

Abstract
We reported previously that homocysteine (Hcy) inhibits endothelial cell (EC) growth by transcriptional inhibition of the cyclin A gene via a hypomethylation-related mechanism. In this study, we examined the effect of Hcy on epigenetic modification of the cyclin A gene and its biologic role in human ECs. Cyclin A mRNA levels were significantly suppressed by Hcy and a DNA methyltransferase inhibitor. The cyclin A promoter contains a CpG island spanning a 477-bp region (−277/200). Bisulfite sequencing followed by polymerase chain reaction (PCR) amplification of the cyclin A promoter (−267/37) showed that Hcy eliminated methylation at 2 CpG sites in the cyclin A promoter, one of which is located on the cycle-dependent element (CDE). Mutation of CG sequence on the CDE leads to a 6-fold increase in promoter activity. Hcy inhibited DNA methyltransferase 1 (DNMT1) activity by 30%, and reduced the binding of methyl CpG binding protein 2 (MeCP2) and increased the bindings of acetylated histone H3 and H4 in the cyclin A promoter. Finally, adenovirus-transduced DNMT1 gene expression reversed the inhibitory effect of Hcy on cyclin A expression and EC growth inhibition. In conclusion, Hcy inhibits cyclin A transcription and cell growth by inhibiting DNA methylation through suppression of DNMT1 in ECs.
Introduction
Hyperhomocysteinemia (HHcy) is an independent risk factor for cardiovascular disease (CVD), but the underlying mechanisms remain unclear. Although numerous studies have established that homocysteine (Hcy) has atherogenic effects on cultured vascular cells and in animal models of HHcy, the biochemical basis by which HHcy contributes to arteriosclerosis remains largely undefined.
We initially proposed hypomethylation as a specific biochemical mechanism by which Hcy induces vascular injury.1,2 Hcy can use adenosine, a normal constituent of all body fluids, to form S-adenosyl-homocysteine (SAH), a potent inhibitor of cellular methylation. We have demonstrated that Hcy, but not cysteine, arrested cell growth and increased cellular SAH concentration in endothelial cells (ECs), but not in vascular smooth muscle cells (VSMCs).3 The hypomethylation hypothesis is supported by clinical studies showing that elevated Hcy levels in patients are linked to increased SAH and impaired erythrocyte membrane protein methylation,4 and by animal studies showing that cystathionine β-synthase (CBS)–deficient mice have increased SAH levels and decreased DNA methylation.5,6 Because damage to and impaired regeneration of ECs is a key feature of arteriosclerosis, growth inhibition of ECs may represent an important mechanism by which Hcy induces atherosclerosis.
Our recent work suggested that the cyclin A gene is an important molecular target that mediates Hcy-induced EC growth inhibition.7 Cyclin A (also named cyclin A2 as opposed to the male germ cell–specific cyclin A1) promotes both G1/S and G2/M transitions of the cell cycle in somatic cells. Cyclin A is expressed in late G1 and throughout S phase, and is controlled mainly at the transcriptional level. We have shown that the cyclin A gene is differentially regulated at a transcriptional level by Hcy in vascular cells. In ECs, clinically relevant concentrations of Hcy (10-50 μM) arrested the cell cycle at G1/S transition via cyclin A transcriptional inhibition.7 In contrast, a supraphysiological concentration of Hcy (1 mM) activated cyclin A and promoted cell proliferation of VSMCs.8 Because cyclin A transcriptional inhibition by Hcy is associated with SAH accumulation in ECs, we hypothesize that DNA hypomethylation may be a key biochemical mechanism responsible for Hcy-induced cyclin A suppression and growth inhibition in ECs.
DNA methylation is an important epigenetic mechanism that selectively regulates gene expression, and is associated with cancer development and cardiovascular disease.9,10 DNA methylation at the promoter region can control gene transcription by recruiting methylcytosine-binding proteins (MBPs), which in turn attract histone deacetylases (HDACs) to bind to the methylated promoters.11 HDAC removes the acetyl group from histone and results in histone deacetylation, which leads to chromatin condensation and inhibits the access of regulatory proteins to the promoter. The molecular and biochemical mechanisms of Hcy-related DNA methylation have not been investigated. In this study, we have examined the key events in the process of DNA methylation and chromatin remodeling on cyclin A promoter in the presence and absence of Hcy, and assessed its biologic significance in cultured primary human vascular cells.
Materials and methods
Cell culture and chemicals
Human umbilical vein endothelial cells (HUVECs) and human aortic smooth muscle cells (HASMCs) (Cambrex, East Rutherford, NJ) were cultured as previously described and used between passages 6 to 8.3,7 The cells were cultured to 80% to 90% confluence and then changed to control medium containing 40 μM adenosine and 10 μM erythro-9-(2-hydroxy-3-nonyl)-adenine hydrochloride, an adenosine deaminase inhibitor to stabilize adenosine, and treated with 50 μM DL-Hcy or other chemicals. Chemicals, if not specified, were purchased from Sigma-Aldrich (St Louis, MO).
Promoter CpG island search
The properties of CpG islands in the proximal region of the human cyclin A promoter (−3740/258) and human cyclin D1 promoter (−5000/212) were examined using a CpG Island Search engine (http://cpgislands.usc.edu; University of Southern California).12 A CpG island is identified as a DNA region of greater than 200-bp stretch with a CG content of 50% or more and CpG ratio of 0.6 or higher.13
Reporter gene construction and promoter activity study
The human cyclin A 5′-flanking region was cloned as described.14 Fragments of cyclin A 5′-flanking region, a 3445-bp SmaI-SmaI restriction fragment (−3200/245), a 761-bp SacI-SmaI fragment (−516/245), and polymerase chain reaction (PCR) fragments of 471 bp (−266/205), 338 bp (−133/+205), and 90 bp (−85/5) were inserted into promoterless luciferase reporter plasmid pGL2-basic (Promega, Madison, WI).15 The ATF consensus sequence TGACGTCA was mutated to TGCCCCCA; the CDE consensus sequence CGCGGG, to ATATTG; and the E2F1 consensus sequence TTTGGCT, to TTTGAAT in the plasmid −133/+205 using a site-directed mutagenesis kit from Invitrogen (Frederick, MD). Promoter activity of the cyclin A constructs was examined in cultured HUVECs by transient transfection and luciferase assay as we described previously.7 Plasmid pCMV-βGAL expressing β-gal was cotransfected to correct for variability in transfection efficiency. The ratio of luciferase activity to β-gal activity in each transfection served as a measure of normalized luciferase activity.
Bisulfate genomic DNA sequencing
HUVECs were plated on 100-mm plates (2-5 × 105 cells/cm2) and cultured to 80% to 90% confluence. The cells were changed to control medium and treated with 50 μM DL-Hcy for 48 hours. Genomic DNA was extracted and modified by bisulfite, which converts all unmethylated cytosines to uracil as described.16 Briefly, 2 μg genomic DNA (21 μL) was incubated with 4 μL of 2 M NaOH at 50°C for 15 minutes to denature the DNA, and mixed with 50 μL low-melting-point agarose (2%; Gibco, Grand Island, NY). The DNA agarose mixture (10 μL) was gently pipetted into 300 μL prechilled mineral oil and left on ice for 30 minutes to form beads. Bisulfite solution (500 μL, 5 M sodium bisulfite and 1 M hydroquinone) was added and incubated at 50°C for 4 hours. The beads were washed subsequently with Tris-EDTA (TE), 0.2 N NaOH, and TE again and water. Bisulfite-modified DNA was amplified with cyclin A–specific primers (primer 1, −131/−107, sense, 5′AGTTTTTTTTGGTTTATTTTTTATT3′; primer 2, 138/119, antisense, 5′ACCCAAACCAACCTACCAAC3′) designed using MethPrimer, a program for designing primers for methylation mapping.17 The PCR products were cloned into the TA cloning vector (Promega). DNA sequencing was performed for 10 plasmid clones from each group.
DNA methyltransferase (DNMT) activity assay
HUVECs were cultured to 80% to 90% confluence on 150-mm plates. The cells were changed to control medium and treated with 50 μM DL-Hcy or 1 mM azacytidine (AZC), a DNMT inhibitor, for 48 hours. Nuclear extract was prepared as described18 with modification. Three standard DNA primers (IDT, Coralville, IA) were used to prepare hemimethylated DNA substrate for DNMT1 and unmethylated DNA substrate for DNMT3: primer 1 (methylation), 5′CCAAGCGCGCGCCTGGCGCCCGGGCCGGCTCAAGCGCGCGCCTGGCGCCCGGATC3′; primer 2 (methylation), 5′GATCCGGGCGCCAGGCGCGCGCTTGAGCCGGCCCGGGCGCCAGGCGCGCGCTTGG3′; primer 3 (methylation), 5′GATCCGGG(5-MethyldC)GCCAGG(5-MethyldC)GCGCGCTTGAGCCGGCCCGGGC-GCCAGG(5-MethyldC)G(5-Methyl dC)GCGCTTGG3′, each 55-bp long.
Primers 1 and 3 were annealed as described19 and used as the hemimethylated DNA substrate for DNMT1, while annealed primers 1 and 2 were used as the unmethylated DNA substrate for DNMT3. DNMT activity was assessed by incubating 20 μg nuclear extract with 500 ng double-stranded DNA substrate and 3 μCi (0.111 MBq) S-adenosyl-L-[methyl-3H]methionine as methyl donor (Perkin Elmer, Boston, MA) in DNMT buffer (100 μL, 10 mM Hepes buffer [pH 7.9], 3 mM MgCl2,100 mM KCl, 5% glycerol, 2 mM EDTA) in a total volume of 100 μL at 37°C for 2 hours. The reaction was terminated by adding 300 mL stop buffer (1% SDS, 2 mM EDTA, 5% butanal, 0.25 mg/mL calf thymus DNA, and 1 mg/mL proteinase K), and the reaction mix was spotted onto a Whatman GF/C filter (Clifton, NJ), which was air dried, washed twice with ice-cold 5% TCA containing 5 mM sodium pyrophosphate, and rinsed with 70% ethanol. Total radioactivity incorporated into DNA was measured by liquid scintillation counting. DNMT activity was expressed as cpm/μg nuclear protein.
Chromatin immunoprecipitation (CHIP) assay
HUVECs and HASMCs were cultured to 80% to 90% confluence on 150-mm plates. The cells were changed to control medium and treated with 50 μM DL-Hcy and 1 mM AZC for 48 hours, or histone deacetylase inhibitors trichostatin A (TSA, 300 nM) and sodium butyrate (NaBr, 5 mM) for 24 hours. CHIP assay was performed as described20 with modification. Cells were treated with 1% formaldehyde for 8 minutes to cross-link proteins to DNA, collected, washed with PBS containing 1 mM phenylmethylsulfonyl fluoride, and ice-cold solution 1 (0.25% Triton X-100, 10 mM EDTA, 0.5 mM EGTA, 10 mM HEPES [pH 7.5]) and thereafter solution 2 (0.2 M NaCl, 1 mM EDTA, 0.5 mM EGTA, 10 mM HEPES, [pH 7.5]). The pellet was resuspended in 0.5 mL of lysis buffer (150 mM NaCl, 25 mM Tris-Cl,] pH 7.5[, 5 mM EDTA, 1% Triton X-100, 0.1% SDS, 0.5% sodium deoxycholate, and protease inhibitors) and sonicated. The lysate was diluted to 1 mL in lysis buffer, and a 20-μL aliquot was saved as input. The rest of the nuclear extracts was precleared with 2.5 μg rabbit IgG (sc20691x; Santa Cruz Biotechnology, Santa Cruz, CA) and then protein A–agarose, and subsequently immunoprecipitated with antibodies against methyl CpG–binding protein 2 (MeCP2), acetylated histone 3 (AcH3), or acetylated histone 4 (AcH4) overnight at 4°C. Immunoprecipitated complexes were collected by adding salmon sperm DNA/protein A–agarose slurry for 1 hour at 4°C, washed sequentially with radioimmune precipitation assay buffer (150 mM NaCl, 50 mM Tris-HCl [pH 8.0], 0.1% SDS, 0.5% sodium deoxycholate, 1.0% Nonidet P-40), high salt wash (500 mM NaCl, 1.0% Nonidet P-40, 0.1% SDS, 50 mM Tris-Cl [pH 8.0]), LiCl wash (250 mM LiCl, 1.0% Nonidet P-40, 0.5% sodium deoxycholate, 1 mM EDTA, 50 mM Tris-Cl [pH 8.0]), and then TE buffer (pH 8.0). The beads were then treated with RNase A (50 μg/mL) and proteinase K (7.5 μL of 20 mg/mL) at 37°C for 30 minutes. Cross-links were reversed at 65°C overnight. DNA was extracted with phenol/chloroform and coprecipitated with glycogen, dissolved in 25 μL TE buffer, and subjected for PCR amplification of cyclin A promoter (−267/37) with primer CY1 (−2676/−249), 5′CGGACAGCCTCGCTCACTA3′, and primer CY2 (20/37), 5′AGCCAAAGACGCCCAGAG3′ (30 cycles; denaturing: 95°C for 30 seconds; annealing: 55°C for 45 seconds; and extension: 72°C for 30 seconds).
Construction of recombinant DNMT1 and DNMT3 adenoviruses and adenoviral transduction
The recombinant adenoviruses expressing human DNMT1 or DNMT3a gene were constructed with replication-defective adenoviral shuttle vector pAdtrack-CMV-GFP and adenoviral backbone plasmid pADEasy-1 (Stratagene, La Jolla, CA) as described. The DNMT1 fragment (Kpn1-Not1) from a DNMT1-pBSKS subclone (EcoR1-Not1) and DNMT3a (Sal1-HindIII) were inserted into pAdtrack-CMV-GFP vector, which was then linearized with Pme1 and transfected by electroporation into BJ5183 E coli. The recombination was confirmed by restriction endonuclease digestion. Finally, the recombinant plasmids pAd-DNMT3 and pAd-DNMT1 were linearized with Pac I and transfected into adenovirus packaging 293 cells. The recombinant adenoviruses were expanded, purified, and titrated as described.7 The recombinant adenovirus encoding green fluorescent protein (Adv-CT) was used as a control. HUVECs at approximately 70% confluence were infected with purified adenovirus at indicated MOI, treated with Hcy after 24 hours of virus infection. Western blotting was carried out to examine ectopic gene expression using antibodies against DNMT1 (Santa Cruz Biotechnology) and DNMT3 (Santa Cruz Biotechnology).
[3H]-thymidine incorporation, and Northern and Western blot analysis
HUVECs at 80% to 90% confluence were treated with dl-Hcy in the control medium, and assessed for proliferation by [3H]-thymidine incorporation, and gene expression by Northern and Western blot analysis as described.7
Statistical analysis
Each experiment was repeated 3 times. Results are expressed as the mean plus or minus standard deviation (± SD). Statistical comparison of single parameters between 2 groups was performed by paired Student t test. Kruskal-Wallis one-way ANOVA was used to compare the means of multiple groups, followed by Dunn test. P values less than or equal to .05 were considered significant.
Results
DNA synthesis and cyclin A mRNA expression are suppressed by Hcy, AZC, TSA, and NaBr
We reported previously that Hcy inhibited EC growth and cyclin A gene transcription in the presence of adenosine. This is associated with an increase in SAH, a potent inhibitor of methyltransferase. To test the susceptibility of DNA synthesis and cyclin A transcription to modulation by DNA methylation, we treated HUVECs with AZC, a potent DNA demethylating agent that can be incorporated into newly synthesized DNA and form covalent complexes between cytosine-specific methyltransferases and the modified DNA, thereby inhibiting DNA methylation. We found that AZC (1 μM) significantly inhibited DNA synthesis and the expression of the cyclin A mRNA, in a similar pattern to that of Hcy (50 μM) (Figure 1). In contrast, neither Hcy nor AZC reduced the mRNA levels of cyclin D1, another key cyclin controlling G1/S transition in the cell cycle. Because DNA methylation can modulate histone acetylation, another epigenetic mechanism regulating chromatin remolding and gene expression, we examined the effect of histone deacetylase inhibitors (TSA and NaBr) on DNA synthesis and cyclin Atranscription. We found that both DNA synthesis and cyclin A mRNA levels were significantly suppressed by TSA and NaBr in HUVECs. These data support the hypothesis that DNA methylation is a critical mechanism mediating cyclin A transcription and cell growth in ECs.
Figure 1.

DNA synthesis and mRNA levels in HUVECs. HUVECs were treated with 50 μM DL-Hcy and 1 mM AZC in control medium for 48 hours, or 300 nM TSA and 5 mM NaBr for 24 hours. (A) Thymidine uptake. Cells were metabolically labeled with 1 μCi (0.037 MBq)/mL [3H]-thymidine during the last 3 hours. Values represent mean (± SD) from 3 separate experiments with triplicates (n = 9). *P < .01 versus non-Hcy and nonviral control. #P < .01 versus Hcy-treated and non–viral-transduced group. (B) Northern blot analysis. Total cellular RNA was used for Northern analysis (10 μg). The blot was hybridized with the probes of human cyclins A and D1, and subsequently with 18S RNA.
A CpG island is located in cyclin A core promoter
DNA methylation occurs at CpG islands, CG-rich regions that frequently coincide with the promoter region. Using a CpG Island Search program, we analyzed the DNA sequence at the 5′-flanking region of the cyclin A and cyclin D1genes, and identified a 477-bp long CpG island that starts at −277 and extends up to +200, with a CG content of 63.31% and a CpG ratio of 0.88, in the core promoter region of the cyclin Agene. There are 2 CpG islands (−4366/−3812 and −3668/−2718) at the far end of the 5′-flanking region of the cyclin D1 gene (Figure 2A). We generated luciferase constructs to test promoter activity of various lengths of the 5′-flanking region of the cyclin Agene. The cyclin A −266/205 fragment, which spans the CpG island, performed the highest promoter activity, which was defined as 100% (Figure 2B). The 3445-bp (3200/245), 761-bp (−516/245), and 338-bp fragments (−133/205) expressed similar promoter activity (81%-87%), while the 88-bp (−83/5) fragment had minimum activity (8.1%). These data indicate that a CpG island is located in the cyclin A core promoter and suggest that methylation may play a role in regulating cyclin Atranscription.
Figure 2.

Promoter analysis. (A) CpG island search. One CpG island is identified in the core promoter region of the cyclin Agene, and 2 in the far 5′-flank region of cyclin D1 gene by computational analysis. (B) Cyclin A promoter activity. Plasmids containing various lengths of the 5′-flanking region of the cyclin A gene were constructed with the luciferase (Luc) gene in PGL2 vector. HUVECs were transiently transfected with 2 μg plasmid DNA by Lipofectin transfection, and harvested 48 hours after transfection. For each construct, 0.5 μg plasmid pCMV-βGAL was cotransfected to correct for differences in the transfection efficiency. The corrected luciferase activity was then divided by that of the cyclin A −266/+205 plasmid and is presented as relative luciferase activity. Values represent mean (± SD) from 3 separate experiments with triplicates (n = 9).
Hcy inhibits the methylation of 2 cytosine residues in cyclin A CpG island
To map methylated cytosine residues in the cyclin A CpG island, we used the bisulfite-sequencing method to convert all unmethylated cytosines to uracil and then sequenced the residues after PCR amplification and cloning. During PCR amplification, uracil pairs with adenine and adenine pairs with thymine. Therefore, following bisulfite modification an unmethylated cytosine will sequence as thymine and a methylated cytosine will sequence as cytosine. Two primers were designed to amplify the CpG island in the cyclin A promoter region and within the primers avoided CG sequence where the cytosine may be converted to uracil. Cyclin A promoter region from −131 to 138 was amplified. The amplified CpG island in the cyclin A promoter contains 23 CpG sites, as indicated in Figure 3A, which involve 3 consensus elements where cis-acting transcription factors bind, including activating transcription factor (ATF)/cyclic AMP-responsive binding protein (CREB), cell cycle–dependent element (CDE)cell-cycle genes homology region (CHR), and E2F1. By bisulfite sequencing, we have identified 2 methylated cytosine residues in the CpG island in the cyclin A promoter (CpG sites 8 and 14, positions 1 and 36) in the control HUVECs, as indicated by frames in Figure 3A and by stalks with oval heads in Figure 3B. These 2 cytosines were not methylated and sequenced as thymine in the Hcy-treated HUVECs. The CpG site 8 is located in the center of the CDE repressor site, and the CpG site 14 is immediately following the E2F1 site. To determine the functional importance of the consensus elements in the CpG island for the cyclin A promoter, we performed transient transfection experiments with mutated cyclin A promoter constructs. In control HUVECs, mutation of the ATF and E2F1 led to a decrease in cyclin A promoter to 28% and 42%, respectively, in comparison with the activity of the wild-type construct (−133/+205) (Figure 3C), whereas the activity of the CDE mutation, where CG was changed to AT, was increased to 622%. In Hcy-treated HUVECs, promoter activity of the wild-type cyclin A promoter was reduced to 30%. The activities of the ATF/CREB, CDE, and E2F1 mutations were not significantly affected by Hcy treatment. These data indicate that Hcy inhibits the methylation of 2 cytosine residues in the cyclin A CpG island and that demethylation of the CDE repressor element may contribute to the inhibition of cyclin A transcription in endothelial cells.
Figure 3.

Cyclin A promoter study. (A) CpG island sequence. Human cyclin A promoter CpG island (−131/138) contains 23 CpG sites (bold) and 3 consensus elements (underlined). Cis-acting transcription factors that bind to cyclin A promoter include ATF/CREB, CDE/CHR, and E2F. (B) Methylation status bisulfate genomic DNA sequencing. HUVECs were treated with 50 μM DL-Hcy for 48 hours. Genomic DNA was extracted and modified by bisulfite to convert all unmethylated cytosines to uracils. Bisulfite-modified DNAs were amplified with cyclin A–specific primers, cloned into the TA cloning vector, and sequenced. Unmethylated cytosines are indicated by stalks. Methylated cytosines are indicated by stalks with oval heads. (C) Promoter activities. Plasmids containing cyclin A promoter (−133/+205) with mutations on each consensus element were constructed with the luciferase (Luc) gene in PGL2 vector. HUVECs were transiently transfected with 2 μg PGL2 plasmid DNA by Lipofectin transfection. Cells were treated with 50 μM DL-Hcy in control medium 24 hours after transfection, and harvested 24 hours later. The corrected luciferase activity for each time point was divided by that of control plasmid in cells not treated with Hcy, and is presented as relative luciferase activity. Values represent the mean (± SD) from 3 separate experiments (n = 9). *P < .01 versus control.
Hcy inhibits DNMT1 activity
DNMT catalyses the transfer of methyl groups to DNA from S-adenosyl-methionine (SAM). In mammals, DNMT1 is the maintenance methyltransferase, which preferentially methylates hemimethylated double-stranded DNA, whereas DNMT3 is known as the de novo methyltransferase, which methylates unmethylated or hemimethylated double-stranded DNA.21 We examined the effect of Hcy on the activities of DNMT1 and DNMT3 by incubating the nuclear extracts with double-stranded unmethylated (for DNMT3) or hemimethylated (for DNMT1) DNA substrates in the presence of [3H]SAM. Activity of DNMT1 was significantly reduced to 71% in Hcy-treated HUVECs in comparison with that of control cells (Figure 4). In contrast, DNMT3 activity was not significantly changed by Hcy treatment. AZC, a specific DNMT inhibitor, served as the positive control and reduced the activities of DNMT1 and DNMT3 to 18% and 23%, respectively. These data support the hypothesis that DNMT1 inhibition may mediate Hcy-induced cyclin A promoter demethylation, cyclin A suppression, and growth inhibition in ECs.
Figure 4.

DNMT activities and protein levels. HUVECs were treated with 50 μM DL-Hcy or 1 mM AZC in control medium for 48 hours. Nuclear extracts (20 μg) were prepared and incubated with hemimethylated double-stranded DNA for DNMT1 activity (A) or with unmethylated double-stranded DNA for DNMT3 activity (B) in the presence of S-adenosyl-L-[methyl-3H] methionine. DNMT activities were determined by the radioactivity level of DNA substrates. Values represent mean (± SD) from 3 separate experiments with triplicated samples (n = 9). *P < .01 versus control. Protein levels of DNMT1 and DNMT3 were examined by Western blotting with antibodies against DNMT1 or DNMT3, respectively, and reblotted with β-actin antibody.
Adenovirus-transduced DNMT1 expression largely restores DNA synthesis and cyclin A expression in Hcy-treated HUVECs
To test the role of DNMT inhibition in mediating Hcy-suppressed growth and cyclin A transcription in ECs, we constructed adenoviruses expressing human DNMT1 and DNMT3. We measured [3H]-thymidine incorporation in adenovirus-infected HUVECs, and examined the effect of viral infection on the growth inhibition induced by Hcy. As shown in Figure 5A, Hcy significantly reduced [3H]-thymidine incorporation to 20% in HUVECs, and this inhibition was not affected by the control adenovirus at 10 to 500 MOI. Cells infected with high concentrations of control virus (500 MOI), showed moderate decreases in [3H]-thymidine incorporation (decreased to 70% and 17% in the absence and presence of Hcy, respectively, possibly reflecting high dose-related nonspecific toxicity). This is similar to what we have previously demonstrated with a different adenoviral construct.7 Adv-DNMT1 and Adv-DNMT3 infections yielded dose-dependent increases in DNMT1 and DNMT3 expression in HUVECs (Figure 5B-D). Adv-DNMT1 infection reversed the Hcy-induced suppression of [3H]-thymidine incorporation, as DNA synthesis recovered to 47% of that of the uninfected control cells at 100 MOI, 71% at 200 MOI, and 86% at 500 MOI (Figure 5B). Adv-DNMT3 infection did not restore Hcy-inhibited DNA synthesis and moderately reduced [3H]-thymidine incorporation at 50 MOI (Figure 5D). Furthermore, with enforced expression of DNMT1 (200 MOI), cyclin A mRNA expression was completely restored (Figure 5C). These data support the hypothesis that Hcy inhibits the activity of DNMT1, results in DNA demethylation of the cyclin Apromoter, and leads to the suppression of cyclin A transcription and EC growth inhibition.
Figure 5.

The effect of adenovirus-transduced DNMT genes on DNA synthesis and cyclin A mRNA levels in Hcy-treated HUVECs. HUVECs were infected with control adenovirus (Ad-CT) (A), adenovirus expressing DNMT1 (Adv-DNMT1) (B,D), and adenovirus expressing DNMT3 (Adv-DNMT3) (C) at the indicated MOI for 24 hours, and then treated with 50 μM DL-Hcy in control medium for another 24 hours. (A-C) Thymidine uptake. Cells were metabolically labeled with 1 μCi (0.037 MBq)/mL [3H]-thymidine during the last 3 hours. [3H]-thymidine incorporation was measured in a liquid scintillation counter. Values represent mean (± SD) from 3 separate experiments with triplicates (n = 9). *P < .01 versus non-Hcy and nonviral control. #P < .01 versus Hcy-treated and non–viral-transduced group. Protein levels of DNMT1 and DNMT3 were examined by Western blotting with antibodies against DNMT1 or DNMT3, and reblotted with β-actin antibody. (D) mRNA levels: HUVECs were infected with Adv-DNMT1 for 24 hours and then treated with DL-Hcy in control medium for another 48 hours. Total cellular RNA (10 μg) was extracted and used for Northern analysis. The blot was hybridized with the probes of human cyclin A and D1, and subsequently with 18S RNA.
Hcy decreases the binding of MeCP2 but increases the bindings of AcH3 and Ach4 to cyclin A promoter CpG island
We performed ChIP assays to examine the association of cyclin A promoter with methyl-CpG–binding protein 2 (MeCP2), the prototype member of the MBP family, which has been reported to bind with higher affinity to methylated CpG site,22 acetylated histone H3 (AcH3), and acetylated histone H4 (AcH4). We found that MeCP2 is associated with the cyclin A promoter −267 to 37 region under control conditions (Figure 6A), which confirmed the status of DNA methylation in this region identified by bisulfite sequencing in Figure 3B. The binding of MeCP2 to the cyclin A promoter was significantly reduced in HUVECs treated with Hcy, AZC, TSA, and NaBr. In contrast, the binding of MeCP2 to the cyclin promoter was not altered by Hcy, TSA, and NaBr, but slightly reduced by AZC in HASMCs. Finally, the binding of AcH3 and AcH4 was absent in the cyclin Apromoter, but significantly induced by treatment with Hcy, AZC, and TSA (Figure 6B). These data are consistent with Hcy-induced DNA hypomethylation of the cyclin A promoter (Figure 3B), and indicate that Hcy-induced DNA hypomethylation leads to decreased binding of MeCP2 and increased binding of acetylated H3 and H4 to the cyclin A promoter in ECs.
Figure 6.
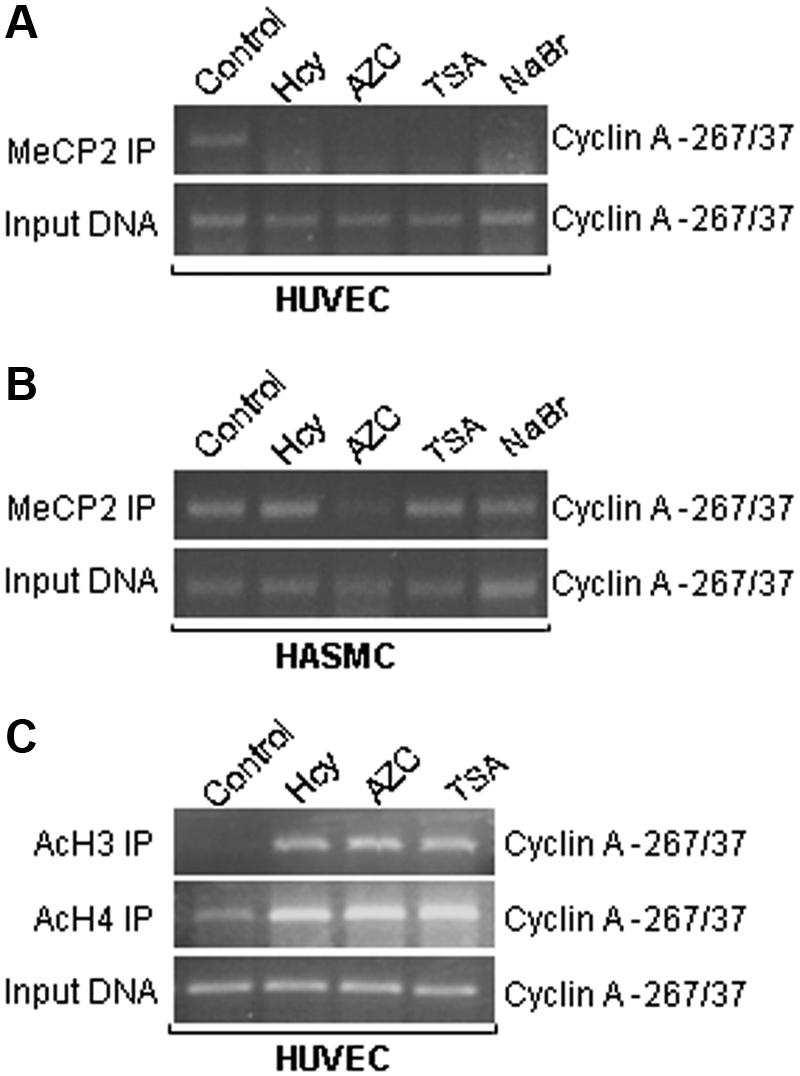
Chromatin remodeling (CHIP assay). HUVECs and HASMCs were treated with 50 μM DL-Hcy and 1 mM AZC in control medium for 48 hours, or 300 nM TSA and 5 mM NaBr for 24 hours. Nuclear extracts were immunoprecipitated with antibody against MeCP2 (A,B), and AcH3 or AcH4 (C). PCR was performed using primers specific for cyclin A promoter from position −267 to 37. Input DNA was amplified to evaluate the total amount of DNA applied to the immunoprecipitation.
Discussion
We have demonstrated in this study that cyclin A mRNA levels are suppressed by Hcy, AZC (a potent DNMT inhibitor), TSA, and NaBr (histone deacetylase inhibitors) in human ECs. We further identified a CpG island that functions as a cyclin A core promoter, wherein 2 CpG sites are demethylated by Hcy. One of these CpG sites is located on the CDE repressor. Mutation of the CG sequence on CDE results in a 6-fold increase in promoter activity. Furthermore, Hcy inhibits DNMT1 activity, and reduces the binding of MeCP2 while increasing the binding of ACH3 and ACH4 to the cyclin Apromoter. Finally, adenovirus-transduced DNMT1 reverses the inhibitory effect of Hcy on cyclin A gene expression and EC growth. These data are consistent with our previous findings that Hcy inhibits cyclin A transcription via a hypomethylation-related mechanism, and are the first to identify a molecular basis of Hcy-related methylation regulation in a pathophysiologically relevant model.
Cyclin A is a key regulator of cell cycle progression and cyclin A–deficient mice die early in embryonic development.23 The synthesis of cyclin A is controlled mainly at the transcription level, and its removal is carried out by ubiquitin-mediated proteolysis. Cyclin A expression is increased in a variety of tumors and atherosclerosis.24 We have previously demonstrated distinct effects of Hcy on different types of vascular cells. Hcy promotes proliferation and cyclin A transcription in VSMCs at a superphysiological concentration (1 mM),8 whereas a clinically relevant concentration of Hcy (50 μM) inhibits growth and cyclin A transcription in ECs (Figure 1), confirming and extending our previous observations.7 The general inhibitory effects of AZC, TSA, and NaBr on cyclin A mRNA expression (Figure 1B) indicate that cyclin A gene expression is subject to DNMT and histone deacetylase–mediated regulation. The DNA methylation hypothesis is supported by an EC-specific SAH accumulation,3 and led us to further hypothesize that DNA hypomethylation is a key biochemical mechanism responsible for Hcy-induced cyclin A suppression and EC growth arrest.
DNA methylation in the CG-rich promoter is a critical mechanism for the regulation of many tumor-specific genes as well as genes involved in early embryonic development. It is known that DNA methylation occurs at CpG islands—DNA regions (> 200-bp) in and near the promoter region that possess a rich CG content (> 50%) compared with the rest of the genome where CG content is 20% or less.25 We have identified a single CpG island within the cyclin A core promoter, which exhibited the highest level of promoter activity (Figure 2). Interestingly, cyclin D1 gene contains 2 CpG islands at the far end of the 5′-flanking region that do not exert core promoter activity. The lack of response of cyclin D1 gene to DNA methylation inhibition induced by Hcy or AZC is likely due to the location of its CpG islands in regions insufficient to control gene transcription.
There were 2 cytosines in the cyclin A CpG island that were methylated under control conditions and were demethylated by Hcy in the cyclin A CpG island (Figure 3B). One of these CpG sites is located in the CDE consensus element. Mutation of the CDE CG leads to a 6-fold increase in cyclin A promoter activity under control conditions and a modest reduction in response to Hcy (Figure 3C). These data support the notion that CDE is a repressor of the cyclin A promoter in ECs, and that the CDE CpG site is responsive to Hcy hypomethylation. The ATF/CREB and E2F1 elements were unmethylated under control conditions and therefore may not be the direct targets of Hcy hypomethylation. However, both the ATF/CREB and E2F1 elements are potent positive regulators of cyclin A transcription in ECs, as ATF/CREB and E2F1 mutations led to a reduction of cyclin A promoter activity to 28% and 42%, respectively (Figure 3C). We have previously reported that in VSMCs, Hcy activates cyclin A transcription through the ATF/CREB element and stimulates proliferation.8 Our present study now supports the hypothesis that Hcy modulates cyclin A transcription differentially in vascular cells; it suppresses cyclin A transcription by CDE site hypomethylation in ECs and activates transcription by enhancing the binding of ATF and CREB to the cyclin A promoter in VSMCs. These distinct mechanisms (ie, CDE hypomethylation and ATF/CREB activation) may be responsible for the opposing actions of Hcy-induced growth inhibition in ECs and growth promotion in VSMCs.
DNA methylation is mediated by DNMT. There are 3 mammalian DNMTs families (1, 2, and 3). All mammalian DNMTs use SAM as a source of methyl groups and transfer the methyl groups to the 5′-position of cytosine residues that are immediately followed by guanine (CpG dinucleotides). The most abundant is DNMT1, which preferentially methylates hemimethylated DNA and is recognized as the “maintenance methyltransferase.” DNMT3 shows no preference for hemimethylated over unmethylated DNA substrates. It is highly expressed during early development stages when most of the de novo methylation occurs, and was known as the “de novo methyltransferase.” DNMT2 is expressed at substantially lower levels in adult tissues and is likely involved in integrated retroviral DNA methylation. We found that Hcy did not change the protein levels of DNMT1 and DNMT3, but selectively reduces the activity of DNMT1 to 71% (Figure 4). The inhibitory effect of Hcy on DNMT1 is likely mediated by the increased levels of SAH in ECs via product feedback inhibition mechanism. We found that AZC treatment reduced DNMT1 level, suggesting a direct inhibitory effect of AZC on DNMT1 expression. Because DNMT1 is consistently expressed in the nucleus, where DNMT3 protein is barely detectable, we believe that DNMT1 is the major DNMT and the target of Hcy signaling in ECs. To demonstrate the role of DNMT1 in Hcy signaling, we generated adenoviral DNMT1 and DNMT3, which induced dose-dependent increases in enzyme mass in ECs (Figure 5). Adenovirus-transduced DNMT1, but not DNMT3, reverses the Hcy-mediated repression of cyclin A gene expression and EC growth. These results indicate that DNMT1 mediates Hcy-induced cyclin A gene suppression and growth inhibition in ECs. Since Hcy results in the EC-selective accumulation of SAH, an established inhibitor for methyltransferase, it is likely that SAH accumulation specifically inactivates DNMT1, in a manner that is reversible by increases in DNMT1 enzyme mass. In addition, adenoviral DNMT1, at 500 MOI, did not inhibit DNA synthesis as other adenoviral constructs did in this and previous studies.7
Recently, DNA methylation has been found to modulate gene transcription by recruiting MeCP2, which in turn attracts HDAC to bind and facilitate the removal of the acetyl group from specific lysine residues on histones H3 and H4. It is possible that acetylation feed back to decrease methylation because TSA and NABr reduced MeCP2 binding to the cyclin A chromatin. We have demonstrated that Hcy largely reduces the binding of MeCP2 and induces the binding of ACH3 and ACH4 to the cyclin A CpG island in ECs (Figure 6). These data suggest that cyclin A demethylation eliminates MeCP2 and HDAC in the cyclin A chromatin. As a result, deacetylase activity of HDAC is reduced, leading to significantly higher levels of acetylated H3 and H4 (ACH3 and ACH4). Acetylation of histones is carried out by histone acetyltransferase (HAT), and is required to maintain chromatin in an open state permitting binding of transcriptional activators or suppressors to regulate gene expression. Our results suggest that the loss of DNA methylation in the CDE repressor site (Figure 3B) and high levels of histone H3 and H4 acetylation (Figure 6) are the primary events in Hcy-induced cyclin A gene silencing, which modulate chromatin structure, facilitating the access of transcriptional suppressors to the promoter and leading to transcriptional inhibition. DNA methylation on the trans-activating sites of the promoter has been demonstrated to mediate transcriptional repression or gene silencing. Thus, our data provide new evidence supporting the hypothesis that DNA methylation of a repressor site on a promoter can activate gene transcription.
In conclusion, our results indicate that DNMT1 inhibition, DNA hypomethylation, histone hyperacetylation, and chromatin remodeling mediate Hcy-induced cyclin A gene silencing and growth inhibition in ECs. These may represent important novel mechanisms for Hcy-induced endothelial injury and impairment of regrowth that may contribute to the development of atherosclerosis.
Acknowledgments
This work was supported in part by National Institutes of Health grants HL67033, HL77288, and HL82774 (H.W.); HL36045 (A.I.S.); and HL74966 (W.D.).
Footnotes
An Inside Blood analysis of this article appears in the front of this issue.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: M.S.J. performed research and wrote the first draft; I.C., F.Y., X.J., M.J., and X.L. performed some research; A.I.S., W.D., and X.Y. participated in experimental design, data analysis, and paper writing; and H.W. designed research, analyzed data, and wrote the paper.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Hong Wang, Temple University School of Medicine, Department of Pharmacology, 3420 North Broad St, Philadelphia, PA 19140; e-mail: hongw@temple.edu.
References
- 1.Lee ME, Wang H. Homocysteine and hypomethylation: a novel link to vascular disease. Trends Cardiovasc Med. 1999;9:49–54. doi: 10.1016/s1050-1738(99)00002-x. [DOI] [PubMed] [Google Scholar]
- 2.Yang F, Tan HM, Wang H. Hyperhomocysteinemia and atherosclerosis. Sheng Li Xue Bao. 2005;57:103–114. [PubMed] [Google Scholar]
- 3.Wang H, Yoshizumi M, Lai K, et al. Inhibition of growth and p21ras methylation in vascular endothelial cells by homocysteine but not cysteine. J Biol Chem. 1997;272:25380–25385. doi: 10.1074/jbc.272.40.25380. [DOI] [PubMed] [Google Scholar]
- 4.Pema AF, Ingrosso D, Zappia V, Galletti P, Capasso G, De Santo NG. Enzymatic methyl esterification of erythrocyte membrane proteins is impaired in chronic renal failure: evidence for high levels of the natural inhibitor S-adenosylhomocysteine. J Clin Invest. 1993;91:2497–2503. doi: 10.1172/JCI116485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dayal S, Bottiglieri T, Arning E, et al. Endothelial dysfunction and elevation of S-adenosylhomocysteine in cystathionine beta-synthase-deficient mice. Circ Res. 2001;88:1203–1209. doi: 10.1161/hh1101.092180. [DOI] [PubMed] [Google Scholar]
- 6.Caudill MA, Wang JC, Melnyk S, et al. Intracellular S-adenosylhomocysteine concentrations predict global DNA hypomethylation in tissues of methyl-deficient cystathionine beta-synthase heterozygous mice. J Nutr. 2001;131:2811–2818. doi: 10.1093/jn/131.11.2811. [DOI] [PubMed] [Google Scholar]
- 7.Wang H, Jiang X, Yang F, et al. Cyclin A transcriptional suppression is the major mechanism mediating homocysteine-induced endothelial cell growth inhibition. Blood. 2002;99:939–945. [PMC free article] [PubMed] [Google Scholar]
- 8.Tsai JC, Wang H, Perrella MA, et al. Induction of cyclin A gene expression by homocysteine in vascular smooth muscle cells. J Clin Invest. 1996;97:146–153. doi: 10.1172/JCI118383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hiltunen MO, Yla-Herttuala S. DNA methylation, smooth muscle cells, and atherogenesis. Arterioscler Thromb Vasc Biol. 2003;23:1750–1753. doi: 10.1161/01.ATV.0000092871.30563.41. [DOI] [PubMed] [Google Scholar]
- 10.Dong C, Yoon W, Goldschmidt-Clermont PJ. DNA methylation and atherosclerosis. J Nutr. 2002;132:2406S–2409S. doi: 10.1093/jn/132.8.2406S. [DOI] [PubMed] [Google Scholar]
- 11.Goll MG, Bestor TH. Eukaryotic cytosine methyltransferases. Annu Rev Biochem. 2005;74:481–514. doi: 10.1146/annurev.biochem.74.010904.153721. [DOI] [PubMed] [Google Scholar]
- 12.Takai D, Jones PA. Comprehensive analysis of CpG islands in human chromosomes 21 and 22. Proc Natl Acad Sci U S A. 2002;99:3740–3745. doi: 10.1073/pnas.052410099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gardiner-Garden M, Frommer M. CpG islands in vertebrate genomes. J Mol Biol. 1987;196:261–282. doi: 10.1016/0022-2836(87)90689-9. [DOI] [PubMed] [Google Scholar]
- 14.Yoshizumi M, Hsieh CM, Zhou F, et al. The ATF site mediates downregulation of the cyclin A gene during contact inhibition in vascular endothelial cells. Mol Cell Biol. 1995;15:3266–3272. doi: 10.1128/mcb.15.6.3266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yoshizumi M, Wang H, Hsieh CM, Sibinga NE, Perrella MA, Lee ME. Down-regulation of the cyclin A promoter by transforming growth factor-beta1 is associated with a reduction in phosphorylated activating transcription factor-1 and cyclic AMP-responsive element-binding protein. J Biol Chem. 1997;272:22259–22264. doi: 10.1074/jbc.272.35.22259. [DOI] [PubMed] [Google Scholar]
- 16.Olek A, Oswald J, Walter J. A modified and improved method for bisulphite based cytosine methylation analysis. Nucleic Acids Res. 1996;24:5064–5066. doi: 10.1093/nar/24.24.5064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li LC, Dahiya R. MethPrimer: designing primers for methylation PCRs. Bioinformatics. 2002;18:1427–1431. doi: 10.1093/bioinformatics/18.11.1427. [DOI] [PubMed] [Google Scholar]
- 18.Yoshizumi M, Lee WS, Hsieh CM, et al. Disappearance of cyclin A correlates with permanent withdrawal of cardiomyocytes from the cell cycle in human and rat hearts. J Clin Invest. 1995;95:2275–2280. doi: 10.1172/JCI117918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tollefsbol TO, Hutchison CA., III Mammalian DNA (cytosine-5-)-methyltransferase expressed in Escherichia coli, purified and characterized. J Biol Chem. 1995;270:18543–18550. doi: 10.1074/jbc.270.31.18543. [DOI] [PubMed] [Google Scholar]
- 20.Chadee DN, Hendzel MJ, Tylipski CP, et al. Increased Ser-10 phosphorylation of histone H3 in mitogen-stimulated and oncogene-transformed mouse fibroblasts. J Biol Chem. 1999;274:24914–24920. doi: 10.1074/jbc.274.35.24914. [DOI] [PubMed] [Google Scholar]
- 21.Siedlecki P, Zielenkiewicz P. Mammalian DNA methyltransferases. Acta Biochim Pol. 2006;53:245–256. [PubMed] [Google Scholar]
- 22.Hendrich B, Bird A. Identification and characterization of a family of mammalian methyl-CpG binding proteins. Mol Cell Biol. 1998;18:6538–6547. doi: 10.1128/mcb.18.11.6538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wolgemuth DJ, Lele KM, Jobanputra V, Salazar G. The A-type cyclins and the meiotic cell cycle in mammalian male germ cells. Int J Androl. 2004;27:192–199. doi: 10.1111/j.1365-2605.2004.00480.x. [DOI] [PubMed] [Google Scholar]
- 24.Chen D, Krasinski K, Sylvester A, Chen J, Nisen PD, Andres V. Downregulation of cyclin-dependent kinase 2 activity and cyclin A promoter activity in vascular smooth muscle cells by p27(KIP1), an inhibitor of neointima formation in the rat carotid artery. J Clin Invest. 1997;99:2334–2341. doi: 10.1172/JCI119414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bird A. DNA methylation patterns and epigenetic memory. Genes Dev. 2002;16:6–21. doi: 10.1101/gad.947102. [DOI] [PubMed] [Google Scholar]