

Abstract
Farnesylation, catalyzed by protein farnesyltransferase (FTase), is an important posttranslational modification guiding cellular localization. Recently predictive models for identifying FTase substrates have been reported. Here we evaluate these models through screening of dansylated-GCaaS peptides, which also provides new insights into the protein substrate selectivity of FTase.
Farnesylation is a posttranslation modification that tags proteins with a farnesyl (C15) isoprenoid supplied by farnesyl diphosphate (FPP) for the purpose of locating the protein to cellular membranes. These lipidated protein substrates are modified by farnesyltransferase (FTase) on their C-terminal FTase recognition sequence called the CaaX box. When it was found that farnesylation occurs on oncogenic Ras proteins1, FTase became a chemotherapeutic drug target.2 Although initially developed based on a simple paradigm where FTIs would target Ras-driven tumors, FTIs have proven to work via a complex mechanism, and their activity is now attributed to the perturbation of a number of cellular proteins.2, 3
The complex and unexpected biology observed with FTIs has made a precise definition of the set of farnesylated proteins in a human cell critically important. It is not known how many proteins in the cell are farnesylated or what are the critical targets of FTIs. Early biochemical studies of Brown and Goldstein4 and the Merck group5 demonstrated that tetrapeptides bearing a cysteine, two amino acids, and the appropriate X residue are farnesylated and serve as the minimum substrate for FTase recognition. Recent modelling studies have provided predictions for FTase Ca1a2X box specificity and thus its protein substrates.6, 7 These models are only predictive and require additional investigation8, 9 to determine cellular protein farnesylation. Using traditional biological tools (radiolabeling and/or western blot analysis), it would be time-consuming to confirm the cellular farnesylation of these hypothetical FTase substrates. Therefore, a screening approach to validate that FTase accepts and modifies the minimal substrate Ca1a2X boxes of a select group of these proteins would be useful (Figure 1).
Figure 1.

Analysis of Dansyl-GCaaX peptides through screening.
As part of our laboratory’s investigation into FTase specificity, we have synthesized a library of Dansyl-GCa1a2S pentapeptides representing FTase substrate candidates. The sequences were identified from a Swissprot database search for carboxyl-terminal Ca1a2S boxes. Sequences were chosen to represent a) biologically important farnesylated proteins, and b) interesting and diverse “aa” amino acid sequences. In view of the current interest in models to define FTase substrate specificity, we now report the substrate ability of these Ca1a2S peptides as an experimental test of these models.
The 27 member Dansyl-GCa1a2S-OH library was synthesized on Wang resin primarily in an automated fashion using a standard Fmoc peptide chemistry, with HBTU/HOBt coupling and piperidine/DMF deprotection (supplementary info). The resin-bound CaaS tetrapeptide was capped by coupling with Dansyl-Gly-OH, followed by cleavage from the resin and side-chain deprotection (90% CF3COOH, 5% iPr3SiH, and 5% H2O). The library was successfully synthesized with yields for each member of the library ranging from 70–100 mg (66–95% overall yield). The purity of each of the peptide sequences was >70%, as confirmed by RP-HPLC analysis. The identity of all peptides was also confirmed by ESI-MS.
The FTase substrate activity of dansylated pentapeptide CaaX boxes can be measured through a fluorescence based assay10, in a 96 well plate format.11 Briefly, 3 μM dansylated-CaaX peptide, 1–9 μM FPP are combined, and farnesylation is initiated by addition of recombinant mammalian FTase (0.05 μM), and the increase in fluorescence intensity is measured at 485 nm and 535 nm emission (supplementary info). To confirm the farnesylation of the dansylated-CaaX peptides, HPLC analysis was performed for each of the dn-GCaaS peptide reactions.11
The results from the screening of the dn-GCaaS peptides are summarized in Table 1. The peptides are presented in descending order of reactivity. Of the 27 peptides screened, 24 were found to be substrates for FTase by fluorescence screening and, in 20 cases, by HPLC analysis (Supplemental info). These peptides did vary widely (~150 fold) in their ability to be farnesylated by FTase. Of the 27 dn-GCaaS boxes screened, 13 were either known substrates, or hypothesized FTase substrates according to the structural analysis-derived “Beese model” by developed by Reid, Casey, and Beese.6 All of the CaaS sequences representing known farnesylated proteins are substrates in our in vitro system, providing support for its use in evaluating FTase substrate selectivity. Note that 11 CaaS sequences whose farnesylation were neither known nor hypothesized were also substrates for FTase. Seven of the 11 sequences are relatively poor substrates, exhibiting less than ten percent of the fluorescence change of dn-GCVLS, and in three cases no product was detected by HPLC. However, four of the sequences – CRQS, CVHS, CKSS, and CFSS – are well accepted by FTase, and are comparable in reactivity to known and hypothesized CaaS boxes. Three naturally-occuring CaaS sequences were not observable as substrates under our experimental conditions: CLRS, CIRS, and CFNS.
Table 1.
Comparison of CaaX Substrate Predictions
| CaaX | Protein | Rel. fluor. increase1 | Beese prediction3 | PREN base4 | PREN base XCVLS5 |
|---|---|---|---|---|---|
| CRIS | Protein phosphatase 1 reg (in hi bi tor) subunit 16 B | 886 | H | + | |
| CKIS | Rab 40 A | 574 | H* | ++ | |
| CQTS | DNA J | 508 | K | + | |
| CVLS | H- Ras | 388 | K | ++ | |
| CVIS | Transducin gamma subunit | 331 | K | ++ | |
| CTIS | Guanylate binding protein 1 interferon - inducible | 325 | K | ++ | |
| CFPS | CCNG 2 : cyclin G2 | 280 | H | − − | − |
| CLIS | Phosphorylase kinase B | 218 | K | ++ | |
| CLVS | G protein coupled receptor kinase 1 | 217 | K | ++ | |
| CSVS | Inositol polyphosphate - 5- phosphatase | 180 | H | + | |
| CRPS | WDTC 1 | 171 | H | + | |
| CTFS | Xylosyl protein beta 1 ,4 -galactosyltransferase | 166 | H | − − | + |
| CRQS | Unknown protein fragment (Q 15693) | 121 | NH | − | ++ |
| CVHS | Galectin - 12 | 100 | NH | − − | + |
| CKSS | Unknown Fragment ( q 12814 ) | 82 | NH | − − | − |
| CFSS | Collagen type V alpha 3 subunit | 60 | NH | − | ++ |
| CDMS | Topo I binding Arg/Ser rich | 42 | H* | − − | + |
| CAKS | Rab 38 | 332 | NH | − − | ++ |
| CEGS | Protein with 8 zinc finger domains | 28 | NH | − − | ++ |
| CQKS | cytidine and deoxycytidylate deaminase | 262 | NH | − | ++ |
| CVES | Unknowm fragment (Q 29856) | 20 | NH | − − | − − |
| CPAS | Ribosomal protein L 12 | 19 | NH | − − | + |
| CGAS | Kinesin family member 22 | 6 | NH | − − | + |
| CAES | G protein –coupled receptor 41 | 62 | NH | − − | ++ |
| CIRS | Rab 3 interacting protein 1 | 0 | NH | − | ++ |
| CLRS | Globoside alpha – 13 – N - acetylgalactosam inyltra nsferase1 | 0 | NH | − − | + |
| CFNS | Unknown Fragment (Q 14922) | 0 | NH | − − | − |
Relative fluorescence increase was determined using a PerkinElmer Fusion plate reader (Ex 335/Em 535; 30 minute values reported) and scaled to the fluorescence increase observed for CVLS.
In these cases, the fluorimetric indication of farnesylated peptide product was not conclusively confirmed by HPLC analysis.
Predictions derived form the FTase substrate model reported by Reid et al. K: known substrate; H: hypothesized substrate; H*: not reported as a substrate by Reid et al., but consistent with their rules for substrate ability; NH: not hypothesized to be a substrate.
PRENbase prediction for the sequence XXXXXXXXCaaS, as described in the text.
PRENbase prediction for the sequence XXXXXXXXCVLS, as described in the text.
The screening data presented in Table 1 provide a qualitative ordering of the substrate ability of the CaaS sequences, and this provides information on the peptide substrate preference of FTase. In general, the reactivity order agrees with the previous observations and predictions that the FTase a1 site is tolerant of many amino acid side chains, while the a2 site has a preference for the larger aliphatic residues as well as methionine, phenylalanine and tyrosine. We have now demonstrated that glutamine, serine, and histidine are also accepted at a2. The a1 position was previously not believed to play a major role in the selectively for FTase, but our data suggest that the amino acid at a1 may modulate reactivity, as indicated by the four-fold variation in the change in fluorescence of the five peptides of the general sequence dn-GCxIS (Table 1). The CRIS and CKIS peptides are the most effective substrates among the 27 examined, suggesting a preference for a positively-charged side chain in the a1position. This is not completely unexpected as the Beese model predicts that solution hydrogen bonding of the a1 side chain to the solvent exposed enzyme is possible.
As with much else in the protein prenylation field, the discovery of farnesylation on Ras in the late 1980’s drove the CaaX box meme, as a sequence where the central residues are aliphatic (H-Ras: CVLS; N-Ras: CVVM; K-Ras4B: CVIM).2 However, it became quickly apparent that this is not strictly the case. A study of inhibitors based on the K-Ras CVIM tetrapeptide found significant flexibility in the CxIM series, but a strong preference for aliphatic residues with CVxM.4 An early site-directed mutagenesis study from the Merck group also demonstrated that certain charged residues (lysine, but not glutamate) are accepted at the a1 position, but not a2•5 Subsequently, a combinatorial screening approach defined the following optimal tetrapeptide FTase substrates: CKQQ and CKQM.12 Based on extensive structural studies, Beese and coworkers6 developed an FTase CaaX selectivity model, where a1 is flexible and a2 is limited to hydrophobic residues, and determined that 61 human proteins are substrates for FTase. Previously, individual CaaX peptides derived from FTase substrate candidates have been investigated as FTase substrates,2 but here we report for the first time a more comprehensive investigation of the ability of a diverse set of human CaaX sequences to act as substrates. Our data suggest that CaaS boxes CRQS, CVHS, CKSS, and CFSS are farnesylated and proteins bearing these C-terminal sequences may be farnesylated in the cell (vide infra). These dnGCa1a2X peptides exhibit a1 and a2 residues that differ from the commonly accepted Ca1a2X box model, where a1 and a2 are aliphatic residues. While it was clear from earlier studies that there are few substrate constraints on the a1 residue, it is surprising, both from known prenylated proteins and from the structure of the FTase a2 binding pocket, that certain polar residues are accepted in the a2 position.
Maurer-Stroh et al. 13 developed a database tool (PRENbase) using sequence conservation across species and other information to predict human proteins that are likely FTase substrates. PRENbase evaluates the twelve C-terminal residues of a protein, and provides a score from ++ (prenylation is very likely) to - - (prenylation is very unlikely). These values are reported in Table 1, along with scores where the CaaX box of the C-terminal sequence was mutated in silico to CVLS, as a control for the effects of the sequence upstream of the CaaS box on FTase recognition and CaaS accessibility. The PRENbase analyses of our 27 selected CaaS proteins indicated that ten of the proteins are likely farnesylation candidates, including the six known farnesylated proteins in the set. Thus, PRENbase is significantly more conservative than the Beese model. While some of this disparity is due to the analysis of the eight residues “upstream” of the CaaX box, there is also an element of conservative analysis of the CaaS motif itself. Note that the xylosyl transferase sequence (CTFS), that is not predicted to be a substrate, is predicted to be one when the CTFS CaaX box is “mutated” to CVLS. The CTFS sequence is acceptable in the Beese model, and a good substrate in our experimental Ca1a2S screen.
Another intriguing divergence between the models involves cyclin G2. The Beese model predicts that its CaaX box CFPS is a substrate, while it is disfavored as a substrate by PRENbase (although this is even maintained with the CVLS “mutant”). Our in vitro screen indicates that CFPS is a very good FTase substrate. Our data, combined with the PRENbase analysis of the CVLS “mutants” to provide information of the suitability of the “upstream” sequence, indicate that an uncharacterized gene product (Q15693) and two unexpected proteins –galectin-12 and collagen type five alpha three subunit – are candidates for farnesylation. The farnesylation of a subunit of the extracellular protein collagen seems unlikely, but galectin-12 is an intriguing, newly reported intracellular protein, whose lipid modification status has not yet been investigated.14
The screening of the dn-GCaaS library of peptides has resulted in the identification of 23 substrate CaaX sequences. Our results essentially validate the model for FTase substrate prediction published by Reid et al, but also indicate that additional a2 residues, including Gln, Ser, and His, are well-tolerated by FTase. Furthermore, these data shed light on the importance of both a1 and a2 positions residues on determining substrate ability for peptide sequences. In particular, our results highlight the preference for Lys and Arg at the a1 position (see summary in Figure 2). With the capability to perform peptide synthesis and rapid biochemical evaluation, CaaX screening provides an important tool for identifying FTase substrate proteins. Together with the previously reported structurally-based model database-derived sequence analysis (e.g. PRENbase), biochemical peptide screening will provide a valuable guide to future proteomic and other biological searches for farnesylated proteins.
Figure 2.
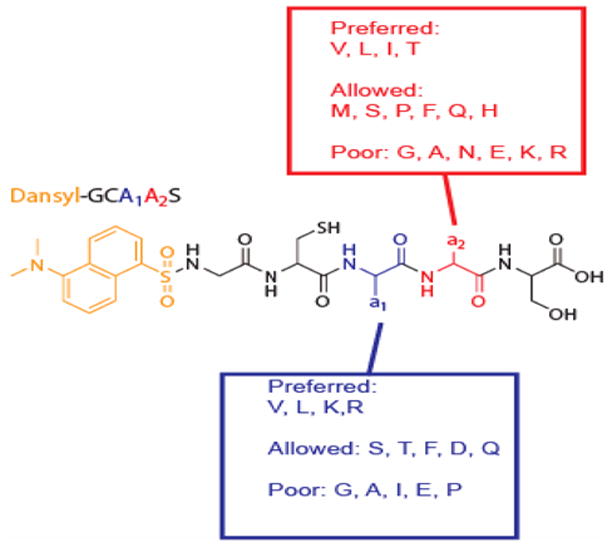
Summary of FTase amino acid specificity.
Supplementary Material
Experimental procedures for solid phase peptide synthesis and the spectrofluorimetric fluorescence assay, and HPLC traces for reaction of each member of the dnGCaaS library with FPP in the presence of FTase. Supplementary data associated with this article can be found in the online version.
Acknowledgments
Supported by NIH grants CA78819 (RAG), GM40602 (CAF), and P30CA21368 (Purdue Cancer Center), and by Purdue Research Foundation fellowships (AJK and SAS).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Literature Cited
- 1.Konstantinopoulos PA, Karamouzis MV, Papavassiliou AG. Nat Rev Drug Discov. 2007;6:541. doi: 10.1038/nrd2221. [DOI] [PubMed] [Google Scholar]
- 2.Basso AD, Kirschmeier P, Bishop WR. J Lipid Res. 2006;47:15. doi: 10.1194/jlr.R500012-JLR200. [DOI] [PubMed] [Google Scholar]
- 3.Sebti SM, Der CJ. Nat Rev Cancer. 2003;3:945. doi: 10.1038/nrc1234. [DOI] [PubMed] [Google Scholar]
- 4.Reiss Y, Stradley S, Gierasch L, Brown M. Proc Natl Acad Sci U S A. 1991;88:732. doi: 10.1073/pnas.88.3.732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Moores SL, Schaber MD, Mosser SD, Rands E, O'Hara MB, Garsky VM, Marshall MS, Pompliano DL, Gibbs JB. J Biol Chem. 1991;266:14603. [PubMed] [Google Scholar]
- 6.Reid TS, Terry KL, Casey PJ, Beese LS. J Mol Biol. 2004;343:417. doi: 10.1016/j.jmb.2004.08.056. [DOI] [PubMed] [Google Scholar]
- 7.Maurer-Stroh S, Eisenhaber F. Genome Biology. 2005;6:R55. doi: 10.1186/gb-2005-6-6-r55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kho Y, Kim SC, Jiang C, Barma D, Kwon SW, Chemg J, Jaunbergs J, Weinbaum C, Tamanoi F, Falck J, Zhao Y. Proc Natl Acad Sci U S A. 2004;101:12479. doi: 10.1073/pnas.0403413101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Suzuki T, Ito M, Ezure T, Shikata M, Ando E, Utsumi T, Tsunasawa S, Nishimura O. Proteomics. 2007;7:1942. doi: 10.1002/pmic.200700237. [DOI] [PubMed] [Google Scholar]
- 10.Cassidy PB, Dolence JM, Poulter CD. Methods Enzymol. 1995;250:30. doi: 10.1016/0076-6879(95)50060-x. [DOI] [PubMed] [Google Scholar]
- 11.Krzysiak AJ, Rawat DS, Scott SA, Pais JE, Handley M, Harrison ML, Fierke CA, Gibbs RA. ACS Chem Biol. 2007;2:385. doi: 10.1021/cb700062b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Boutin JA, Marande W, Petit L, Loynel A, Desmet C, Canet E, Fauchere JL. Cell Signalling. 1999;11:59. doi: 10.1016/s0898-6568(98)00032-1. [DOI] [PubMed] [Google Scholar]
- 13.Maurer-Stroh S, Koranda M, Benetka W, Schneider G, Sirota FL, Eisenhaber F. PLoS Comput Biol. 2007;3:e66. doi: 10.1371/journal.pcbi.0030066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yang RY, Liu FT. Galectins in cell growth and apoptosis. Cell Mol Life Sci. 2003;60:267. doi: 10.1007/s000180300022. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Experimental procedures for solid phase peptide synthesis and the spectrofluorimetric fluorescence assay, and HPLC traces for reaction of each member of the dnGCaaS library with FPP in the presence of FTase. Supplementary data associated with this article can be found in the online version.