeat can form a four-stranded parallel G-quadruplex. Strands containing two or more G-rich regions can form G-G hairpins and dimerize in different orientations to form a two-stranded quadruplex, while a sequence with four G-rich repeats can fold upon itself to form a intramolecular quadruplex (12).
The stabilization of the G-quadruplex structure of the 3′ single strand overhang of telomeric DNA by ligands and metal cations has been demonstrated to inhibit telomerase activity in vivo, thus stimulating interest in the study of telomeric quadruplex formation and the development of G-quadruplex selective ligands and anticancer compounds (13-15). Electrospray ionization mass spectrometry (ESI-MS) has emerged as a useful tool for the analysis of quadruplex DNA and its non-covalent complexes with ligands due to its low sample consumption and fast analysis time. During the ESI process, non-covalent complexes in solution are transferred to the gas-phase with minimal internal energy, allowing many of the binding interactions to be maintained. The preservation of these non-covalent complexes allows information about binding stoichiometry and selectivity to be elucidated from the mass spectra Our group (16,17) and others (18-24) have focused on developing ESI-MS based techniques to assess whether the binding of quadruplex interactive ligands observed in the gas-phase can be correlated to known solution behavior with the ultimate goal of developing ESI-MS as a screening tool for drug/DNA complexes.
There have been several ESI-MS studies aimed at examining structural features of duplex (25-27) and hairpin (28,29) DNA in solution and the gas-phase, primarily based on correlation of relative abundances of ions observed in ESI mass spectra as a function of solution conditions or based on collisional activated dissociation (CAD) patterns. CAD is a versatile technique that has been used to characterize gas-phase stabilities and fragmentation pathways of DNA duplexes (26,27,30,31). For example, the dissociation profiles of a series of 6-, 8- and 12-base pair self-complementary duplexes have been shown to correspond to the known solution melting behavior of DNA, demonstrating the utility of CAD to provide relative comparisons of the gas-phase stabilities of DNA duplexes (30). Fewer studies have been undertaken to elucidate the gas-phase structures or stabilities of quadruplex DNA (32-37). One recent study focused on using ion mobility mass spectrometry and molecular dynamics simulations to examine gas-phase conformations of quadruplexes with different lengths of the telomeric repeat d(TTAGGG)n where n = 1, 2, 4, and 6 (33). The experimental cross sections of the quadruplex structures determined by ion mobility matched those determined by molecular modeling, with the gas-phase structures also corresponding to solution structures unveiled by circular dichroism. Only one study has been devoted to the CAD fragmentation of DNA quadruplexes, and it found that [d(TGGGT)]4 ions dissociated via the loss of a single strand ion, producing a triplex species (32).
Molecular modeling techniques comprise another promising tool for the study of nucleic acid structures in a solvent free environment. Molecular dynamics (MD) simulations are now widely used for the detailed study of the structural, energetic, and dynamic properties of macromolecules, including proteins and nucleic acids. Recently, several research groups have studied G-quadruplexes with MD simulations in a solvent-free environment (18,24, 34-36). An interesting conclusion of those studies was that, in the proper charge state, G-quadruplex structures could be maintained with some distortion during the solvent-free MD simulations. With respect to G-quadruplex DNA, Rueda et al. found that quadruplexes not only remained stable in very extended (0.5 to 1μs) MD simulations in the presence of suitable cations, but also displayed structures that closely resembled those found in extended MD trajectories in aqueous solution (36). The additional finding that the collision cross sections of MD snapshots were in good agreement with those measured by ion mobility experiments (18, 24,33) indicates that both ion mobility and MD simulations are viable techniques for the evaluation of strand orientation patterns for G-quadruplexes. Baker et al. also found this approach to be useful for determining the binding patterns of ligands with G-quadruplexes (18). However, it should be noted that similar collision cross sections can be associated with extremely different conformations; therefore this method may not be used to measure how well a G-quadruplex maintains its G-quartets in the solvent-free environment of ESI-MS experiments.
In the present study, we use the capabilities of both ESI-MS/MS and molecular dynamics simulations to assess the relative stabilities of different quadruplex structures in the gas phase. First, tandem mass spectrometry, specifically collisional activation dissociation, was used to evaluate the fragmentation patterns of gas-phase quadruplex ions and create energy-variable dissociation curves which provide a way to compare the relative kinetic stabilities of different quadruplex structures. Second, the relative thermodynamic stabilities of G-rich sequences were assessed by using a molecular mechanical force field approach. Although molecular mechanics is capable of predicting the relative free energy difference between two conformations (such as different strand orientation patterns of a G-quadruplex) or the binding free energy for non-covalent binding, it is a very difficult to make free energy comparisons between different molecules because no common reference structure exists. To make this challenging problem tractable, a simple hypothesis was applied to qualitatively assess the relative stabilities of a set of G-rich DNA sequences. The basic strategy entails undertaking MD simulations in a solvent free environment with the starting structures of the “ideal” G-quadruplexes, either from the crystal or NMR structures or from models constructed with experimental structures as templates. Then both structural and energetic analyses were undertaken on the collected MD snapshots. We intended to investigate how well the ideal G-quadruplex structures were maintained in the solvent-free environment of ESI-MS experiments. The ability of G-rich sequences to form G-quadruplexes was then estimated by the root-mean square displacements (RMSD) as well as the free energy differences between the MD snapshots and the “ideal” G-quadruplexes. Although the above hypothesis is rather simple, it worked well for the G-rich sequences in this study.
Materials and Methods
Chemicals
Single strand oligodeoxynucleotides (ODNs), custom synthesized as ammonium salts on the 1.0 μmole scale with purification by HPLC, were obtained from Integrated DNA Technologies (Coralville, IA) and used without further purification. The sequences of the quadruplex-forming ODNs are shown in Table 1. Stock solutions of d(G4T4G4) (G3), d(T2AG3)2 (G4), d(T2AG3)4 (G5), d(T2G4)4 (G6), and d(G2T4)4 (G7) were prepared at 0.8 mM concentration in 150 mM ammonium acetate buffer while d(TG4T) (G1) and d(T2G3T) (G2) were prepared at 1 mM in 150 mM ammonium acetate buffer. Each quadruplex structure was annealed by heating the stock solutions to 90 °C for 10 min. followed by cooling to 25 ° C during a period of 7 h. The ammonium acetate buffer was chosen for the annealing solutions since quadruplexes form in the presence of the NH4+ ion, and NH4+ is more suitable than Na+ or K+ for ESI-MS studies due to its greater lability (32). Stock solutions of the duplex forming ODNs, d(GCG3A2T2G3CG) and d(CGC3A2T2C3GC) were prepared at 2 mM in 250 mM ammonium acetate. Equal volumes of each single strand were combined and the duplex was annealed using the same procedure described above for the quadruplex DNA. A 500 μM stock solution of the single strand ODN d(TTGGGGGT) was prepared in deionized water. N,N′-bis(2-morpholinylpropyl)-3,4,9,10-perylenetetracarboxylic acid diimide (Tel01) was prepared as previously reported (38). A 1 mM stock solution of the ligand was prepared in 0.1% aqueous trifluoroacetic acid.
Table 1.
Quadruplex sequences used in this study.
| Quadruplex | Sequence | Type of Quadruplex | Number of Tetrads | Molecular Weight (Da) |
|---|---|---|---|---|
| G1 | [d(TG4T)]4 | parallel 4-stranded24 | 4 | 7453.2 |
| G2 | [d(T2G3T)]4 | parallel or anti-parallel 4-stranded | 3 | 7353.0 |
| G3 | [d(G4T4G4)]2 | antiparallel 2-stranded32 | 4 | 7577.0 |
| G4 | [d(T2AG3)2]2 | parallel or antiparallel 2-stranded24 | 3 | 7513.0 |
| G5 | d(T2AG3)4 | anti-parallel intramolecular24 | 3 | 7575.0 |
| G6 | d(T2G4)4 | anti-parallel intramolecular | 4 | 7639.0 |
| G7 | d(G2T4)4 | anti-parallel intramolecular | 2 | 7438.9 |
Mass Spectrometry
ESI-MS experiments were undertaken on a ThermoFinnigan LCQ Duo mass spectrometer (San Jose, CA). Analytical solutions of the quadruplex were prepared at 10 μM concentration in 50 mM ammonium acetate buffer with 25% (v/v) methanol to aid in the desolvation of the DNA. Equimolar (10 μM) concentrations of Tel01 with each quadruplex, duplex or single strand ODN were prepared in the ammonium acetate buffer for the ligand binding study. The samples were directly infused into the mass spectrometer at 3 μL/min using a Harvard Syringe Pump (Holliston, MA). Ions were generated in the negative ion mode using a needle voltage of 3.5 kV, a heated capillary temperature of 110 °C and sheath and auxiliary gas flows of 40 and 10 arbitrary units, respectively. An ion accumulation time of 100 ms was used and spectra were acquired by summing 300 scans.
For collisional activated dissociation (CAD) experiments, the desired precursor ion was isolated in the trap using resonance ejection, followed by fragmentation induced by increasing the resonance voltage applied to the trap. An activation time of 30 ms was used for all experiments. To obtain the energy-variable dissociation curves, the applied CAD voltage, a parameter which influences the collision energy, was increased from 0.70 to 1.70 V in increments of 0.11 V. The data presented represent an average of three experiments done on three different days to account for possible instrument instability. Raw collisional activation voltages were extracted from the LCQTune software in which the normalized collision, along with parent mass, and tickle amplitude parameters, are used to calculate the amplitude of the resonance excitation voltage. Aside from the energy variable experiments, CAD was performed by increasing the collision voltage to the amplitude that resulted in dissociation of the precursor ion to 10% of its initial abundance.
Circular Dichroism
CD spectra were collected on a Jasco J-815 Circular Dichroism Spectrometer using a 1.0 cm pathlength quartz cell at 25 °C. The ellipticity of the quadruplex DNA was monitored from 220 to 330 nm. Each CD spectrum was averaged for four scans. The spectral contribution of ammonium acetate was subtracted using the software provided with the instrument. The quadruplex concentration was 10 uM in 50 mM NH4OAc.
Molecular Modeling
Starting Geometries
For the gas-phase MD simulations, the initial structure of G1 was taken from a parallel-stranded crystal structure resolved at 0.95 Å (PDB: 1S45) (39), and the starting structure of G3 was taken from an antiparallel-stranded crystal structure resolved at 1.86 Å (PDB: 1JB7) (40). For G4 - G7, a parallel-stranded crystal structure resolved at 2.1 Å (PDB 1KFI) (41) and an antiparallel-stranded NMR structure (PDB 143D) (42) were applied as templates to construct the initial structures of both strand orientations for each G-quadruplex. An additional starting structure for G7 was modeled by mutating G to T and T to G on the model structure of antiparallel G6. It should be noted that for a particular quadruplex our decision on which strand orientations to model was based on both experimental and theoretical findings. To investigate the role of cations in stabilizing the G-quartet, two models were built for G1, one with and one without ammoniums residing in the central channel. In total, twelve model structures, namely, G1-ammonium, G1-no-cation, G3-antiparallel, G4-antiparallel, G4-parallel, G5-antiparallel, G5-parallel, G6-antiparallel, G6-parallel, G7-antiparallel, G7-parallel, G7-T-tetrad, were used for the MD simulations. Modelling was not undertaken for quadruplex G2 due to computational time limitations. As demonstrated later, the omission of G2 does not change the conclusions drawn from this study, and the first six quadruplexes that were modelled were selected to span the range of potential structures.
Charge States
Although the most abundant charge state observed in ESI-MS experiments can be readily identified from the mass spectra, the exact locations of the deprotonation sites are not clear. Rueda et al. proposed two neutralization protocols to model the desired charge state for a G-quadruplex: a distributed model and a more realistic localized model (34-36). In the present study, we adopted a modified version of the localized model. Instead of maximizing the sum of the distances between the charged phosphates, we calculate electrostatic energies of all possible ways of assigning deprotonation sites and the assignment that gave the most favorable electrostatic energy was chosen for the subsequent molecular mechanical studies. It is noted that this assignment is among the top assignments determined by maximizing the sum of the distances between the charged phosphates. If a G-quadruplex has ammonium adducts, a set of three-dimensional fine grids (0.2 Å) that enclosed the quadruplex was constructed. Grid points that were within 2.0 Å of any atom of the quadruplex were omitted. A +1 charge probe was placed at each grid point to calculate the electrostatic energy between the point charge and the quadruplex. Finally, an ammonium ion was placed at the grid point that gave the optimum electrostatic energy. This procedure was repeated n times to incorporate n ammonium ions. According to the recent work of Rueda et al. (34-36) and Baker et al. (24,33), quite similar results were obtained for different assignments of charged nucleotides for specific charge states. In the present work, only the best deprotonation assignment predicted by our localized model is subjected to further studies for each G-quadruplex model.
Force field
The molecular mechanical force field is the cornerstone of MD simulations. Many force fields are parameterized to reproduce ab initio gas-phase energies, but are mainly used to study molecular systems in aqueous solution explicitly or implicitly. It is therefore not a surprise that many successful gas-phase MD studies have been conducted with those force fields. For example, AMBER force fields have been extensively applied by Rueda et al. in their gas phase nucleic acid simulations (34-36). In this work, all of the minimizations, MD simulations and subsequent structural and energetic analyses were carried out with the AMBER 8 suite of programs (43) using the Parm99 force field (44). Considering that the protonated nucleotides are not “standard” residues in the force field library, we constructed a model molecule (two deoxyribose groups linked by a neutral phosphate group at positions 3′ and 5′, respectively), which mimics the chemical environment of phosphate in neutral nucleotides, to re-derive the charges of phosphate functional group atoms (P, O1P, H1P, O2P, O3′ and O5′). We followed the same AMBER restrained electrostatic potential charge (RESP) procedure to reassign the charges of the neutral phosphate group. Finally, the HF/6-31G* RESP point charges of the neutral phosphate group is listed as the follows: P – 1.1659, O1P – -0.7003, H1P – 0.4705, O2P – -0.7294, O5′ – -0.1973, O3′ – -0.6549 for 3′-hydroxyl nucleotides and -0.5232 for other types of nucleotides, respectively.
Minimizations and MD simulations
A delicate minimization protocol was designed to minimize the initial structures step by step. (1) Only hydrogen atoms were optimized for 1000 steps. (2) A 1000-step minimization was carried out with the G-quartet guanine nucleosides restrained by a 50 kcal/molÅ2 harmonic potential. (3) The G-quartet guanine bases were restrained by a 50, 10, 5 and 1 kcal/molÅ2 harmonic potentials in a set of subsequent 1000-step minimizations. (4) 1000-step full minimization was carried out for the complete molecules. The fully minimized structures were then subjected to a set of MD simulations. Similar to the restrained minimizations, the G-quartet guanine bases were restrained by a 10, 5, 1, 0.5, 0.1 kcal/molÅ2 harmonic potential in a set of subsequent 100 picosecond restrained MD simulations. In each step, the last snapshot was taken as the starting structure for the next simulation run. Finally, a completely unrestrained 50 nanosecond MD simulation was performed with a time step of 1 femtosecond. All the simulations were carried out at 300 K using a weak-coupling algorithm (45). The translational and rotational center-of-mass motion was removed every 1000 steps. No non-bonded cutoff was applied and the SHAKE algorithm was applied for all the bonds involving hydrogen atoms (46). We found these minimization and MD simulation schemes were able to avoid substantial conformational changes triggered by unbalanced forces.
Structural and Energetic Analysis
A large variety of geometrical analyses including root-mean-square displacements, hydrogen bonding, stacking between two guanines, and free energy analysis, were performed for 50 snapshots evenly sampled from the last 30 nanosecond simulations. Hydrogen bonds were formed when the heteroatom-heteroatom distance was less than 4.0 Å and the donor-hydrogen/donor-acceptor angle was deviated less than 60° from linearity (the default criteria of hydrogen-bond formation in the Carnal program of AMBER 8 (43)). Vertical base stacking is a significant stabilizing interaction of DNA structures. Although the stabilization of base stacking involves both van der Waals and electrostatic interaction within the adjacent rings, a geometrical definition, proposed by Gabb et al. (47) is widely used to detect stacking interaction. In this work, a slightly relaxed criteria of stacking interaction in the Gabb's method was applied: a stacking interaction was recognized when the distance between two base rings was less than 5.5 Å, the angle between the two normals to the base planes was less than 30°, and the angle between the normal of any base plane and the vector between the centers of the two bases was less than 50°. The numbers of stacking pairs were 4, 8 and 12 for an ideal G-quadruplex with two, three and four tetrads, respectively.
The free energy of a molecule was composed of two parts: the molecular mechanical energy (EMM) and the conformational entropy (TS). The EMM energies were calculated with the Anal program of AMBER 8 on the minimized structure of each snapshot (43), while the conformational entropies were estimated by normal model analysis on a thoroughly minimized structure with the Nmode program of AMBER 8 (43). As discussed above, free energy analysis was essential in interpreting the CAD experimental data.
Results and Discussion
Mass Spectra of G-quadruplexes
The quadruplex structures selected for this study represent a variety of quadruplex-forming sequences and strand orientations. The variation allows one to evaluate which structures are most stable in the gas-phase and thus most suitable for future ESI-MS studies of quadruplex DNA. Sequences G1 and G2 were selected to form four-stranded quadruplex structures with four and three quartets, respectively. Previous circular dichroism (CD) studies performed on G1 in an ammonium acetate buffer indicate that it forms a parallel, four stranded quadruplex structure (33). Both G3 and G4 contain two G-rich regions per strand and form two-stranded quadruplex structures. CD studies have found that G3 forms an antiparallel structure (32) while the CD spectrum for G4 has characteristics of both parallel and antiparallel orientations (33). The intramolecular quadruplex-forming sequences selected for this study, G5, G6, and G7, contain four repeats each of G-rich sequences capable of forming three, four, and two quartets, respectively. The major species in the CD spectrum of G5 in an ammonium acetate buffer is the antiparallel quadruplex, with evidence that the parallel quadruplex is also present to a lesser degree (33). The CD spectra of the other quadruplexes were measured in the present study and indicated that they are: G2: parallel or anti-parallel; G6 and G7: both anti-parallel. All quadruplexes in the present study contain 24 nucleobases to ensure that the molecular weights and degrees of freedom of the DNA molecules were comparable for the energy-variable dissociation studies.
Full scan ESI mass spectra of solutions containing each quadruplex were obtained to evaluate the annealing efficiencies. Representative spectra are shown in Figure 1 for the four-stranded quadruplex G1 (Figure 1a), the two-stranded structure G3 (Figure 1b), and the intramolecular quadruplex G5 (Figure 1c). The 5- charge state proved to be the most abundant for all of the quadruplex structures, and thus was used for all subsequent CAD and variable-energy dissociation experiments. The 5- ions of intermolecular quadruplexes G1 (Figure 1a) and G2 (spectra not shown) can also be assigned unambiguously as four stranded structures, while G3 (Figure 1b) and G4 can be defined as two-stranded structures for G3 and G4 based on the m/z values which are distinctive for species containing one, two, three or four strands.
Figure 1.

ESI mass spectra of solutions containing 10 μM of (a) G1, (b) G3, and (c) G5.
Ammonium adducts are also associated with the four-stranded quadruplexes G1 and G2. The dominant 5- ion of G1 contained three ammoniums (Figure 1a) while the corresponding ion of G2 contained two adducted ammoniums (spectra not shown). These results are consistent with the previous observation that n-1 ammonium cations are incorporated in quadruplex structures containing n tetrads (32). As shown in Figure 1b and 1c, no ammoniums are associated with the most abundant ions for the two-stranded G3 and single stranded G5 quadruplex. Similar results were obtained for other two-stranded quadruplex G4 and the single stranded G6 and G7 (spectra not shown). In addition, an ion corresponding to the two-stranded G3 structure with two ammoniums is present in the mass spectra but it is approximately one third the abundance of the dominant ion without adducted ammonium (Figure 1b). These results are consistent with previous studies of quadruplexes by ESI-MS that found adducted ammoniums are only retained by the dominant quadruplex species for the four-stranded quadruplex G1, suggesting tighter binding of the ammonium ions by this structure (32,33).
While Figure 1 confirms that ions containing strand stoichiometries consistent with quadruplex structures for G1 and G3 are present, it does not verify that the quadruplex structures are maintained in the gas-phase nor does it indicate whether G5 is in the folded quadruplex conformation or a denatured state. ESI-MS studies of quadruplexes have been largely limited to using multiple stranded quadruplexes because the m/z of an intramolecular quadruplex is the same as a denatured single strand structure, thereby precluding direct confirmation that the quadruplex is maintained in the gas-phase. While recent studies have confirmed that quadruplex structures are conserved in the gas-phase, they did not provide information about the relative stabilities of the structures (33). Here, molecular dynamics studies were carried out to demonstrate that quadruplex structures can be maintained in the gas-phase, while further calculations and CAD experiments were used to assess quadruplex stability.
Ligand Binding of the Quadruplexes
The binding of the quadruplexes to one quadruplex-selective ligand was used to demonstrate that the quadruplexes were present in the annealed solutions. In a previous study we demonstrated that that the perylene diimide N,N′-bis(2-morpholinylpropyl)-3,4,9,10-perylenetetracarobxylic acid diimide (Tel01) (38) demonstrated binding selectivity for quadruplexes over duplex and single strand DNA by forming significantly lower abundance complexes with duplex DNA and exhibited no binding to single strand DNA (17). Here, we examined the binding of Tel01 to the quadruplexes, one duplex, and one single strand oligonucleotide by ESI-MS. Solutions containing equimolar concentrations of the DNA and ligand were prepared and analyzed by ESI-MS. The fraction of bound DNA values were determined from the mass spectra as described previously (17), and the results are summarized in Table 2. Tel01 formed abundant complexes with each quadruplex, with fraction of bound DNA values ranging from 0.80 for G6 to 0.97 for G2. These values are on par with the results of our previous study (17). The binding of Tel01 to a duplex d(GCGGGAATTGGGCG/CGCCCAATTCCCGC) (DS1) was indicated by the presence of Tel01/DS1 complexes in the mass spectrum, but they are significantly lower in abundance than the complexes formed between the ligand and the quadruplexes. Tel01 also did not form any complexes with the single strand d(TTGGGGGT) (SS1). This sequence was selected since it is G-rich like many of the quadruplexes, however it was not annealed and ESI-MS experiments were done to confirm that only the single strand species was present in solution (spectra not shown). Had the quadruplexes not properly annealed and remained in the denatured single strand conformation, little or no Tel01 binding would be expected. These results confirm that the quadruplexes are present in solution and that the ESI-MS results reflect the anticipated quadruplex selectivity.
Table 2.
Fraction of bound DNA values for equimolar concentrations (10 μM) of each DNA with Tel01.
| DNA Sequence | Fraction DNA bound to Tel01a |
|---|---|
| G1 | 0.95 |
| G2 | 0.97 |
| G3 | 0.84 |
| G4 | 0.93 |
| G5 | 0.93 |
| G6 | 0.80 |
| G7 | 0.95 |
| DS1 | 0.20 |
| SS1 | 0.00 |
All error values less than or equal to 0.05.
Molecular dynamics trajectories of the quadruplexes in the gas-phase
Molecular dynamics simulations were undertaken to demonstrate that the quadruplex conformations are maintained under conditions used for the ESI-MS/MS experiments. A net charge of 5- was assigned to the twelve model quadruplexes since this was the most abundant charge state observed in the ESI-MS experiments. Plots of the RMSD values of the tetrad forming guanines versus simulations time were constructed using the final minimized structure as a reference point. In most cases, the trajectories fluctuated vigorously during the first few nanoseconds and then became quite stable after 20 nanoseconds as demonstrated by the plots for G1 shown in Figure 2. Similar plots for the other quadruplexes are provided in the supporting material (Figure S1). The reference structures for the comparisons are the last structures of a series of minimizations which have their G-quartets well maintained. This behavior is consistent with what Rueda et al. found in their simulations of DNA in the gas phase (34-36).
Figure 2.

Root-mean-square displacements (Å) of G-tetrad guanines over 50 nanosecond MD simulations (compared to minimized reference structures) for two G1 models: (a) G1 with three ammoniums residing in the central cavity between the stacked tetrads, and (b) G1 in the absence of ammoniums in the central cavity).
The final snapshots of the G1 simulations with and without the ammonium ions are depicted in Figure 3. From Figures 3a and 3b, it is clear that three ammonium cations (blue) reside in the centers of two adjacent tetrad planes. Furthermore, a comparison of Figures 3b and 3d shows the impact of the ammonium ions on the G-tetrad stabilization as the deviations in the tetrad position are more pronounced when the ammoniums are absent. The final snapshots of the structures with the most energetically favorable strand orientations of G3-G7 are presented in Figure 4 (snapshots of the less favorable structures are shown in the supporting material Figure S2). The antiparallel structures of G3, G4, G5, and G7 and the parallel structure of G6 were identified as the most favorable strand orientations of the quadruplexes in the gas-phase. One conclusion of those MD simulations is that the G-quartets of the more favorable strand orientations or stoichiometries of G1, G3, G4, G5, G6 and G7 are maintained with large or small distortions in the solvent-free environment within the 50 ns MD simulation time frame. Although running much longer MD simulations can be helpful to make more reliable judgments on the stability of the G-quadruplexes, in our opinion, it is unlikely to lead to strikingly different conclusions.
Figure 3.
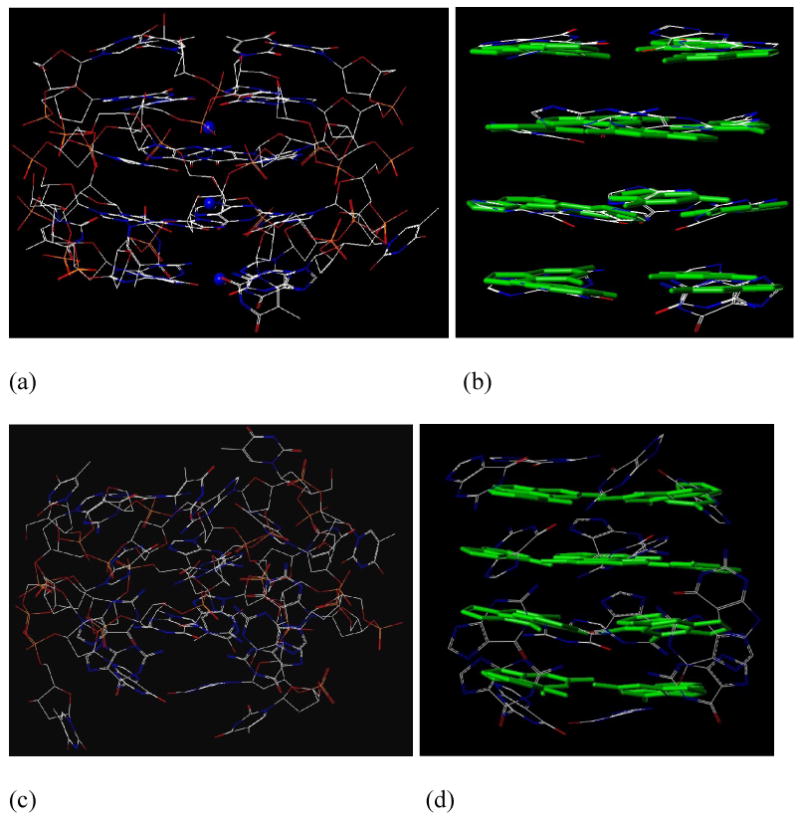
The final snapshot of a 50 nanosecond MD simulations for G1: (a) an all heavy atom model with three ammoniums residing in the central channel between two adjacent planes (G1-ammonium), (b) G-quartets of the final snapshot of G1-ammonium (the green capped sticks of the G-quartets in the reference structure are presented for comparison purposes), (c) an all heavy atom model in the absence of ammonium ions (G1-no-ion), and (d) G-quartets of the final snapshot of G1-no-ion (the green capped sticks of the G-quartets in the reference structure are presented for comparison purposes).
Figure 4.
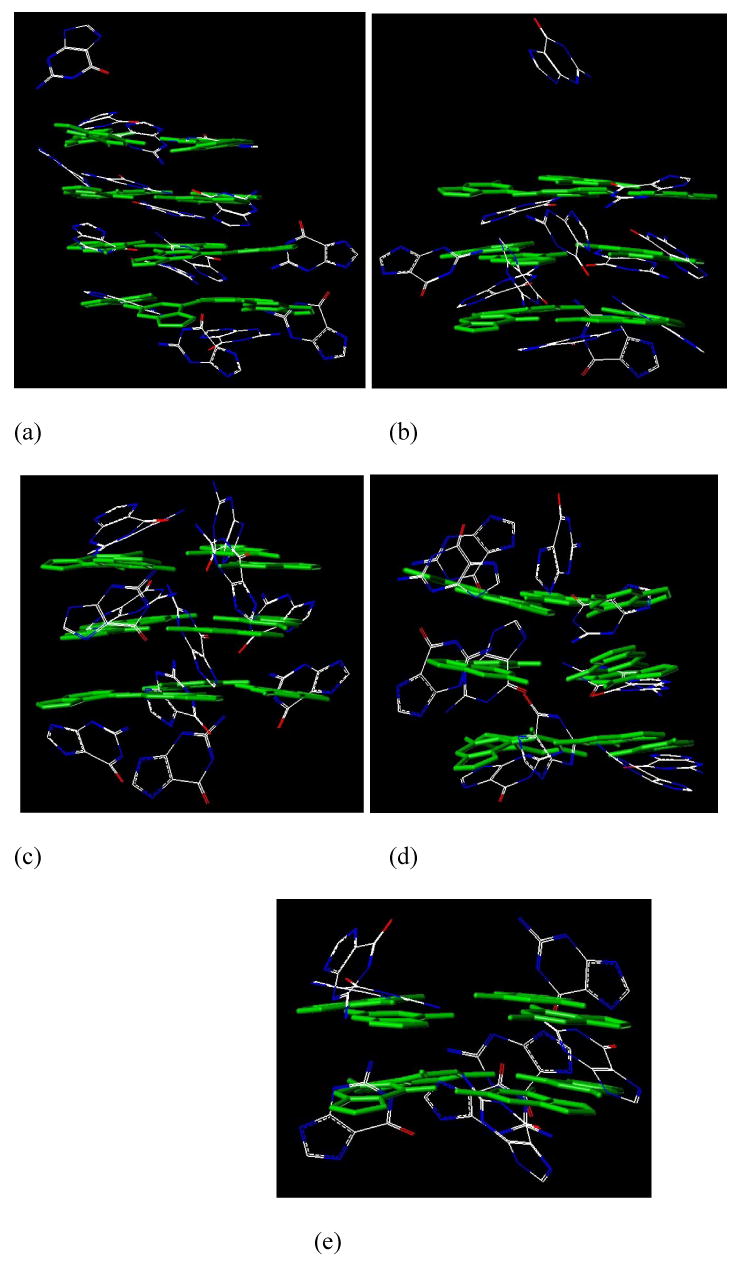
The G-quartets of G3, G4, G5, G6, and G7 in the final snapshots of 50 nanosecond MD simulations. Only the energetically more favorable strand orientations are presented: (a) G3-antiparallel, (b) G4-antiparallel, (c) G5-antiparallel, (d) G6-parallel, and (e) G7-antiparallel. The green capped sticks of the G-quartets in the reference structure are presented for comparison purposes.
CAD Fragmentation Pathways of the Quadruplexes
While significant attention has been devoted to examining the fragmentation pathways of DNA duplexes of different length, sequence and base pair composition (26,30,51,52), fewer studies have focused on quadruplexes. Thus, CAD spectra of G1, G2, G3, G4, G5, G6, and G7 (5- charge state) were obtained to determine how different quadruplex strand stoichiometries and sequences affect the fragmentation patterns. The quadruplex ions dissociate via the loss of a nucleobase, strand separation, or a combination of the two (Figure 5). The primary fragmentation route of the four-stranded quadruplexes G1 and G2 was strand separation to produce a triplex ion and single strand species in the 3- and 2- charge states, respectively. Guanine base loss from the resulting triplex species was also observed as demonstrated by the CAD spectrum of [(G1)4]5- shown in Figure 5a.
Figure 5.

CAD spectra of the 5- charge state of: (a) [(G1)4], collision energy 1.55V, (b) [(G3)2], collision energy 1.25 V, (c) [(G4)2], collision energy 1.25 V, and (d) [G5] collision energy 1.40 V.
In contrast to the four-stranded quadruplexes, the two-stranded quadruplexes dissociate primarily through guanine base loss from the precursor ion, although some strand separation products are also present. Interestingly, different degrees of strand separation are observed in the CAD spectra of G3 (Figure 5b) and G4 (Figure 5c) even though an applied collision voltage of 1.25 V was used for each ion. The relative abundances of the single strand ions in the CAD spectrum of G3 are significantly lower than those of the single strand ions in the spectrum of G4, suggesting the different sequences or orientations of the quadruplexes impact the fragmentation pathways. G3 has two repeats of four guanine nucleobases, allowing it to form a quadruplex containing four G-tetrads, while the sequence of G4 allows it to form only three tetrads. With a greater number of tetrads, there are more Hoogsteen hydrogen bonds holding the single strands of G3 together, making covalent cleavage of guanines favored over non-covalent strand separation. This result is reminiscent of an increase in nucleobase loss observed in the CAD spectra of DNA duplexes that parallels the total number of interstrand hydrogen bonds, either by increasing the GC content of the sequence and/or lengthening the duplex (26,30,51,52).
Guanine nucleobase loss is also the primary fragmentation pathway of the intramolecular quadruplexes, as demonstrated by the CAD spectrum of G5 in Figure 5d. Similar spectra were acquired for G6 and G7 (data not shown). This result was expected since the quadruplex is single stranded, and thus cannot undergo strand separation. The CAD spectra of the intramolecular quadruplexes resemble those of single strand ions that have not formed a quadruplex, and thus give no diagnostic information about differences in the binding interactions between the quadruplexes.
Energy-variable dissociation studies
While there are some general differences in the CAD spectra of the four, two and single stranded quadruplexes, the fragmentation pathways alone do not provide insight into the relative stabilities of the quadruplexes, nor can they distinguish the intramolecular structures (G5 G6, and G7) or the four-stranded structures (G1 and G2). To probe the relative kinetic stabilities of the quadruplexes in the gas-phase, energy-variable dissociation experiments were undertaken. The quadruplexes that dissociate via the same fragmentation pathway may require different amounts of energy to induce their dissociation as a result of different strand orientations and/or binding interactions. For this part of the study, the quadruplex ions in the 5- charge state were isolated and subjected to increasing collision energy based on the applied collisional activation voltage. The abundance of the precursor ion relative to the abundance of the product ions was monitored as a fraction of 1.0 and plotted as a function of CAD voltage. The reported E1/2 values are calculated by determining the CAD voltage at which the relative abundance of the precursor ion is reduced to 50% (27,30,51). A greater E1/2 value implies a more kinetically stable ion, one that requires greater energization to induce dissociation. A similar approach has been used for in-source CAD and thermal denaturation experiments of DNA duplexes (31).
The four-stranded quadruplexes G1 and G2 both dissociate via strand separation to produce single strand ions in the 2- charge state and triplex species with a 3- charge. However, the CAD voltages (E1/2 values) required to produce these ions are significantly different as demonstrated by the representative dissociation profiles shown in Figure 6. The E1/2 value for G1 was calculated to be 1.33 ± 0.05 V, while that of G2 was 1.14 ± 0.05 V, indicating G1 is substantially more kinetically stable in the gas phase than G2. This result is unsurprising since the sequence of G1 contains a single repeat of four guanines, allowing it to form four tetrads, while G2 can only form three tetrads. With four tetrads, there are more hydrogen bonds between the four strands (16 for G1 versus 12 for G2) and greater stacking interactions between the tetrads. The differences in binding interactions between the single strands in G1 versus G2 are reflected in the E1/2 values of the quadruplexes.
Figure 6.

Energy-variable dissociation curves for [(G1)4]5- and [(G2)4]5-.
The gas-phase stabilities of the intramolecular quadruplexes G5, G6, and G7 are also distinguished based on different E1/2 values. Unlike G1 and G2, the strand separation pathway is not possible for the intramolecular quadruplexes. Therefore, the emergence of base loss ions and the formation of an-B and w ions is diagnostic for the disruption of the quadruplex structures. In order to cleave the covalent bond between the guanine and the ribose and disassemble the quadruplex structure, both Hoogsteen hydrogen bonding as well as tetrad stacking interactions must be broken. The dissociation curves obtained for G5 and G6 (Figure S3) demonstrate that G5 and G6 have similar gas-phase stabilities, with E1/2 values of 1.20 ± 0.02 V and 1.18 ± 0.01 V, respectively, while G7 is significantly less kinetically stable with an E1/2 value of 1.10 ± 0.02. The differences in the gas-phase stabilities may be partially explained by the number of tetrads per structure. The least stable intramolecular quadruplex, G7, contains four repeats of two guanines, allowing for only two tetrads in the quadruplex structure, compared to three tetrads in G5 or four tetrads in G7. However the E1/2 values of G5 and G6 are approximately the same, even though G6 can form four G-tetrads and G5 can only form three. These results indicate there are also other factors governing the kinetic stabilities of the quadruplexes. As shown below, the structural and free energy analysis on the MD trajectories shed light on the trend of E1/2 values of the G-quadruplexes.
Energy-variable dissociation curves were also constructed for the two-stranded quadruplexes G3 and G4, both of which dissociate via strand separation and guanine base loss from the parent complex (Figure S4). Their E1/2 values are virtually the same within the margin of errors of the measurements. While both quadruplexes are two-stranded, G3 is able to form four G-tetrads while G4 can only form three, so one would expect a greater E1/2 value for G3. These results again suggest that other factors influence the apparent gas-phase stabilities of the quadruplexes, possibly strand orientation because G3 forms an antiparallel quadruplex, while G4 likely forms a mixture of parallel and antiparallel structures.
In addition to comparing E1/2 values amongst quadruplexes that have the same strand stoichiometries and thus fragmentation patterns, the E1/2 values of all quadruplexes were also compared relative to each other (Table 3). The E1/2 values for the quadruplexes fall into three main groups. G1, the parallel four-stranded quadruplex with four tetrads and three retained ammonium ions, has by far the largest E1/2. The quadruplexes with the next highest E1/2 values are two of the intramolecular quadruplexes with three and four tetrads, G5 and G6, respectively. The next grouping based on E1/2 values include the multistranded quadruplexes with three tetrads, G2 with four strands, and G4 with two strands, followed by the intramolecular quadruplex with two tetrads G7, and finally the two stranded quadruplex with four tetrads.
Table 3.
Summary of E1/2 values for the quadruplexes.
| Quadruplex | E1/2 Value | Number of Tetrads |
|---|---|---|
| G1 | 1.33 +/- 0.05 | 4 |
| G2 | 1.16 +/- 0.02 | 3 |
| G3 | 1.11 +/- 0.03 | 4 |
| G4 | 1.07 +/- 0.03 | 3 |
| G5 | 1.20 +/- 0.02 | 3 |
| G6 | 1.18 +/- 0.01 | 4 |
| G7 | 1.10 +/- 0.02 | 2 |
It is apparent that the E1/2 values are not solely artifacts of the DNA sequence. For example, G4 and G5 both contain four d(T2AG3) repeats, with all four on one strand for G5 and two on two different strands for G4. However, their E1/2 values are significantly different, with G5 having a greater E1/2 value. Base content alone also does not account for the trend apparent in Table 3, as both G1 and G6 both contain 16 guanines and 8 thymines in the quadruplex, but their E1/2 values are significantly different. It has also been demonstrated in the discussion above that quadruplexes with the same strand stoichiometries have different E1/2 values. For example, G5 and G7 could be distinguished by their E1/2 values, even though both form intramolecular quadruplexes. Therefore, we ascribe the differences in E1/2 values of the quadruplexes to differences in gas-phase stabilities of different quadruplex structures.
Molecular dynamics structural and energetic analysis
To provide insight into the relative stabilities of the quadruplexes determined by the energy-variable dissociation experiments, structure and free energy analysis were performed on the snapshots collected from the 50 ns MD simulations for twelve quadruplex models. As we pointed out in the introduction section, the root mean square displacements and the relative free energies between the “ideal” and the MD structures are two key parameters for evaluating the stabilities of G-rich DNA sequences. Since we were interested in the relative stabilities of the G-tetrads in the gas-phase MD simulations, only the G-tetrad atoms were used in least-square fittings to calculate the RMSD values, using the finally minimized structures as references (the “ideal” G-quadruplexes).
The RMSD of G-tetrads of the 50 MD snapshots in comparison to the finally minimized structures are listed in Table 4. The G1-ammonium quadruplex has the most stable MD trajectory and its mean RMSD to the ideal structure is only 0.66 Å, which is significantly smaller than all other model molecules including the G1-no-cation quadruplex. The molecular mechanical energies and the entropies, as well as the free energies of the finally minimized and the 50 MD snapshots of the twelve G-quadruplex models, are also listed in Table 4. There is more than one strand orientation for G4, G5, G6, and G7. According to the calculated free energies, the antiparallel strand orientation is more favorable for G5 and G7, and only slightly more favorable for G4. On the other hand, the free energy of parallel G6 is virtually the same as the antiparallel orientation, indicating that G6 probably forms a mixture of parallel and antiparallel structures.
Table 4.
Root mean square displacements of G-tetrad guanines and the free energies and their components of the finally minimized structures and 50 snapshots collected from the MD simulations. For a quadruplex, the energetically more favorable stoichiometry or strand orientation according to the free MD free energies is shown in bold.
| Quadruplex | Mean RMSD (Å) | Energy (kcal/mol) | Entropy (cal/mol/K) | Free Energy (kcal/mol) | |||
|---|---|---|---|---|---|---|---|
| Minimization | MD | Minimization | MD | Minimization | MD | ||
| G1-ammonium | 0.66 | -1840.9 | -1962.6 | 2132.7 | 2044.9 | -2480.7 | -2576.0 |
| G1-no-cation | 2.32 | -1680.6 | -1795.6 | 2112.4 | 2038.9 | -2314.3 | -2407.3 |
| G3-antiparallel | 3.22 | -1779.3 | -1868.3 | 2091.2 | 2049.5 | -2406.7 | -2483.2 |
| G3-antiparallel | 3.82 | -1593.8 | -1782.1 | 2210.2 | 2041.7 | -2256.9 | -2394.6 |
| G4-parallel | 2.96 | -1625.7 | -1782.5 | 2098.6 | 2016.8 | -2256.3 | -2387.5 |
| G5-antiparallel | 3.09 | -1645.2 | -1818.0 | 2212.0 | 2034.5 | -2308.8 | -2428.4 |
| G5-parallel | 2.23 | -1668.4 | -1803.1 | 2113.2 | 2038.0 | -2302.4 | -2414.5 |
| G6-antiparallel | 3.96 | -1719.8 | -1917.8 | 2194.2 | 2053.1 | -2378.1 | -2533.4 |
| G6-parallel | 2.56 | -1773.4 | -1921.9 | 2125.9 | 2041.8 | -2411.1 | -2534.4 |
| G7-antiparallel | 3.58 | -1174.6 | -1534.1 | 2259.3 | 2042.1 | -1852.4 | -2146.7 |
| G7-parallel | 2.70 | -1225.2 | -1514.6 | 2313.0 | 2034.9 | -1919.1 | -2125.1 |
| G7-T-tetrad | 4.44 | -1332.7 | -1515.8 | 2083.9 | 2026.7 | -1957.9 | -2123.8 |
The relative free energies between the finally minimized and the MD structures are listed in Table 5. The hydrogen bonding and stacking interaction analysis on the MD trajectories were performed, and the results are presented in Table S1 in the supporting material. The overall conclusion is that the number of stacking interactions and the number of hydrogen bonds among guanine tetrads slightly decrease in comparison to the final minimized structures, while the number of hydrogen bonds formed between guanine tetrads and other atoms increases significantly.
Table 5.
The relative free energies between the last minimized and MD structures. For each quadruplex, only the energetically more favorable stoichiometry or strand orientation is considered.
| Quadruplex | Relative Free Energy (kcal/mol) |
|---|---|
| G1-ammonium | 95.3 |
| G3-antiparallel | 93.0 |
| G4-antiparallel | 137.7 |
| G5-antiparallel | 119.6 |
| G6-parallel | 123.3 |
| G7-antiparallel | 165.9 |
Relative Stability Prediction of G-rich DNA Sequences
In theory, there is almost no way to evaluate the relative stabilities of different DNA sequences with the molecular mechanical approach. To make this challenging problem tractable, we proposed a simple hypothesis to qualitatively assess the relative stability of the six G-quadruplexes. In short, the smaller the RMSD values and the relative free energies of the MD snapshots compared to the finally minimized structures, then the smaller the distortion of the G-quartets and the more stable the G-quadruplex and the higher the E1/2 value.
If only the mean RMSD values are considered, the rank order of stability is: G1 > G6 > G5 > G3 > G7 > G4, whereas the rank order becomes G3 > G1 > G5 > G6 > G4 > G7 when the relative free energies are taken into account. We thus conclude that G1 and G3 are more stable than G5 and G6, and G5 and G6 are more stable than G4 and G7. It should be noted that the above comparisons were made only for the energetically more favorable stoichiometry or strand orientation of each quadruplex, and thus the rank order may change if one considers the contribution of other strand orientations. It is still debatable whether RMSD can be applied to assess the stability of a protein or DNA sequence. Some correlations are found between the RMSD and free energy for a set of protein decoys, especially when the decoys are not drifted away from the experimental structures (<5 Å). As for our case, all RMSD values are smaller than 4.0 Å except for the G7-T-tetrad, for which the reference structure is not a G-quadruplex at all. Moreover, the overall ranks predicted by the two kinds of parameters are consistent. Therefore we believe that both RMSD and relative free energy are useful parameters to assess the relative stability of G-quadruplexes.
The conclusions about the relative quadruplex stabilities determined by RMSD and relative free energy results correlate well with the rankings determined by the energy variable CAD experiments. One exception to the general agreement is the result for G3. This quadruplex had the lowest E1/2 value; however, it was determined to be the most stable quadruplex according to the free energy analysis and ranked fourth based on the RMSD comparison. This discrepancy is attributed to the fragmentation pathways of this particular structure in the gas phase. Previous studies of the fragmentation mechanisms of single strand and duplex oligonucleotides have found that base loss and subsequent back bone fragmentation is initiated by protonation of the nucleobase, followed by covalent cleavage (51). In duplex DNA, the terminal ends of the duplex must unzip to accommodate this nucleobase loss. When high-proton affinity bases such as guanine are located at the terminal positions of a duplex, more extensive nucleobase loss and further unzipping will occur, leading to decreased stability of the quadruplexes. If a similar fragmentation mechanism is assumed for quadruplex DNA, G3 can undergo more facile nucleobase loss and thus has decreased gas-phase stability since it is the only quadruplex containing G-tracts on both the 5′ and 3′ ends of the strands forming the quadruplex. The gas-phase stabilities of the other quadruplexes do not appear to be affected by this anomaly.
Aside from G3, the results of the molecular dynamics simulation support the general conclusions of the energy-variable dissociation study. Of the quadruplexes assessed, the parallel four stranded structure G1 was the most stable, likely due to the four-tetrads and stabilizing NH4+ adducts that did not remain bound to the other structures. The next most stable structures were the intramolecular quadruplexes G5 and G6 which have approximately equal gas-phase stabilities. While G6 has more tetrads than G5 with four compared to three, the quadruplexes have different strand orientations as determined by the molecular dynamics simulations. The parallel strand orientation of G6 may negate the stabilizing effects of the additional tetrad compared to the antiparallel orientation of G5. The least stable quadruplexes are the double stranded antiparallel G4 with three tetrads and the intramolecular quadruplex G7 with only two tetrads. G7 has fewer tetrads than both G5 and G6 so it is unsurprisingly less stable. G4 and G5 have the same number of tetrads, however the antiparallel intramolecular orientation of G5 apparently increases its gas-phase stability relative to G4. It is expected that more accurate predictions could be achieved with more extensive MD sampling and improved molecular mechanical force fields (such as Parmbsc0, a modified version of AMBER Parm99, recently described by Orozco et al. (55)).
Conclusion
The CAD fragmentation patterns and E1/2 values for a series of quadruplexes have been acquired to assess their relative gas-phase stabilities. State of the art molecular dynamics simulations have also been applied to investigate the structural and energetic properties of G-quadruplexes in a solvent-free environment. It is concluded that G-quadruplexes can maintain their structures in the gas phase although the G-quartets are distorted to some degree. The ammonium ions were also shown to play an important rule in stabilizing the G-quartets for a four-stranded parallel quadruplex. Free energy analysis is useful not only for identifying the most favorable stoichiometry or strand orientation, but also for ranking the relative stability of a set of quadruplexes. The relative gas-phase stabilities of the quadruplexes determined by the molecular dynamics simulations correlate well with the ranking determined from E1/2 values of the quadruplexes, suggesting energy-variable dissociation curves are also useful as a means for rapidly screening the kinetic stability of quadruplexes.
Supplementary Material
Supporting Information Available
Root-mean-square deviations (Å) of G-tetrad guanines of 50 nanosecond simulations of G3-antiparallel, G4-antiparallel, G4-parallel, G5-antiparallel, G5-parallel, G6-antiparallel, G6-parallel, G7-antiparallel, G7-parallel, G7-antiparallel, the final snapshots of 50 nanosecond MD simulations of G4-parallel, G5-parallel (3)-G6-antiparallel (4) G7-parallel (5) G7-antiparallel, and summary of the number of stacking pairs and hydrogen bonding (HB) pairs in the last 100 picoseconds restrained and 50 nanoseconds non-restrained MD simulations of the quadruplexes. Energy-variable dissociation curves for [G5]5-, [G6]5-, and [G7]5-. Energy-variable dissociation curves for [(G3)2]5- and [(G4)2]5-.
This material is available free of charge via the Internet.
Acknowledgments
Funding from the Robert A. Welch Foundation (F-1155) and the National Institutes of Health (RO1 GM65956) and computational time from NCSA (MCB000013 to J.W.) is gratefully acknowledged.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Rezler EM, Bearss DJ, Hurley LH. Telomeres and telomerases as drug targets. Curr Opin in Pharmacol. 2002;2:415–423. doi: 10.1016/s1471-4892(02)00182-0. [DOI] [PubMed] [Google Scholar]
- 2.Bearss DJ, Hurley LH, Von Hoff DD. Telomere maintenance mechanisms as a target for drug development. Oncogene. 2000;19:6632–6641. doi: 10.1038/sj.onc.1204092. [DOI] [PubMed] [Google Scholar]
- 3.Rezler EM, Bearss DJ, Hurley LH. Telomere inhibition and telomere disruption as processes for drug targeting. Annu Rev Pharmacol Toxicol. 2003;43:359–379. doi: 10.1146/annurev.pharmtox.43.100901.135733. [DOI] [PubMed] [Google Scholar]
- 4.Wellinger RJ, Sen D. The DNA structures at the ends of eukaryotic chromosomes. Eur J Cancer. 1997;33:735–749. doi: 10.1016/S0959-8049(97)00067-1. [DOI] [PubMed] [Google Scholar]
- 5.Harley CB, Futcher AB, Greider CW. Telomeres shorten during aging of human fibroblasts. Nature. 1990;345:458–460. doi: 10.1038/345458a0. [DOI] [PubMed] [Google Scholar]
- 6.Kim NW, Piatyszek MA, Prowse KR, Harley CB, West MD, Ho PLC, Coviello GM, Wright WE, Weinrich SL, Shay JW. Specific association of human telomerase activity with immortal cells and cancer. Science. 1994;266:2011–2015. doi: 10.1126/science.7605428. [DOI] [PubMed] [Google Scholar]
- 7.Nakamura TM, Morin GB, Chapman KB, Weinrich SL, Andrews WH, Lingner J, Harley CB, Cech TR. Telomerase catalytic subunit homologs from fission yeast and human. Science. 1997;277:955–959. doi: 10.1126/science.277.5328.955. [DOI] [PubMed] [Google Scholar]
- 8.Moyzis RK, Buckingham JM, Cram LS, Dani M, Deaven LL, Jones MD, Meyne J, Ratliff RL, Wu JR. A highly conserved repetitive DNA-sequence, (TTAGGG)n, present at the telomeres of human-chromosomes. Proc Natl Acad Sci U S A. 1988;85:6622–6626. doi: 10.1073/pnas.85.18.6622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Blackburn EH, Gall JG. Tandemly repeated sequence at termini of extrachromosomal ribosomal-RNA genes in Tetrahymena. J Mol Biol. 1978;120:33–53. doi: 10.1016/0022-2836(78)90294-2. [DOI] [PubMed] [Google Scholar]
- 10.Haider S, Parkinson GN, Neidle S. Crystal structure of the potassium form of an Oxytricha nova G-quadruplex. J Mol Biol. 2002;320:189–200. doi: 10.1016/S0022-2836(02)00428-X. [DOI] [PubMed] [Google Scholar]
- 11.Tohl J, Eimer W. Interaction of a G-DNA quadruplex with mono- and divalent cations - A force field calculation. Biophys Chem. 1997;67:177–186. doi: 10.1016/s0301-4622(97)00037-9. [DOI] [PubMed] [Google Scholar]
- 12.Kerwin SM. G-quadruplex DNA as a target for drug design. Curr Pharm Des. 2000;6:441–471. doi: 10.2174/1381612003400849. [DOI] [PubMed] [Google Scholar]
- 13.Mergny JL, Mailliet P, Lavelle F, Riou JF, Laoui A, Helene C. The development of telomerase inhibitors: The G-quartet approach. Anti-Cancer Drug Des. 1999;14:327–339. [PubMed] [Google Scholar]
- 14.Hurley LH, Wheelhouse RT, Sun D, Kerwin SM, Salazar M, Fedoroff OY, Han FX, Han HY, Izbicka E, Von Hoff DD. G-quadruplexes as targets for drug design. Pharmacol Ther. 2000;85:141–158. doi: 10.1016/s0163-7258(99)00068-6. [DOI] [PubMed] [Google Scholar]
- 15.Han HY, Hurley LH. G-quadruplex DNA: A potential target for anti-cancer drug design. Trends Pharmacol Sci. 2000;21:136–142. doi: 10.1016/s0165-6147(00)01457-7. [DOI] [PubMed] [Google Scholar]
- 16.David WM, Brodbelt J, Kerwin SM, Thomas PW. Investigation of quadruplex oligonucleotide-drug interactions by electrospray ionization mass spectrometry. Anal Chem. 2002;74:2029–2033. doi: 10.1021/ac011283w. [DOI] [PubMed] [Google Scholar]
- 17.Mazzitelli CL, Kern JT, Rodriguez M, Brodbelt JS, Kerwin SM. Evaluation of binding of perylene diimide and benzannulated perylene diimide ligands to DNA by electrospray ionization mass spectrometry. J Am Soc Mass Spectrom. 2006;17:593–604. doi: 10.1016/j.jasms.2005.12.011. [DOI] [PubMed] [Google Scholar]
- 18.Baker ES, Lee JT, Sessler JL, Bowers MT. Cyclo[n]pyrroles: Size and site-specific binding to G-quadruplexes. J Am Chem Soc. 2006;128:2641–2648. doi: 10.1021/ja0564968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rosu F, De Pauw E, Guittat L, Alberti P, Lacroix L, Mailliet P, Riou JF, Mergny JL. Selective interaction of ethidium derivatives with quadruplexes: An equilibrium dialysis and electrospray ionization mass spectrometry analysis. Biochemistry. 2003;42:10361–10371. doi: 10.1021/bi034531m. [DOI] [PubMed] [Google Scholar]
- 20.Guittat L, Alberti P, Rosu F, Van Miert S, Thetiot E, Pieters L, Gabelica V, De Pauw E, Ottaviani A, Riou JF. Interactions of cryptolepine and neocryptolepine with unusual DNA structures. Biochimie. 2003;85:535–547. doi: 10.1016/s0300-9084(03)00035-x. [DOI] [PubMed] [Google Scholar]
- 21.Guittat L, De Cian A, Rosu F, Gabelica V, De Pauw E, Delfourne E, Mergny JL. Ascididemin and meridine stabilise G-quadruplexes and inhibit telomerase in vitro. Biochim Biophys Acta. 2005;1724:375–384. doi: 10.1016/j.bbagen.2005.04.023. [DOI] [PubMed] [Google Scholar]
- 22.Carrasco C, Rosu F, Gabelica V, Houssier C, De Pauw E, Garbay-Jaureguiberry C, Roques B, Wilson WD, Chaires JB, Waring MJ, Bailly C. Tight binding of the antitumor drug ditercalinium to quadruplex DNA. Chem Bio Chem. 2002;3:1235–1241. doi: 10.1002/1439-7633(20021202)3:12<1235::AID-CBIC1235>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 23.Rosu F, Gabelica V, Shin-ya K, De Pauw E. Telomestatin-induced stabilization of the human telomeric DNA quadruplex monitored by electrospray mass spectrometry. Chem Commun. 2003;21:2702–2703. doi: 10.1039/b309394h. [DOI] [PubMed] [Google Scholar]
- 24.Gabelica V, Shammel Baker E, Teulade-Fichou MP, De Pauw E, Bowers MT. Stabilization and Structure of Telomeric and c-myc Region Intramolecular G-Quadruplexes: The Role of Central Cations and Small Planar Ligands. J Am Chem Soc. 2007;129:895–904. doi: 10.1021/ja065989p. [DOI] [PubMed] [Google Scholar]
- 25.Gidden J, Ferzoco A, Baker ES, Bowers MT. Duplex formation and the onset of helicity in poly d(CG)(n) oligonucleotides in a solvent-free environment. J Am Chem Soc. 2004;126:15132–15140. doi: 10.1021/ja046433+. [DOI] [PubMed] [Google Scholar]
- 26.Gabelica V, De Pauw E. Collision-induced dissociation of 16-mer DNA duplexes with various sequences: evidence for conservation of the double helix conformation in the gas phase. Int J Mass Spectrom. 2002;219:151–159. [Google Scholar]
- 27.Pan S, Sun XJ, Lee JK. Stability of complementary and mismatched DNA duplexes: Comparison and contrast in gas versus solution phases. Int J Mass Spectrom. 2006;253:238–248. [Google Scholar]
- 28.Guo XH, Bruist MF, Davis DL, Bentzley CM. Secondary structural characterization of oligonucleotide strands using electrospray ionization mass spectrometry. Nucleic Acids Res. 2005;33:3659–3666. doi: 10.1093/nar/gki671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Guo XH, Liu ZQ, Liu SY, Bentzley CM, Bruist MF. Structural features of the L-argininamide-binding DNA aptamer studied with ESI-FTMS. Anal Chem. 2006;78:7259–7266. doi: 10.1021/ac060606r. [DOI] [PubMed] [Google Scholar]
- 30.Wan KX, Gross ML, Shibue T. Gas-phase stability of double-stranded oligodeoxynucleotides and their noncovalent complexes with DNA-binding drugs as revealed by collisional activation in an ion trap. J Am Soc Mass Spectrom. 2000;11:450–457. doi: 10.1016/S1044-0305(00)00095-7. [DOI] [PubMed] [Google Scholar]
- 31.Gabelica V, De Pauw E. Comparison between solution-phase stability and gas-phase kinetic stability of oligodeoxynucleotide duplexes. J Mass Spectrom. 2001;36:397–402. doi: 10.1002/jms.141. [DOI] [PubMed] [Google Scholar]
- 32.Rosu F, Gabelica V, Houssier C, Colson P, De Pauw E. Triplex and quadruplex DNA structures studied by electrospray mass spectrometry. Rapid Commun Mass Spectrom. 2002;16:1729–1736. doi: 10.1002/rcm.778. [DOI] [PubMed] [Google Scholar]
- 33.Baker ES, Bernstein SL, Gabelica V, De Pauw E, Bowers MT. G-quadruplexes in telomeric repeats are conserved in a solvent-free environment. Int J Mass Spectrom. 2006;253:225–237. [Google Scholar]
- 34.Rueda M, Kalko SG, Luque FJ, Orozco M. The structure and dynamics of DNA in the gas phase. J Am Chem Soc. 2003;125:8007–8014. doi: 10.1021/ja0300564. [DOI] [PubMed] [Google Scholar]
- 35.Rueda M, Luque FJ, Orozco M. Nature of minor-groove binders - DNA complexes in the gas phase. J Am Chem Soc. 2005;127:11690–11698. doi: 10.1021/ja0422110. [DOI] [PubMed] [Google Scholar]
- 36.Rueda M, Luque FJ, Orozco M. G-quadruplexes can maintain their structure in the gas phase. J Am Chem Soc. 2006;128:3608–3619. doi: 10.1021/ja055936s. [DOI] [PubMed] [Google Scholar]
- 37.Gros J, Rosu F, Amrane S, De Cian A, Gabelica V, Lacroix L, Mergny JL. Guanines are a quartet's best friend: impact of base substitutions on the kinetics and stability of tetramolecular quadruplexes. Nuc Acid Res. 2007;35:3064–3075. doi: 10.1093/nar/gkm111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kerwin SM, Chen G, Kern JT, Thomas PW. Perylene diimide G-quadruplex DNA binding selectivity is mediated by ligand aggregation. Bioorg Med Chem Lett. 2002;12:447–450. doi: 10.1016/s0960-894x(01)00775-2. [DOI] [PubMed] [Google Scholar]
- 39.Caceres C, Wright G, Gouyette C, Parkinson G, Subirana JA. A thymine tetrad in d(TGGGGT) quadruplexes stabilized with Tl+/Na+ ions. Nucleic Acids Res. 2004;32:1097–1102. doi: 10.1093/nar/gkh269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Horvath MP, Schultz SC. DNA G-quartets in a 1.86 angstrom resolution structure of an Oxytricha nova telomeric protein-DNA complex. J Mol Biol. 2001;310:367–377. doi: 10.1006/jmbi.2001.4766. [DOI] [PubMed] [Google Scholar]
- 41.Muller S, Diederichs K, Breed J, Kissmehl R, Hauser K, Plattner H, Welte W. Crystal structure analysis of the exocytosis-sensitive phosphoprotein, pp63/parafusin (phosphoglucomutase), from Paramecium reveals significant conformational variability. J Mol Biol. 2002;315:141–153. doi: 10.1006/jmbi.2001.5168. [DOI] [PubMed] [Google Scholar]
- 42.Wang Y, Patel DJ. Solution structure of the human telomeric repeat d[(AG3T2AG3)3] G-Tetraplex. Structure. 1993;1:263–282. doi: 10.1016/0969-2126(93)90015-9. [DOI] [PubMed] [Google Scholar]
- 43.Case DA, Darden TA, Cheatham TE, Simmerling CL, Wang J, Duke RE, Luo R, Merz KM, Wang B, Pearlman DA, et al. AMBER 8. University of California; San Francisco: 2005. [Google Scholar]
- 44.Wang JM, Cieplak P, Kollman PA. How well does a restrained electrostatic potential (RESP) model perform in calculating conformational energies of organic and biological molecules? J Comput Chem. 2000;21:1049–1074. [Google Scholar]
- 45.Berendsen HJC, Postma JPM, Van Gunsteren WF, Dinola A, Haak JR. Molecular dynamics with coupling to an external bath. J Chem Phys. 1984;81:3684–3690. [Google Scholar]
- 46.Ryckaert JP, Ciccotti G, Berendsen HJC. Numerical-integration of cartesian equations of motion of a system with constraints - molecular-dynamics of N-alkanes. J Comput Phys. 1977;23:327–341. [Google Scholar]
- 47.Gabb HA, Sanghani SR, Robert CH, Prévost C. Finding and visualizing nucleic acid base stacking. J Mol Graphics. 1996;14:6–11. doi: 10.1016/0263-7855(95)00086-0. [DOI] [PubMed] [Google Scholar]
- 48.Srinivasan J, Cheatham TE, Cieplak P, Kollman PA, Case DA. Continuum solvent studies of the stability of DNA, RNA, and phosphoramidate - DNA helices. J Am Chem Soc. 1998;120:9401–9409. [Google Scholar]
- 49.Kollman PA, Massova I, Reyes C, Kuhn B, Huo SH, Chong L, Lee M, Lee T, Duan Y, Wang W, et al. Calculating structures and free energies of complex molecules: Combining molecular mechanics and continuum models. Acc Chem Res. 2000;33:889–897. doi: 10.1021/ar000033j. [DOI] [PubMed] [Google Scholar]
- 50.Wang J, Hou T, Xu X. Recent advances in free energy calculations with a combination of molecular mechanics and continuum models. Curr Computer-Aided Drug Design. 2006;2:287–306. [Google Scholar]
- 51.Keller KM, Zhang JM, Oehlers L, Brodbelt JS. Influence of initial charge state on fragmentation patterns for noncovalent drug/DNA duplex complexes. J Mass Spectrom. 2005;40:1362–1371. doi: 10.1002/jms.927. [DOI] [PubMed] [Google Scholar]
- 52.Gabelica V, De Pauw E. Comparison of the collision-induced dissociation of duplex DNA at different collision regimes: Evidence for a multistep dissociation mechanism. J Am Soc Mass Spectrom. 2002;13:91–98. doi: 10.1016/s1044-0305(01)00335-x. [DOI] [PubMed] [Google Scholar]
- 53.Pan S, Verhoeven K, Lee JK. Investigation of the initial fragmentation of oligodeoxynucleotides in a quadrupole ion trap: Charge level-related base loss. J Am Soc Mass Spectrom. 2005;16:1853–1865. doi: 10.1016/j.jasms.2005.07.009. [DOI] [PubMed] [Google Scholar]
- 54.Gale DC, Smith RD. Characterization of noncovalent complexes formed between minor groove binding molecules and duplex DNA by electrospray ionization mass spectrometry. J Am Soc Mass Spectrom. 1995;6:1154–1164. doi: 10.1016/1044-0305(95)00530-7. [DOI] [PubMed] [Google Scholar]
- 55.Perez A, Marchan I, Svozil D, Sponer J, Cheatham TE, III, Laughton CA, Orozco M. Refinement of the AMBER force field for nucleic acids: improving the description of a, g conformers. Biophys J. 2007;92:3817–3829. doi: 10.1529/biophysj.106.097782. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information Available
Root-mean-square deviations (Å) of G-tetrad guanines of 50 nanosecond simulations of G3-antiparallel, G4-antiparallel, G4-parallel, G5-antiparallel, G5-parallel, G6-antiparallel, G6-parallel, G7-antiparallel, G7-parallel, G7-antiparallel, the final snapshots of 50 nanosecond MD simulations of G4-parallel, G5-parallel (3)-G6-antiparallel (4) G7-parallel (5) G7-antiparallel, and summary of the number of stacking pairs and hydrogen bonding (HB) pairs in the last 100 picoseconds restrained and 50 nanoseconds non-restrained MD simulations of the quadruplexes. Energy-variable dissociation curves for [G5]5-, [G6]5-, and [G7]5-. Energy-variable dissociation curves for [(G3)2]5- and [(G4)2]5-.
This material is available free of charge via the Internet.