

Abstract
Heterotrimeric G protein signaling is important for cell-proliferative and glucose-sensing signal transduction pathways in the model plant organism Arabidopsis thaliana. AtRGS1 is a seven-transmembrane, RGS domain-containing protein that is a putative membrane receptor for d-glucose. Here we show, by using FRET, that d-glucose alters the interaction between the AtGPA1 and AtRGS1 in vivo. AtGPA1 is a unique heterotrimeric G protein α subunit that is constitutively GTP-bound given its high spontaneous nucleotide exchange coupled with slow GTP hydrolysis. Analysis of a point mutation in AtRGS1 that abrogates GTPase-accelerating activity demonstrates that the regulation of AtGPA1 GTP hydrolysis mediates sugar signal transduction during Arabidopsis development, in contrast to animals where nucleotide exchange is the limiting step in the heterotrimeric G protein nucleotide cycle.
Keywords: d-glucose, G protein-coupled receptor, guanine nucleotide cycle, RGS protein, GTPase-accelerating protein
G protein-coupled receptors (GPCRs), also known as seven-transmembrane domain (7TM) receptors, compose a large superfamily of cell membrane proteins that convert extracellular signals from environmental cues to intracellular responses (1–3). Both genetic and biochemical data firmly support a role for G proteins in sugar-regulated plant cell proliferation, yet plants have a limited repertoire of heterotrimeric G protein-signaling components (4–10). Arabidopsis thaliana has one canonical G protein α (Gα) subunit (AtGPA1), one Gβ subunit, and two Gγ subunits, but, as yet, no bona fide GPCR (although candidate GPCRs have been proposed) (10, 11). The Arabidopsis regulator of G protein signaling-1 (AtRGS1) protein contains seven transmembrane-spanning domains and a C-terminal RGS domain (5). Thus, AtRGS1 has the membrane topology and structural characteristics of a GPCR, and genetic evidence is consistent with AtRGS1 being a receptor or coreceptor for d-glucose at the plasma membrane (4, 5). The RGS domain of AtRGS1 has GTPase-accelerating activity toward AtGPA1 in vitro and AtRGS1 functions in the AtGPA1-regulated, sugar-sensing, and cell-proliferation pathways in vivo (4, 5, 12).
Based on these previous findings, it has been proposed that AtRGS1 could be a guanine-nucleotide exchange factor (GEF) and/or a GTPase-accelerating protein (GAP) for AtGPA1 (3, 13). A ligand for AtRGS1 that modulates GEF or GAP activity has not been identified. However, based on the altered sugar responsiveness of Atrgs1-null mutants, it has been suggested that d-glucose is a candidate ligand (4–8). Here we examined the requirement for G protein signaling in the Arabidopsis glucose-sensing pathway. Conventional GPCRs serve as nucleotide exchange factors controlling the rate-limiting step in heterotrimeric G protein cycling: the release of GDP (14). We found that the Arabidopsis AtRGS1 protein serves as a 7TM GAP, and it is GTP hydrolysis, not GDP release, by AtGPA1 that is rate-limiting in Arabidopsis d-glucose signal transduction.
Results and Discussion
Growth arrest during Arabidopsis development can be induced by high concentrations of d-glucose and quantified by the fraction of seedlings with green cotyledons (4, 15). At 1% d-glucose, individuals of all Arabidopsis genotypes that were tested grew normally (100% green seedlings) (data not shown), whereas at 6% glucose, approximately half of the individual wild-type seedlings arrested (Fig. 1A). As expected (4), AtRGS1-deficient plants were tolerant to 6% d-glucose, whereas AtGPA1-deficient plants were hypersensitive to glucose-induced growth arrest (Fig. 1A). Likewise, Arabidopsis overexpressing wild-type AtRGS1 were hypersensitive to glucose-induced growth arrest (Fig. 1A). Plants lacking both AtGPA1 and AtRGS1 also were hypersensitive to glucose (Fig. 1A), phenocopying the Atgpa1 mutant phenotype of glucose hypersensitivity. This finding indicates that AtGPA1 and AtRGS1 work in the same glucose signal transduction pathway, and that the null Atgpa1 allele is epistatic to the null Atrgs1 allele in this pathway. These data suggest that, in the presence of ligand (high concentrations of glucose), a signal transduction pathway inhibits plant growth and development. This pathway is modulated by heterotrimeric G protein signal transduction, and AtGPA1-GTP most likely attenuates an antiproliferative pathway (6). We hypothesize that this process is modulated by AtRGS1 acting as a glucose sensor and regulating the GTPase activity of AtGPA1. Therefore, based on this hypothesis, glucose should alter the interaction between AtRGS1 and AtGPA1, thereby coupling receptor activation to the modulation of AtGPA1's activity on seedling developmental processes.
Fig. 1.

Signal transduction by d-glucose is mediated by AtRGS1 and AtGPA1. (A) Seedlings of different genotypes were grown on 6% d-glucose, and the percentage of seedlings with green cotyledons was quantified. All genotypes had 100% green seedlings when grown on 1% d-glucose. Genotypes: Col, wild-type Columbia ecotype; Atrgs1–2, Atrgs1-null mutant; Atgpa1–4, Atgpa1-null mutant; 35S:AtRGS1(wild type: Ox9, Ox10, Ox16), three independent wild-type AtRGS1 constitutive overexpression lines; Atrgs1–2 Atgpa1–4, double-null mutant. Statistical significance was determined by Dunnett's test (*, P < 0.05 vs. Col). (B–G) d-glucose-induced FRET between AtRGS1-YFP and AtGPA1-CFP in Arabidopsis roots was measured in vivo. Fluorescence emission for CFP excitation/YFP emission (B and E) and CFP excitation/CFP emission (C and F) were captured 5 min (B and C) and 8 min (E and F) after the addition of d-glucose. The normalized net FRET (nF/I) at 5 min (D) and 8 min (G) after d-glucose addition is shown. PM, plasma membrane. (H) Levels of nF/I were calculated every 30 s from 5 to 30 min after addition of 6% (wt/vol) d-glucose, 6% (wt/vol) l-glucose, or no treatment controls. Red and blue lines show the observed FRET for the regions of interest (ROI) 1 and ROI 2, denoted in G. Black lines indicate other independent FRET efficiency measurements. Arrows indicate image capture time points of t = 5 and t = 8 min, as denoted in B–G.
To test this prediction in vivo, we performed FRET studies on plants coexpressing AtGPA1-CFP and AtRGS1-YFP fusion proteins (e.g., Fig. 1 B–G). Within 6 min after the addition of exogenous d-glucose, FRET increased (Fig. 1G, arrows). The change in FRET was transient, peaking at 8–10 min and decaying with a half-life of ≈2 min. Interestingly, FRET signals were exclusively observed on or around plastids that were in close proximity to the plasma membrane. The observed FRET was specific to d-glucose because it was not observed upon the addition of l-glucose (Fig. 1H) or mannitol [supporting information (SI) Fig. 4]. Thus, both genetic and imaging data are consistent with the hypothesis that AtRGS1 is a membrane receptor or coreceptor for d-glucose that interacts with AtGPA1 upon glucose binding. However, the imaging data do not preclude a mechanism in which indirect modulation of FRET may occur by an alternative glucose-sensing pathway. The imaging data further suggest that downstream signal transduction may be spatially localized where plastids abut the plasma membrane. This finding is consistent with evidence showing that the plastid protein THF1 directly interacts with AtGPA1 at the plastid/plasma membrane interface, and that the thf1-null allele is epistatic to the Atgpa1-null allele in the sugar-sensing pathway (6).
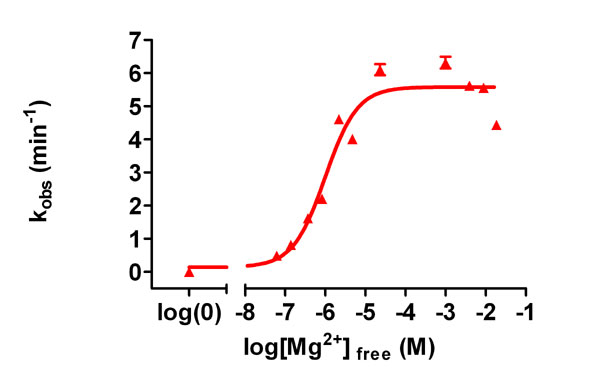
Based on its chimeric structure (N-terminal 7TM and C-terminal RGS domains), we previously hypothesized that AtRGS1 could be a d-glucose-regulated GAP, GEF, or dual GAP and GEF for AtGPA1 (3). Our present analysis of the biochemical properties of AtGPA1 is consistent with the former (glucose-regulated GAP) and suggests that the AtGPA1 GTPase cycle does not require GEF activity. Recombinant AtGPA1 had a high specific activity, binding [35S]GTPγS with a stoichiometry of 0.91 mol of GTPγS/mol of protein. Equilibrium competition binding assays with a variety of purine and pyrimidine nucleotide triphosphates were conducted. Only guanine nucleotide triphosphates were able to compete with [35S]GTPγS for binding to AtGPA1 (SI Table 1), indicating that AtGPA1 is selective toward guanine nucleotides and is a bona fide GTP-binding protein. The observed rate of [35S]GTPγS binding to AtGPA1 was fast (kobs = 1.4 min−1). However, this value is likely an underestimate because the first time point shows >75% binding in this assay format (Fig. 2A). Human GαoA had a kobs value of 0.09 min−1 in this GTPγS-binding assay (Fig. 2A), consistent with it being one of the fastest exchanging mammalian Gα subunits described (SI Table 2). To obviate the poor temporal resolution of the [35S]GTPγS-binding assay, we used fluorescence spectroscopy to quantitate the kinetics of AtGPA1 binding to BODIPYFL-GTPγS (Fig. 2B). By using this real-time assay, the kobs value for GTPγS binding by AtGPA1 was 14.4 min−1, a value 22-fold faster than the most rapidly exchanging Gα subunits previously described (SI Table 2). To verify this result by using an independent approach, we measured GTPγS binding to AtGPA1 by using intrinsic tryptophan fluorescence, and comparable kinetic data were obtained (kobs = 8.7 min−1) (Fig. 2C). To ensure that the high rate of nucleotide exchange observed was not the result of nonphysiological Mg2+ concentrations, we measured GTPγS-binding rates at different concentrations of free magnesium. The rate of GTPγS binding was independent of [Mg2+]free over a wide range (2 μM–20 mM) (SI Fig. 5), indicating that the nucleotide exchange rate observed in vitro is likely to be similar in vivo. We also directly measured GDP release by using [β-32P]GDP-bound AtGPA1 (Fig. 2D). The rate of GDP dissociation from AtGPA1 (koff = 12.6 min−1) was highly concordant with the measured GTPγS-binding rate.
Fig. 2.

Biochemical properties of AtGPA1. (A) Time course of [35S]GTPγS binding to 100 nM AtGPA1 or GαOA at 20°C. Data are the mean ± SEM of duplicate samples. Observed association rate constants (kobs) were: AtGPA1 1.44 [95% confidence interval (C.I.), 0.89–2.0] min−1 and GαOA 0.088 (95% C.I., 0.076–0.1) min−1. (B) Time course of BODIPYFL-GTPγS binding to 200 nM AtGPA1 at 20°C. Data are plotted as the mean ± SEM of three experiments. kobs = 14.4 (95% C.I., 12.5–16.2) min−1. RFU, relative fluorescence units. (C) The tryptophan fluorescence of 100 nM AtGPA1 or buffer alone (control) was measured at 20°C. At 100 s, GTPγS was added to a final concentration of 1 μM (arrow). Data are presented as the mean ± SEM of duplicates. kobs = 8.7 min−1 (SEM ± 1.5 min−1; n = 3). (D) Time course of [β-32P]GDP dissociation from AtGPA1 at 20°C. Data are the mean ± SEM of duplicates. Observed dissociation rate constant (koff) was 12.6 (95% C.I., 4.4–21.0) min−1. (E) Time course of single turnover [γ-32P]GTP hydrolysis by 200 nM AtGPA1 or GαOA at 20°C. Data are the mean ± SEM of duplicates. Rate constants for GTP hydrolysis (kcat) were: AtGPA1 0.12 (95% C.I., 0.11–0.13) min−1 and GαOA 0.97 (95% C.I., 0.70–1.4) min−1. (F) Time course of steady-state [γ-32P]GTP hydrolysis by 200 nM AtGPA1 in the presence or absence of 1 μM AtRGS1 at 20°C. Results are the mean ± SEM of duplicate samples. Rates of Pi production were AtGPA1 2.3 (95% C.I., 2.0–2.5) × 102 cpm/min, AtGPA1+AtRGS1 8.1 (95% C.I., 7.6–8.5) × 103 cpm/min, and AtRGS1 alone 6.9 (95% C.I., 4.0–9.8) × 10 cpm/min.
GDP dissociation is the rate-limiting step of the heterotrimeric G protein cycle as described in animals (14). To understand the exceptionally rapid nucleotide exchange exhibited by AtGPA1 in the context of its complete nucleotide cycle, we also measured the rate of GTP hydrolysis by AtGPA1 by using a single turnover GTPase assay. The kcat value for AtGPA1 at 20°C was 0.12 min−1 (Fig. 2E), making AtGPA1 among the slowest heterotrimeric GTPases described (SI Table 2). The kcat value of GαoA was determined in parallel (Fig. 2E) and was consistent with published values (SI Table 2). This observation demonstrates that the rate of GTP hydrolysis (kcat) by AtGPA1 is over two orders of magnitude slower than the rate of nucleotide exchange (koff). Thus, GTP hydrolysis (rather than GDP release) is the rate-limiting step in the guanine nucleotide cycle of AtGPA1. Two predictions follow from these observations: (i) The steady-state rate of GTP hydrolysis should approximate kcat, and (ii) RGS domain-mediated GAP activity should accelerate steady-state GTP hydrolysis.
We tested these two predictions by performing steady-state GTPase assays by using [γ-32P]GTP. The rate of GTP hydrolysis at steady state (ks) at 20°C was 0.063 min−1 (±0.015 min−1; n = 4) (Fig. 2F). Thus, the observed ks approximated the rate of nucleotide hydrolysis, not nucleotide exchange, therefore satisfying the first prediction. We observed that a 5-fold molar excess of AtRGS1 gave a 35-fold increase in steady-state GTPase activity (Fig. 2F), satisfying the second prediction and validating that GTP hydrolysis is the rate-limiting step in the Arabidopsis G protein cycle in vitro. Under steady-state conditions, the fraction of G protein in the active state can be approximated by kobs/(kobs + kcat) (SI Table 2) (16). In the case of Gαo, for example, the percentage of protein bound to GTP at steady state is predicted to be 10%. In stark contrast, the calculated value for AtGPA1 is 99% (SI Table 2). Furthermore, AtGPA1 appears to have a higher affinity for GTP than GDP (SI Table 1), further supporting the hypothesis that AtGPA1 is constitutively present in the GTP-bound (and presumed activated) form in vivo. This finding suggests that AtGPA1 may not require GPCR-mediated GEF activity to accomplish signal transduction as do all other known Gα subunits. In total, the biochemical properties exhibited by AtGPA1 are consistent with this Gα subunit being constitutively GTP-bound and thus a substrate for d-glucose-regulated AtRGS1 GAP activity.
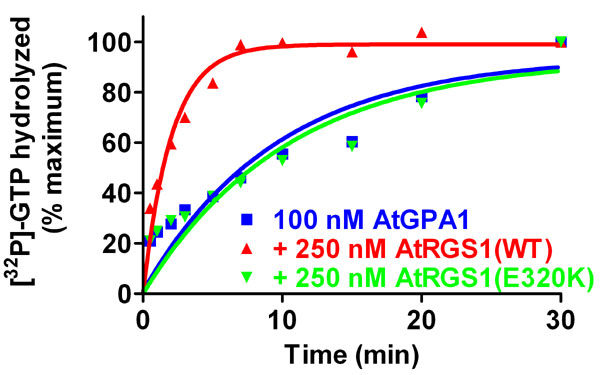
To test the hypothesis that GAP activity is the essential regulatory function of AtRGS1 in d-glucose signaling, we engineered a loss-of-function mutant in the RGS domain of AtRGS1 (Glu-320 to Lys) based on structural information of the RGS domain–Gα interface (SI Fig. 6). Unlike the GAPs for Ras-family GTPases, RGS proteins do not contribute a catalytic residue per se to the chemistry of GTP hydrolysis that is intrinsic to Gα, but instead bind most avidly to the Gα transition state (i.e., between GTP and GDP/Pi-bound states) (17). Hence, loss-of-function mutations to RGS domains such as E320K are designed to eliminate GAP activity by disrupting the Gα-binding interface (18). We analyzed the in vitro biochemical properties of AtRGS1(E320K) by using a GST fusion of AtRGS1 (amino acids 249–459). AtGPA1 GTPase activity was accelerated by substoichiometric amounts of AtRGS1(wild type), whereas AtRGS1(E320K) was ineffective over a wide range of concentrations. The E320K substitution reduced AtRGS1 GAP activity by at least three orders of magnitude (Fig. 3A). Complementary data were obtained by using single turnover and steady-state time course assays (SI Figs. 7 and 8). To test that AtRGS1(E320K) is deficient in its interaction with AtGPA1 as well as in GAP activity, we measured the AtGPA1–AtRGS1 interaction by using surface plasmon resonance spectroscopy. Consistent with published data (12), AtRGS1(wild-type) bound AtGPA1 in the transition state for GTP hydrolysis (GDP-AlF4−), but not the ground (GDP; Fig. 3B) or activated (GTPγS) states (data not shown). Binding of AtGPA1 to AtRGS1(E320K) could not be detected in any nucleotide state, thus verifying that AtRGS1(E320K) is a true loss-of-function mutant. Equivalent results were obtained by using GST coprecipitation assays (SI Fig. 9).
Fig. 3.

In vitro and in vivo characterization of AtRGS1(E320K) as a GAP-dead mutant. (A) Dose–response analysis of AtGPA1 steady-state GTPase acceleration by wild type (WT) and E320K AtRGS1. GTPase assays were conducted for 20 min with 200 nM AtGPA1, and EC50 values were determined by regression for WT as 2.7 (95% C.I., 2.4–3.1) × 10−8 M and for the E320K mutant as 4.5 (95% C.I., 4.3–4.7) × 10−5 M. For these EC50 regression analyses, it was assumed that the E320K mutant has the same efficacy as WT protein. (B) Surface plasmon resonance was used to measure the interaction between AtRGS1 and AtGPA1. WT or E320K GST-AtRGS1 was immobilized on a biosensor surface. Then 1 μM AtGPA1 in the GDP or GDP-AlF4−-bound form was injected over the biosensor surface. Nonspecific binding to GST was subtracted from all curves. (C–E) AtRGS1(E320K)-GFP localization was visualized in Arabidopsis hypocotyls (C), cotelydons (D), and roots (E) by using epifluorescence microscopy. (F) Hypocotoyl lengths of 2-day-old, dark-grown seedlings were measured. Genotypes: 35S:AtRGS1(WT or E320K) (wild-type or E320K AtRGS1 constitutive overexpression lines). Multiple independent overexpression lines were generated and analyzed; individual lines are denoted Ox. Statistical significance was determined by Bonferroni's test (*, P < 0.05 vs. Col). (G) Experiments were performed as in F, but Arabidopsis-expressing AtRGS1-GFP-fusion proteins were used. Statistical significance was determined by Bonferroni's test (*, P < 0.05 vs. Col; #, P > 0.05 vs. Atrgs1–2). (H) Seedlings of the indicated genotypes were grown on 6% glucose, and the percentage of seedlings with green cotyledons was quantified. All genotypes had 100% green seedlings when grown on 1% glucose. Genotypes are described in F. Statistical significance was determined by Bonferroni's test (*, P < 0.05 vs. Col; #, P > 0.05 vs. Atrgs1–2).
We generated Arabidopsis lines expressing either untagged or C-terminal GFP-fused AtRGS1(E320K). AtRGS1(E320K)-GFP was observed to be plasma membrane-localized in the hypocotyls, cotyledons, and roots in an identical pattern to AtRGS1(wild-type)-GFP (Fig. 3 C–E) (5), indicating that AtRGS1(E320K) is properly expressed, folded, and membrane-targeted in vivo. Studies from mutants in the Arabidopsis G protein pathway indicate a role for G protein signaling in regulating cell growth. Dark-grown AtGPA1-deficient lines exhibit reduced cell proliferation and consequently have a shortened hypocotyl (19), whereas AtRGS1-null seedlings have longer hypocotyls because of increased cell elongation (5). In contrast, seedlings overexpressing wild-type AtRGS1 have shortened hypocotyls (5). Unlike AtRGS1(wild-type)-overexpressing plants, AtRGS1(E320K)-expressing plants had wild-type-length hypocotyls (Fig. 3F). Data obtained by using AtRGS1-GFP fusion proteins expressed in AtRGS1-deficient plants confirm that AtRGS1(E320K) cannot complement the Atrgs1-null mutation (Fig. 3G). These results indicate that GAP activity is the essential determinant in AtRGS1-mediated cell proliferative signaling.
We also measured the contribution of AtRGS1 GAP activity to glucose-mediated growth arrest. Expression of the AtRGS1(E320K) mutant was unable to complement Atrgs1-null alleles and did not elicit the glucose-hypersensitive phenotype typical of AtRGS1(wild-type)-overexpressing plants (compare Fig. 1A with Fig. 3H). This result suggests that the influence of AtRGS1 on developmental responses to environmental glucose is directly dependent on its GAP activity. This finding is consistent with the hypothesis that AtRGS1 is a glucose-regulated GAP for GTP-bound AtGPA1. Furthermore, the intrinsic biochemical properties of high spontaneous nucleotide exchange and low GTPase activity suggest that AtGPA1 is predominantly in the GTP-bound state, and this state is the likely substrate for AtRGS1 in vivo. The action of AtRGS1 is likely to be catalytic because biochemical experiments demonstrate that AtRGS1 is a potent accelerator of AtGPA1 GTPase activity at substoichiometric concentrations in vitro (Fig. 3A and SI Fig. 8). The in vitro biochemical properties of AtGPA1 imply that the Arabidopsis G protein cycle is distinct from the mammalian G protein cycle in two critical aspects: GEF activity is not required, and GDP-bound AtGPA1 may be the active signaling species in some cases. These observations are concordant with data showing that AtGPA1-GDP interacts with AtPLDα1 to regulate phosphatidic acid production and inhibition of stomatal opening (20).
In addition to AtRGS1, two other putative Arabidopsis GPCRs, GCR1 and GCR2, have been described (21, 22). We recently argued that GCR2 is not a transmembrane receptor, but rather a plant homolog of bacterial lanthionine synthetases that was misidentified as a GPCR (11). GCR1 is a predicted 7TM protein reported to interact with AtGPA1 (23). Physiological data suggest that GCR1 may be involved in the AtGPA1 signaling pathway (23). However, GCR1 also regulates hormonal signaling in a G protein-independent manner (24). These reports on GCR1 and GCR2 are entirely consistent with our findings presented here, in that, to date, none of these plant proteins has been shown to possess GEF activity toward the sole Arabidopsis Gα subunit, AtGPA1. Thus, in light of the unique biochemical activities of AtGPA1 as a rapid spontaneous exchanger and poor GTPase, putative plant membrane receptors such as GCR1 also may regulate AtGPA1 activity by GEF-independent mechanisms.
Arabidopsis Gβγ subunits also are implicated in both cell proliferation and sugar-sensing pathways (6, 19). The canonical function for Gβγ subunits in metazoan organisms is to participate directly in GPCR-mediated GEF activity and regulate effector pathways (1, 3). Although our data would appear to exclude the former, it does not exclude the latter. Gβγ subunits also are required for the proper membrane targeting and stability of mammalian Gα subunits (25), and this finding appears to be the case in Arabidopsis as well (26). Moreover, FRET studies suggest that the Arabidopsis Gαβγ heterotrimer does not dissociate upon GTP binding, but merely changes conformation (26). This result is in line with recent studies on mammalian G protein signaling by nondissociated heterotrimers (27), as well as those of the regulatory protein AGS8 shown to facilitate signal transduction properties of a nondissociated heterotrimer in complex with phospholipase Cβ (28). Mammalian Gβγ subunits also attenuate spontaneous nucleotide exchange on Gα subunits by ≤5-fold (29, 30). It is possible that AtGPA1 constitutive activity is dampened by the Arabidopsis Gβγ complex in vivo. However, to have a significant physiological effect on AtGPA1 nucleotide cycling kinetics, Arabidopsis Gβγ would need to attenuate GDP release by an improbable three orders of magnitude (i.e., leading to a predicted GTP occupancy of ≈10%), whereas a reduction in AtGPA1 GDP release rate by two orders of magnitude is predicted to maintain GTP occupancy level at >50% (data based on calculations similar to those of SI Table 2).
We described genetic, biochemical, and cellular data on AtRGS1 and AtGPA1 in support of a unique paradigm for heterotrimeric G protein action. This AtRGS1-coupled, high-glucose-sensing mechanism appears to be distinct from the most thoroughly characterized plant glucose sensor, hexokinase (4). This finding is not surprising because multiple sugar-sensing mechanisms exist in plants (9, 31). Further studies should be directed toward an unequivocal demonstration of glucose binding to AtRGS1 and glucose-mediated modulation of AtRGS1 GAP activity. This system is not unique to Arabidopsis because we have identified AtRGS1 orthologs in several plant species (SI Figs. 10–12). Moreover, we identified putative 7TM RGS proteins in both fungi and protozoa as well (SI Figs. 10–12). These findings suggest that many organisms may use 7TM RGS proteins for glucose sensing and the control of cell proliferation.
Methods
Materials and Data Analysis.
[35S]GTPγS and [γ-32P]GTP were from PerkinElmer (Wellesley, MA). [β-32P]GDP was from MP Biomedicals (Solon, OH). BODIPYFL-GTPγS was from Molecular Probes (Eugene, OR). XTP and UTP were from Rob Nicholas (University of North Carolina), and all other nucleotides were obtained from Sigma–Aldrich (St. Louis, MO). Unless otherwise specified, all other chemicals were of the highest purity obtainable from Sigma–Aldrich or Fisher Scientific (Pittsburgh, PA). Nonlinear regression and statistical analyses were performed in Prism version 4.0 (GraphPad, San Diego, CA). All data presented are representative of three or more independent experiments. Multiple comparison tests were calculated by ANOVA by using either Dunnett's or Bonferroni's post-test at the 95% significance level (Prism).
Protein Purification/Enzymology.
His6-AtGPA1-GDP and GST-AtRGS1 (amino acids 249–459) were purified as described (12). Site-directed mutagenesis was conducted by using QuikChange (Stratagene, La Jolla, CA). [35S]GTPγS binding and [γ-32P]GTP steady-state hydrolysis assays were conducted as described (32). SPR assays using an anti-GST biosensor were conducted as described (12, 18). GST, GST-AtRGS1(wild type), and GST-AtRGS1(E320K) were immobilized to 240, 250, and 290 resonance units (RU), respectively. Bulk buffer refractive index change upon AtGPA1 injection was observed to increase response units on all sensor surfaces equally by ≈200 RU. Bulk shift and nonspecific binding were accounted for by subtraction of simultaneous sensorgram curves derived from the GST-only surface.
Intrinisic Tryptophan Fluorescence.
Tryptophan fluorescence of Gα was used as a probe for G protein activation (33). Structural and mutagenic analyses indicate that a tryptophan residue in the α2-helix (switch-II), equivalent to W207 in Gαt (34), shifts from a solvent-exposed area to a hydrophobic pocket upon Gα activation, resulting in an increased fluorescence quantum yield (35). AtGPA1 also has this α2-helix tryptophan (W229). Tryptophan fluorescence of AtGPA1 was measured at 20°C in 10 mM Tris·HCl (pH 8.0), 1 mM EDTA, and 10 mM MgCl2 by using a PerkinElmer LS55 spectrophotometer. Excitation and emission wavelengths were 282 and 340 nm, respectively, with slits widths of 5 nm.
GDP Release.
First, 100 nM AtGPA1 was preloaded with 3.2 nM [β-32P]GDP [3,000 Ci/mmol (1 Ci = 37 GBq)] for 10 min at 20°C in 20 mM Tris·HCl (pH 8.0), 2 mM EDTA, 25 mM MgCl2, 100 mM NaCl, 1 mM DTT, and 0.1% (wt/vol) C12E10. Then, samples were placed on ice, and aliquots were taken to assess total [β-32P]GDP bound. To quantify release of bound [β-32P]GDP, 100 μM GTPγS was added, and reactions again were incubated at 20°C. Aliquots were taken at indicated times, vacuum-filtered onto HA45 nitrocellulose (Millipore, Billerica, MA), washed, and analyzed by liquid scintillation. Data were plotted as amount [β-32P]GDP bound, with the initial preloaded aliquot serving as zero time point, and fit to a single exponential decay function.
Single Turnover GTP Hydrolysis.
The rate of GTP hydrolysis by AtGPA1 was measured by a single turnover. Mg2+ is a crucial cofactor for GTP hydrolysis (16), and thus it is typically excluded from the [γ-32P]GTP loading phase to prevent hydrolysis before initiation of the single turnover reaction (36). However, AtGPA1 was observed to be highly dependent on Mg2+ for GTP binding (data not shown). Thus, we modified our standard method (32) to account for [32Pi] release during preloading, as detailed in SI Fig. 7.
Arabidopsis.
All experiments were conducted by using Columbia ecotype A. thaliana. Generation and characterization of the majority of Arabidopsis lines containing T-DNA insertions and transgenic alleles used in these studies are described (4, 5, 13). Isolation of Atrgs1–2/Atgpa1–4 double mutants also has been described (13). AtGPA1-L-CFP consists of enhanced cyan fluorescent protein inserted into the loop between the predicted αA and αB helices (between Ala-97 and Gln-98) of AtGPA1 as described (6, 13).
Plant Growth Assays.
For phenotypic analyses, wild-type and mutant seeds were sterilized; sown in Petri dishes containing 1/2 Murashige and Skoog basal medium with Gamborg's vitamins (ICN Biomedicals, Aurora, OH), 1% (wt/vol) sucrose, and 0.5% (wt/vol) phytoagar (Research Products International, Mt. Prospect, IL); adjusted to pH 5.7; and treated at 4°C in the dark for 3 days and then moved to a growth chamber with 23°C and light intensity of 100 μmol per m2/s. For the phenotypic analysis of 2-day-old etiolated seedlings, Petri dishes were wrapped in aluminum foil and placed in the dark at 23°C. Etiolated hypocotyls were measured by ruler.
AtRGS1 Transgenic Plants.
The AtRGS1 ORF (At3g26090) was PCR-amplified from cDNA made from seedlings grown in light for 10 days, cloned into the pENTR/D-TOPO vector (Invitrogen, Carlsbad, CA), and then subcloned into Gateway plant transformation destination binary vector pB2GW7 or pGWB42 for the AtRGS1-yellow fluorescent protein (YFP) fusion by attL/attR site recombination reactions. The E320K mutation was created by using QuikChange mutagenesis. In these constructs, expression was driven by the 35S promoter of the cauliflower mosaic virus. All constructs were transformed into Arabidopsis plants of the indicated genotypes by Agrobacterium-mediated transformation (37), and expression of all transgenes was verified by RT-PCR (SI Fig. 13).
Green Seedling Assay.
The green seedling assay was performed largely according to published protocols (4). Briefly, wild-type (Col-0), mutant, and transgenic seeds were sown, chilled, light treated, and grown under identical conditions until maturation. Seeds from matched lots were sterilized with 80% (vol/vol) ethanol for 2 min, followed by 30% (vol/vol) bleach with 0.1% (vol/vol) Tween-20 for 10 min, and then washed with sterile deionized water six times under sterile conditions. Sterilized seeds were sown on plates consisting of 1/2 Murashige and Skoog basal medium with vitamins (Plantmedia, Dublin, OH) (pH adjusted to 5.7 with 1 N KOH), 0.5% (wt/vol) phytoagar (Plantmedia) and different concentrations of d-glucose (Sigma–Aldrich) and stratified at 4°C in the dark for 48 h. Then plates were moved to a 23°C growth chamber, under 16/8-h photo period at 100 μmol/m2/s, and placed horizontally. Ten days later, the percentage of green seedling was scored as the number of green seedlings divided by the total number of seeds. Each experiment was repeated three times. Minimally, 50 seeds were scored for each treatment of each genotype.
FRET Microscopy.
Fluorescence images of AtRGS1-YFP/AtGPA1-L-CFP seedlings were captured by using an Olympus IX81 inverted microscope (Center Valley, PA) controlled by IPlab software version 3.6 (BD Biosciences, Rockville, MA). Images of CFP, YFP, and the YFP/CFP ratio were observed through a 60× water immersion objective and simultaneously captured by a cooled charge-coupled device Photometrics Cascade Digital Camera (Roper Scientific, Tucson, AZ) equipped with CFP/YFP FRET emission filter OI-05-EM in a dual-view mounting tube. Filter sets used were YFP (excitation, 500/20 nm; emission, 535/30 nm), CFP (excitation, 436/20 nm; emission, 480/40 nm), and FRET (505dcxr; excitation, 436/20 nm; emission, 480/30 nm and 535/40 nm). Normalized net FRET was calculated in IPlab version 3.6 software using established algorithms for two-filter FRET with fluorescence microscopy (38).
Supplementary Material
Acknowledgments
We thank Dr. Robert Nicholas (University of North Carolina) for providing nucleotides. This work was supported by National Institute of General Medical Sciences Grant R01GM065989 (to A.M.J.), Department of Energy Grant DE-FG02–05er15671 (to A.M.J.), and National Science Foundation Grant MCB-0209711 (to A.M.J.); Natural Sciences and Engineering Research Council of Canada, Canadian Foundation for Innovation, and British Columbia Knowledge Development Fund grants (to J.-G.C.); National Institute of General Medical Sciences Grant R01GM074268 (to D.P.S.); National Research Service Award Fellowship GM076944 (to C.A.J.); and the China Scholarship Council (Y.G.).
Abbreviations
- 7TM
seven-transmembrane domain
- C.I.
confidence interval
- GAP
GTPase-accelerating protein
- GEF
guanine-nucleotide exchange factor
- GPCR
G protein-coupled receptor.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/cgi/content/full/0704751104/DC1.
References
- 1.Birnbaumer L. Annu Rev Pharmacol Toxicol. 1990;30:675–705. doi: 10.1146/annurev.pa.30.040190.003331. [DOI] [PubMed] [Google Scholar]
- 2.Pierce KL, Premont RT, Lefkowitz RJ. Nat Rev Mol Cell Biol. 2002;3:639–650. doi: 10.1038/nrm908. [DOI] [PubMed] [Google Scholar]
- 3.McCudden CR, Hains MD, Kimple RJ, Siderovski DP, Willard FS. Cell Mol Life Sci. 2005;62:551–577. doi: 10.1007/s00018-004-4462-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen JG, Jones AM. Methods Enzymol. 2004;389:338–350. doi: 10.1016/S0076-6879(04)89020-7. [DOI] [PubMed] [Google Scholar]
- 5.Chen JG, Willard FS, Huang J, Liang J, Chasse SA, Jones AM, Siderovski DP. Science. 2003;301:1728–1731. doi: 10.1126/science.1087790. [DOI] [PubMed] [Google Scholar]
- 6.Huang J, Taylor JP, Chen JG, Uhrig JF, Schnell DJ, Nakagawa T, Korth KL, Jones AM. Plant Cell. 2006;18:1226–1238. doi: 10.1105/tpc.105.037259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen Y, Ji F, Xie H, Liang J, Zhang J. Plant Physiol. 2006;140:302–310. doi: 10.1104/pp.105.069872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pandey S, Chen JG, Jones AM, Assmann SM. Plant Physiol. 2006;141:243–256. doi: 10.1104/pp.106.079038. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 9.Wang HX, Weerasinghe RR, Perdue TD, Cakmakci NG, Taylor JP, Marzluff WF, Jones AM. Mol Biol Cell. 2006;17:4257–4269. doi: 10.1091/mbc.E06-01-0046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Moriyama EN, Strope PK, Opiyo SO, Chen Z, Jones AM. Genome Biol. 2006;7:R96. doi: 10.1186/gb-2006-7-10-r96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Johnston CA, Temple BR, Chen JG, Gao Y, Moriyama EN, Jones AM, Siderovski DP, Willard FS. Science. 2007 doi: 10.1126/science.1143230. in press. [DOI] [PubMed] [Google Scholar]
- 12.Willard FS, Siderovski DP. Methods Enzymol. 2004;389:320–338. doi: 10.1016/S0076-6879(04)89019-0. [DOI] [PubMed] [Google Scholar]
- 13.Chen JG, Gao Y, Jones AM. Plant Physiol. 2006;141:887–897. doi: 10.1104/pp.106.079202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ferguson KM, Higashijima T, Smigel MD, Gilman AG. J Biol Chem. 1986;261:7393–7399. [PubMed] [Google Scholar]
- 15.Arenas-Huertero F, Arroyo A, Zhou L, Sheen J, Leon P. Genes Dev. 2000;14:2085–2096. [PMC free article] [PubMed] [Google Scholar]
- 16.Higashijima T, Ferguson KM, Smigel MD, Gilman AG. J Biol Chem. 1987;262:757–761. [PubMed] [Google Scholar]
- 17.Berman DM, Kozasa T, Gilman AG. J Biol Chem. 1996;271:27209–27212. doi: 10.1074/jbc.271.44.27209. [DOI] [PubMed] [Google Scholar]
- 18.Willard FS, Kimple AJ, Johnston CA, Siderovski DP. Anal Biochem. 2005;340:341–351. doi: 10.1016/j.ab.2005.02.015. [DOI] [PubMed] [Google Scholar]
- 19.Ullah H, Chen JG, Young JC, Im KH, Sussman MR, Jones AM. Science. 2001;292:2066–2069. doi: 10.1126/science.1059040. [DOI] [PubMed] [Google Scholar]
- 20.Mishra G, Zhang W, Deng F, Zhao J, Wang X. Science. 2006;312:264–266. doi: 10.1126/science.1123769. [DOI] [PubMed] [Google Scholar]
- 21.Liu X, Yue Y, Li B, Nie Y, Li W, Wu WH, Ma L. Science. 2007;315:1712–1716. doi: 10.1126/science.1135882. [DOI] [PubMed] [Google Scholar]
- 22.Plakidou-Dymock S, Dymock D, Hooley R. Curr Biol. 1998;8:315–324. doi: 10.1016/s0960-9822(98)70131-9. [DOI] [PubMed] [Google Scholar]
- 23.Pandey S, Assmann SM. Plant Cell. 2004;16:1616–1632. doi: 10.1105/tpc.020321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen JG, Pandey S, Huang J, Alonso JM, Ecker JR, Assmann SM, Jones AM. Plant Physiol. 2004;135:907–915. doi: 10.1104/pp.104.038992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Marrari Y, Crouthamel M, Irannejad R, Wedegaertner PB. Biochemistry. 2007;46:7665–7677. doi: 10.1021/bi700338m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Adjobo-Hermans MJ, Goedhart J, Gadella TW., Jr J Cell Sci. 2006;119:5087–5097. doi: 10.1242/jcs.03284. [DOI] [PubMed] [Google Scholar]
- 27.Digby GJ, Lober RM, Sethi PR, Lambert NA. Proc Natl Acad Sci USA. 2006;103:17789–17794. doi: 10.1073/pnas.0607116103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yuan C, Sato M, Lanier SM, Smrcka AV. J Biol Chem. 2007;282:19938–19947. doi: 10.1074/jbc.M700396200. [DOI] [PubMed] [Google Scholar]
- 29.Brandt DR, Ross EM. J Biol Chem. 1985;260:266–272. [PubMed] [Google Scholar]
- 30.Higashijima T, Ferguson KM, Sternweis PC, Smigel MD, Gilman AG. J Biol Chem. 1987;262:762–766. [PubMed] [Google Scholar]
- 31.Rolland F, Baena-Gonzalez E, Sheen J. Annu Rev Plant Biol. 2006;57:675–709. doi: 10.1146/annurev.arplant.57.032905.105441. [DOI] [PubMed] [Google Scholar]
- 32.Afshar K, Willard FS, Colombo K, Johnston CA, McCudden CR, Siderovski DP, Gonczy P. Cell. 2004;119:219–230. doi: 10.1016/j.cell.2004.09.026. [DOI] [PubMed] [Google Scholar]
- 33.Higashijima T, Ferguson KM, Sternweis PC, Ross EM, Smigel MD, Gilman AG. J Biol Chem. 1987;262:752–756. [PubMed] [Google Scholar]
- 34.Faurobert E, Otto-Bruc A, Chardin P, Chabre M. EMBO J. 1993;12:4191–4198. doi: 10.1002/j.1460-2075.1993.tb06103.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lan KL, Remmers AE, Neubig RR. Biochemistry. 1998;37:837–843. doi: 10.1021/bi972122i. [DOI] [PubMed] [Google Scholar]
- 36.Krumins AM, Gilman AG. Methods Enzymol. 2002;344:673–685. doi: 10.1016/s0076-6879(02)44748-9. [DOI] [PubMed] [Google Scholar]
- 37.Bechtold N, Pelletier G. Methods Mol Biol. 1998;82:259–266. doi: 10.1385/0-89603-391-0:259. [DOI] [PubMed] [Google Scholar]
- 38.Gordon GW, Berry G, Liang XH, Levine B, Herman B. Biophys J. 1998;74:2702–2713. doi: 10.1016/S0006-3495(98)77976-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}