

Cocaine‐induced brain damage can be divided into primary neurotoxic effects causing toxic encephalopathy, secondary effects of compromised cerebral blood flow in ischaemic and haemorrhagic stroke, cerebral vasculitis and vasospasm, and tertiary effects due to hypoxia as a result of cardiopulmonary collapse. Toxic leucoencephalopathy mainly affects white matter (WM) tracts serving higher cerebral function, thereby leading to altered personality, attention deficits and memory impairment in mild cases and to dementia, coma and brain death in severe cases. There are numerous legal and illegal substances provoking toxic encephalopathy, which could develop gradually or acutely.1,2,3,4 This is, to our knowledge, the first report of fatal cocaine‐associated encephalopathy, in a patient who had not taken cocaine previously, assessed by proton nuclear magnetic resonance spectroscopy (1H‐MRS).
Case report
A 21‐year‐old man had recurrent episodes of depression. He was found breathing and deeply comatose in a hotel room with injection marks in the left cubital vein. A syringe and a farewell letter were beside him; 14 empty packages with tracks of white powder, later identified as cocaine, were found close by.
On admission he was unresponsive and intubated with intact brain stem reflexes. There was bilateral hyperreflexia and moderately increased muscle tone, but normal plantar reflexes. Blood assays showed mild metabolic acidosis (pH 7.30) with initially elevated liver enzyme concentrations (alanine aminotransferase 530 U/l); rhabdomyolysis (myoglobin 38 000 μg/l) and leucocytosis (17 billions/l); dehydration with increased creatinine (0.32 mmol/l); and hyperkalaemia (5.7 mmol/l). Urine toxicology screening was positive only for cocaine. The patient was treated for aspiration pneumonia with normalisation of all blood assays during the next days. The initial CT examination of brain without contrast was normal. EEG after 3 and 12 days revealed diffuse 2–4 Hz activity with periodically 2–30 s long episodes of EEG flattening and lack of response to visual or tactile stimulation. MRI of the brain on day 21 showed diffuse symmetrical WM changes in the cerebrum with hyperintense signal on the T2‐weighted images (fig 1). Brain stem and cerebellum appeared normal. No contrast enhancement was observed. Magnetic resonance angiography was unremarkable.
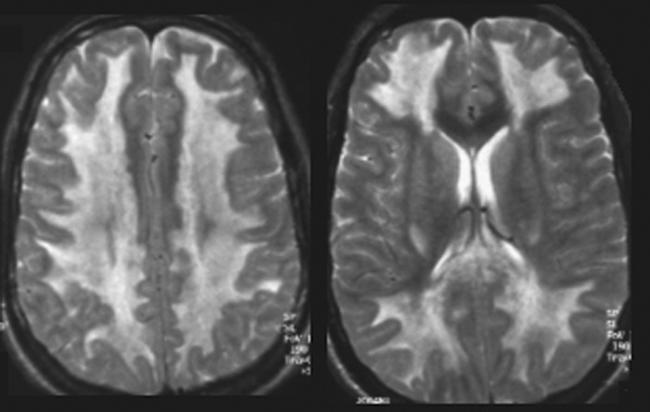
Figure 1 T2‐weighted images 21 days after a single overdose of intravenous cocaine reveal marked white matter hyperintensities with preservation of U‐fibres.
The patient had 1H‐MRS on day 21. Two volumes of interest, 25×25×20 mm in occipitoparietal WM and 21×27×20 mm in mid‐occipital grey matter (GM), were examined using stimulated echo acquisition mode (STEAM) at 1.5 T; echo time 20 ms, TM interval 30 ms, repetition time 1500 ms and 128 averages (fig 2).

Figure 2 Proton nuclear magnetic resonance spectroscopy on day 21 after admission in (A) occipitoparietal white matter (WM) and (B) mid‐occipital grey matter (GM) shows severe metabolic derangement with substantial decreases in N‐acetyl aspartate (NAA), myoinositol (MI) and creatine (Cr) in both GM and WM, and demyelination with increased lactate, lipids and macromolecules in WM. Cho, Choline; Glx, glutamine+glutamate.
In WM (fig 2A), N‐acetylaspartate (NAA) was <10% and total creatine (Cr) was <20% of the values in healthy controls. Highly reduced NAA and Cr indicated major neuroaxonal injury and loss of tissue integrity. Myoinositol (MI) and glutamine+glutamate (Glx) were decreased to a third of normal values, whereas lactate, lipids and macromolecules were increased. Lactate concentration was approximately 3 mM. Total choline (Cho) was normal. The WM spectrum was consistent with severe toxic encephalopathy and demyelination.
In GM (fig 2B), NAA reached 40% of control values, once again indicating impaired neuronal function. Cr was reduced to about 80%, and MI to 66% of control levels. There were normal amounts of Glx and Cho. Lipids and macromolecules were not detected, and lactate was <0.5 mM.
Due to the lack of clinical improvement, highly abnormal MRI and severe axonal and neuronal injury indicated by magnetic resonance spectroscopy, treatment was reduced to palliative care. At 24 days after admission, the patient died of pneumonia without having regained consciousness. Autopsy revealed severe leucoencephalopathy with pronounced demyelination, lipid‐loaded macrophages and liquefying of the central cerebral WM. However, the cortex, subcortical WM including U‐fibres, brain stem, cerebellum and notably the hippocampus appeared relatively intact on gross examination and histology. There was no inflammatory infiltration of the cerebral vessels.
Discussion
Toxic leucoencephalopathy has been described with intravenous and inhalative consumption of cocaine, heroine and other drugs of misuse. Whereas the clinical picture varies extensively,2,3,4,5 the radiological and histological findings are quite characteristic. Typically, T2‐weighted MRI shows diffuse bihemispheric WM lesions, often with preservation of U‐fibres2,3,4,5 such as in our case. Although, in our patient, cocaine‐induced cerebral vasculitis and vasospasms can be excluded from the differential diagnoses due to normal appearance of the cerebral vessels at histology and the diffuse symmetric manifestation of the WM lesions, the contribution of eventual hypoxic brain damage is more uncertain. However, CT examination of the brain 24 h after admission was normal and no episode of respiratory arrest had been registered at the time MRI and spectroscopy were performed. Also, sparing the hypoxia‐sensitive hippocampi on histological examination makes a severe hypoxic brain injury improbable. It is also unlikely that the initial aspiration pneumonia, dehydration and hyperkalaemia should have caused leucoencephalopathy, since these conditions were quickly reversed and generally do not provoke such a clinical picture.
Although 1H MRS has been used extensively in children with a variety of brain disorders and in adults mainly with traumatic brain injury, studies on acute toxic encephalopathy secondary to drug misuse are sparse.1,3 The 1H MRS spectrum obtained from our patient was coherent with an intense demyelination process, but differed from other demyelinating WM disorders such as multiple sclerosis in that a substantial lactate signal was detected. However, this corroborates earlier findings in toxic encephalopathy provoked by heroine3 and simultaneous intoxication with amphetamine and morphine.1 The increase in lactate points towards a conversion of aerobic metabolism to anaerobic energy production, but whether this is due to mitochondrial impairment secondary to hypoxia, due to macrophage invasion, or due to a direct consequence of cocaine‐induced neuroaxonal toxicity remains unknown. Although the GM volume contained a smaller fraction of WM, not all the NAA reduction in the GM can be explained by partial volume effects. Instead, the reduction in cortical NAA could be interpreted as secondary neuronal impairment after leucoencephalopathy. In conclusion, the virtual total loss of NAA, severely decreased creatine and significant elevation of lactate, lipids and macromolecules in the WM were consistent with massive demyelination and neuronal damage, which was confirmed by histopathological findings. It can be stated that the poor clinical picture and prognosis were mirrored by abnormalities revealed by 1H‐NMR spectroscopy, which therefore, might serve as a diagnostic tool in toxic encephalopathy secondary to overdose of drugs of misuse.
Acknowledgements
The John and Birthe Meyer Foundation kindly donated the MRI system.
Footnotes
Competing interests: None declared.
Consent was obtained for publication of the patient's details described in this report.
References
- 1.Gottfried J A, Mayer S A, Shungu D C.et al Delayed posthypoxic demyelination. Association with arylsulfatase A deficiency and lactic acidosis on protein MR spectroscopy. Neurology 1997491400–1404. [DOI] [PubMed] [Google Scholar]
- 2.Maschke M, Fehlings T, Kastrup O.et al Toxic leukoencephalopathy after intravenous consumption of heroin and cocaine with unexpected clinical recovery. J Neurol 1999246850–851. [DOI] [PubMed] [Google Scholar]
- 3.Vella S, Kreis R, Lovblad K O.et al Acute leukoencephalopathy after inhalation of a single dose of heroin. Neuropediatrics 200334100–104. [DOI] [PubMed] [Google Scholar]
- 4.Ryan A, Molloy F M, Farrell M A.et al Fatal toxic leukoencephalopathy: clinical, radiological, and necropsy findings in two patients. J Neurol Neurosurg Psychiatry 2005761014–1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bartlett E, Mikulis D J. Chasing “Chasing the dragon” with MRI: leukoencephalopathy in drug abuse. Br J Radiol 200578997–1004. [DOI] [PubMed] [Google Scholar]