

Summary
Endoglin is an auxiliary receptor for TGFβ signalling. Heterozygous germline Endoglin mutations have been identified in patients with the vascular abnormality, Hereditary Haemorrhagic Telangiectasia. Endoglin is upregulated in endothelial cells during angiogenesis and loss of Endoglin in the mouse results in embryonic lethality at mid-gestation. This phenotype points to an important role of Endoglin in new blood vessel formation but precludes analysis at later stages in development and in postnatal life. To bypass this limitation and allow further investigations of the function of Endoglin we have generated a floxed Endoglin allele in which loxP sites flank exons 5 and 6. Mice homozygous for this allele are normal and in the presence of appropriate Cre lines will allow time and cell specific Endoglin deletion for in vivo analysis of function in cardiovascular development and disease.
Keywords: angiogenesis, hereditary, haemorrhagic, telangiectasia, cardiovascular, development
Endoglin is a transmembrane protein which is expressed primarily in endothelial cells. It interacts with receptors of the TGFβ family of ligands and promotes signaling through activin like kinase 1(ALK1) in response to TGFβ or BMP9 (David et al., 2007; Lebrin et al., 2004). Patients heterozygous for mutations in Endoglin mutations develop the familial vascular disorder Hereditary Haemorrhagic Telangiectasia (McAllister et al., 1995). Large arteriovenous malformations may develop in major organs such as the lung or liver and bleeding often occurs from small telangiectases in the nasal, oral, and gastrointestinal mucosa (Guttmacher et al., 1995). This inherited disorder is also associated with mutations in ALK1 (Johnson et al., 1996), leading to the idea that Endoglin and ALK1 have common regulatory functions in the vasculature. Endoglin is dynamically regulated and is expressed at higher levels during developmental, physiological, or pathological angiogenesis compared with quiescent blood vessels (Duff et al., 2003; Jonker and Arthur, 2002; Torsney et al., 2002). To study the role of Endoglin, mice homozygous for null Endoglin alleles have been generated independently in three different laboratories. All Endoglin null mice show early embryonic lethality at 10.5–11.5 days post coitum (dpc) characterized by angiogenesis defects of the yolk sac and abnormal heart development (Arthur et al., 2000; Bourdeau et al., 1999; Li et al., 1999). The formation of the primitive vascular plexus in the yolk sac appears to occur normally but the subsequent branching and remodeling associated with angiogenesis fails to proceed. Further analysis of the Endoglin null embryos has shown that Endoglin is required for downstream TGFβ signaling from the endothelial cell to the adjacent smooth muscle cell to promote smooth muscle cell differentation (Carvalho et al., 2004). This has raised the interesting hypothesis that Endoglin is required for the stabilization of neo-vessels, but further work is hampered by the early embryonic lethal phenotype of the Endoglin null mouse. In parallel, the heterozygous Endoglin null mouse has been developed as a model of HHT. Clinical symptoms similar to those of HHT patients can occur in up to 72% of mice but are highly dependent on the presence or absence of modifier genes in the background strain (Bourdeau et al., 2001; Torsney et al., 2003). To bypass some of these difficulties and to determine the role of Endoglin in late developmental and adult life, we have generated a floxed Endoglin mouse for conditional knockout studies.
The targeting construct was designed to allow conditional deletion of exons 5 and 6 of the Endoglin gene which would also lead to a frameshift mutation in exon 7 and generate a null allele. The targeting strategy, illustrated in Figure 1, shows the generation of a tri-floxed Endoglin allele in which loxP sites flank a PGK-Neo cassette and exons 5 and 6 of the Endoglin gene. Transfection of ES cells and subsequent PCR and southern blot screening of G418 resistant colonies, led to the derivation of two independent ES clones (clones 6 and 19) (Fig. 1). Mice generated from these clones were used as founders to breed mice that were homozygous for the tri-floxed allele (Eng3fl). We were initially surprised to observe that these mice had an embryonic lethal phenotype, indistinguishable from the conventional Endoglin KO, and Endoglin protein was undetectable in homozygous Eng3fl/3fl embryos at 9.5 dpc (not shown). This is consistent with the disruption of translation of the Endoglin gene by an abberant splicing event, which has been reported to be up to 100% efficient when the PGK-Neo cassette is in the reverse orientation, as in our design (Nagy, 2000).
FIG. 1.
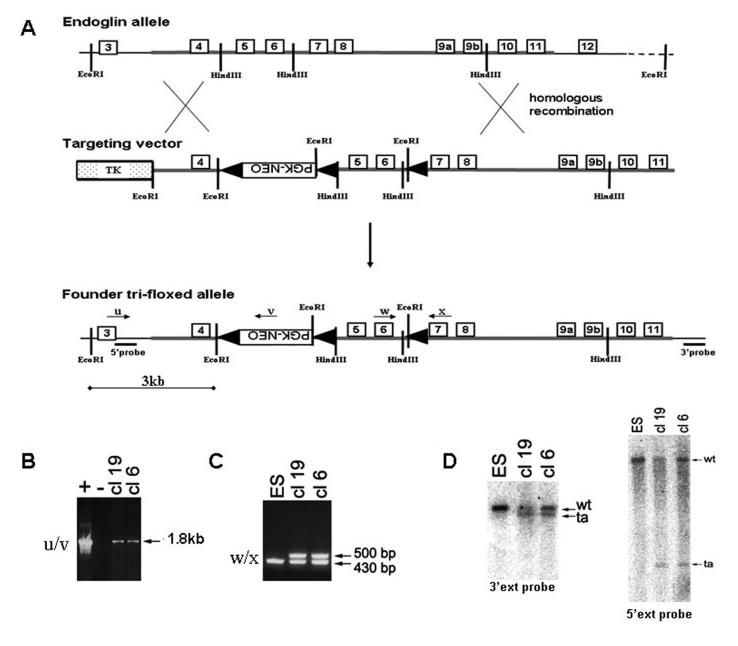
Generation of founder tri-floxed endoglin mice. (A) Targeting the endoglin gene with trifloxed (3fl) eng vector by homologous recombination in ES cells. Endoglin exons 5 and 6 and a PGK-neo selection cassette are flanked by loxP sites (black arrowheads). The DNA used in the targeting vector is drawn as a thick line and carries 1.3 and 5.7 kb homologous sequences in the left and right arm, respectively. (B–D) Two successfully targeted ES clones (cl-6 and cl-19) were recognized by genomic PCR using primers u and v to amplify a 1.8 kb product (B) and (C) primers w and x to amplify a 500 bp product. Successful targeting was confirmed by Southern Blotting using EcoRI digested genomic DNA hybridized with radiolabelled 3′ and 5′ external probes. The wild type (WT) EcoRI fragment (∼16 kb) is reduced in the targeted allele (ta) to 3 kb at the 5′ end and ∼13 kb at the 3′ end.
We therefore proceeded to remove the neo cassette from heterozygous Eng3fl/+ mice in vivo using the Meu-Cre40 transgenic mouse (Leneuve et al., 2003). This line produces a ubiquitous low level of Cre activity allowing all three possible recombinant alleles to be derived from a triple floxed allele. The breeding strategy is summarized in Figure 2A. Essentially mosaic F1 male mice, heterozygous for both the tri-floxed allele and the Meu-Cre40 transgene are identified by PCR genotyping of genomic DNA prepared from sperm or ear tissue biopsy. These mice are recognized by their mosaicism-all 3 possible recombinant alleles are present within the tissues and can be detected by PCR. The required recombination event, the bifloxed Endoglin allele with the neo cassette removed was detected using PCR primers y and z (Fig. 2B, upper panel, lane 3). These mosaics were then crossed with wild type C57Bl/6 females to generate F2 mice and all possible recombination events were observed in the F2 generation. To summarize, a total of 47 F2 mice had inherited the floxed Endoglin allele, but not the Meu-Cre40 transgene, and of these, 5 mice (∼10%) had the required bifloxed Endoglin allele (Eng2fl) without the neo cassette (shown in Fig. 2C). These Eng2fl/+ mice were then intercrossed to generate homozygous Endoglin bifloxed mice (Eng2fl/2fl), which are viable and fertile, with no obvious phenotype. This suggests that the loxP sites in introns 4 and 6 do not interfere with normal expression of the Endoglin gene. Furthermore, compound heterozygotes carrying the bifloxed allele and our previously published null Endoglin allele were also viable, showing that the bifloxed allele was able to functionally rescue the Endoglin null allele.
FIG. 2.
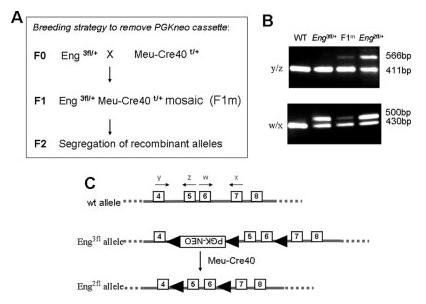
Removing the floxed neo cassette in vivo to generate bifloxed endoglin mice. (A) Breeding strategy showing how the neo cassette was removed from the trifloxed mice using the Meu-Cre40 transgenic line. (B) The bifloxed endoglin allele - with the neo cassette removed- was recognized by PCR for a 566 bp product with primers y and z and readily discriminated from the 411 bp product corresponding to the WT allele (upper panel). (The presence of the 1.8 kb neo cassette in the tri-floxed allele precludes the formation of a detectable PCR product.) The lower panel shows PCR products amplified with primers w and x to produce a 500 bp product from all alleles with a loxP site in intron 6. PCR products corresponding to the bifloxed allele can be seen at low levels in the mosaic (F1m) mouse, but increase to ∼50% in the F2 Eng2fl/+ mouse. (C) Diagram summarizing conversion of the trifloxed allele (Eng3fl) to the required bifloxed allele (Eng2fl), including position of diagnostic primers.
The purpose of conditional alleles is only achieved if they are readily converted to a null allele in the presence of Cre recombinase. Therefore, to formally show that removal of exons 5 and 6 from the bifloxed Endoglin allele generated an Endoglin null allele, we first crossed the bifloxed mice with the ubiquitously expressed Cre line, PGK-Cre (Lallemand et al., 1998). In PGK-Cre females, Cre recombinase is present in all oocytes irrespective of the presence of the Cre transgene on the haploid chromosome set. When PGK-Cre females were crossed with Eng2fl/+ males, the bifloxed allele recombined at 100% efficiency in the oocyte, to delete exons 5 and 6 and generate the EngΔex5–6 allele in the progeny (Fig. 3A). We then examined the genotypes of progeny from an interheterozygous cross of EngΔex5-6/+ mice. A similar number of EngΔex5-6/Δex5-6 and wild type embryos were observed at 9.5 dpc consistent with expected Mendelian ratios. However, there were no EngΔex5-6/Δex5-6 offspring among 54 neonates examined (Table 1). This highly significant (P < 0.0001) deviation from the expected number indicates that the EngΔex5-6/Δex5-6 offspring had an embryonic lethal phenotype. Further analysis showed that embryos homozygous for the EngΔex5-6 allele die in mid-embryogenesis with a phenotype indistinguishable from the conventional Endoglin knockout mice (Fig. 3). In addition, immunohistochemical analysis shows that CD31 positive endothelial cells are present in the EngΔex5-6/Δex5-6 9.5 dpc embryos but that Endoglin protein is not detected with the MJ7/18 anti-Endoglin antibody (Fig. 3). If the EngΔex5-6 allele was translated, it would be expected to produce a truncated peptide containing the extracellular domain of Endoglin encoded by exons 1-4 with only 2 further amino acids encoded by a frameshifted sequence in exon 7 before reaching a stop codon. Any truncated protein that may be present has no transmembrane or C-terminal tail domain and previous data suggests truncated proteins of this small size do not get processed to the cell surface and are unlikely to interfere with function (Raab et al., 1999). Therefore EngΔex5-6 likely represents a null allele.
FIG. 3.
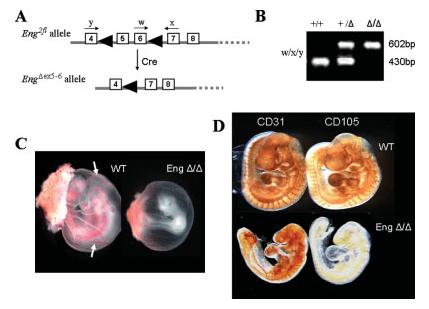
Mice homozygous for the Eng Δex5-6 allele have an embryonic lethal phenotype and do not express endoglin protein. (A) Diagram showing generation of the Eng Δex5-6 allele following deletion of exons 5 and 6 in the presence of Cre, and position of primers w, x, and y. (B) Multiplex PCR using primers w, x, and y produce a 602 bp product from the Eng Δex5-6 allele and a 430 bp product from the wildtype allele. (C) EngΔ/Δ embryos at 9.5 dpc show angiogenesis defects in the yolk sac compared with normal vessels (arrows) in their wild type littermates. (D) Wholemount immunohistochemistry of wild type and EngΔ/Δ showing CD31 and endoglin expression. There is clear evidence of CD31 positive endothelial cells in the EngΔ/Δembryos but endoglin protein was not detected.
Table 1.
Homozygous EngΔex5-6/Δex5-6 Offspring Have an Embryonic Lethal Phenotype
| Age | +/+ | +/Δ | Δ/Δ | Total | χ2 | P |
|---|---|---|---|---|---|---|
| Embryo (9.5 dpc) | 11 (14) | 35 (28) | 10 (14) | 56 | 3.5 | NS |
| Neonates | 16 (13.5) | 38 (27) | 0 (13.5) | 54 | 18.4 | <0.0001 |
χ2 analysis of the numbers of each genotype in offspring from an inter heterozygous Eng+/Δ cross. Expected numbers of each genotype, based on Mendelian inheritance, are given in brackets.
Endoglin is critical for cardiovascular development and its importance continues into adult life during tissue remodelling and angiogenesis. For example, Endoglin is upregulated in healing wounds in endothelial cells during angiogenesis and in myofibroblasts during collagen synthesis (Torsney et al., 2002). It is expressed in endothelial progenitor cells where it has been shown to be important in mediating cardiac repair (van Laake et al., 2006). It is upregulated in tumor angiogenesis, where it represents a potentially valuable antiangiogenesis target (Duff et al., 2003). It is also associated with the regulation of two important mediators of vascular tone, enos, and cox-2, which may contribute to the pathobiology of HHT (Jerkic et al., 2006; Toporsian et al., 2005). Endoglin therefore appears to be an important and multifunctional protein critical for the development and function of the cardiovasculature. The bifloxed Endoglin mouse described here provides a valuable tool for investigating the role of Endoglin in vivo and for investigating its potential as a therapeutic target.
MATERIALS AND METHODS
Construction of the Targeting Vector
The targeting vector was assembled in pPNTlox2 which contains a floxed phosphoglycerol kinase neomycin-resistance gene cassette (PGK-neo) for positive selection of recombinants and a Thymidine kinase (Tk) gene to allow negative selection of ES cells that contain random integration of the construct. The short arm containing exon 4 and part of intron 3 of the Endoglin gene was prepared by PCR and cloned into the EcoRI site of pPNTlox2. The long arm containing exons 5 to 11 was cloned into the NotI site of pPNTlox2. A third loxP site was introduced into the HindIII site in intron 6 of the Endoglin gene. This cloning strategy leads to the floxed PGK-neo cassette being present in the reverse orientation relative to the Endoglin sequence (Fig. 1).
Electroporation of ES Cells and Generation of Chimeric Mice
Mouse E14 (129/ola) ES cells were electroporated with linearised targeting vector and selected in G418-containing medium. The genotypes of G418-resistant ES clones were first analyzed by PCR using the following primers (all DNA sequences are written in the 5′ to 3′ direction and approximate locations are illustrated in the Figures). Primer-u (ATGTGGCAGGTCTCAAGGTG) and Primer-v (ATCGCCTTCTATCGCCTTC) amplify a 1.8 kb product specific to the targeted recombinant allele, whilst primer-w (GACGCCATTCTCATCCTGC) and primer-x (CCACGCCTTTGTCCTTGC) amplify products of 430 bp from the wild type Endoglin allele and 500 bp from the floxed Endoglin allele. Amplification was performed for 35 cycles with 15 s at 96°C, 30 s at 58°C, and 60 s at 74°C. Further Southern blot analysis used 3′ and 5′ external probes to hybridize to EcoRI digested genomic DNA, as previously described (Arthur et al., 2000). Ultimately two of 40 (5%) G418-resistant ES clones were found to be homologous recombinants and microinjected into C57BL/6 blastocysts. The resulting chimeras were bred to C57BL/6 females, and germline transmission of the floxed Endoglin allele was obtained.
Genotype Analysis
Ear biopsy (or sperm from mosaic males, taken directly from the uterus of a euthenased female on the day following coitus) was used to prepare genomic DNA as previously described (Arthur et al., 2000). Mouse genotypes were determined by PCR analysis using primers w and x above, Cre primers Cre-F (GATCGCTGCCAGGATATACG) and Cre-R (AATCGCCATCTTCCAGCAG) which gives a PCR product of 574 bp when a Cre transgene is present; Primer-y (GGTCAGCCAGTCTAGCCAAG) and primer-z (GTGGTTGCCATTCAAGTGTG) amplify products of 411 bp for the wild type allele and 566 bp for the bi-floxed Endoglin allele. PCR using primer-x with primer-y gives a product of 602 bp only in the presence of the EngΔex5-6 allele, and no detectable PCR product is amplified from a wild-type allele.
Immunohistochemistry
For whole-mount immunohistochemistry, embryos were dissected from decidua and fixed in 4% paraformaldehyde/PBS at 4°C overnight before rinsing in 0.5%NP40/PBS and dehydration to methanol. Embryos were bleached in 5% hydrogen peroxide in methanol for 1–2 h at room temperature, before rehydration to PBS and blocking in PBSST (5% rabbit serum, 0.1% Triton X-100, PBS). The embryos were incubated with primary antibody diluted in PBSST at 4°C overnight. Primary antibodies, rat monoclonal MEC13.3 and MJ7/18 (BD Biosciences), were used to detect CD31 and Endoglin protein, respectively. Embryos were extensively washed with PBSST at 4°C and then incubated with biotinylated rabbit anti-rat secondary antibody (Vector Labs) in PBSST at 4°C overnight. Embryos were washed extensively in PBSST at 4°C, and finally in PBT (0.2% BSA, 0.1% Triton X-100, PBS) for 20 min at room temperature. Staining was visualized by addition of streptavidin-HRP complex (Vector ABC elite) and DAB substrate (Biogenex).
ACKNOWLEDGMENTS
The authors thank Dr Martin Holzenberger and the late Professor Peter Lonai for providing the Meu-Cre40 mice and the Pk-Cre mice, respectively.
Contract grant sponsor: the Wellcome Trust, British Heart Foundation, (EU QLG1-CT-2001-01032), Borwick Trust, Cookson Trust, and the Dutch Platform for Tissue Engineering (Bsik).
LITERATURE CITED
- Arthur HM, Ure J, Smith AJ, Renforth G, Wilson DI, Torsney E, Charlton R, Parums DV, Jowett T, Marchuk DA, Burn J, Diamond AG. Endoglin, an ancillary TGFβ receptor, is required for extra-embryonic angiogenesis and plays a key role in heart development. Dev Biol. 2000;217:42–53. doi: 10.1006/dbio.1999.9534. [DOI] [PubMed] [Google Scholar]
- Bourdeau A, Dumont DJ, Letarte M. A murine model of hereditary hemorrhagic telangiectasia. J Clin Invest. 1999;104:1343–1351. doi: 10.1172/JCI8088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourdeau A, Faughnan ME, McDonald ML, Paterson AD, Wanless IR, Letarte M. Potential role of modifier genes influencing transforming growth factor-β1 levels in the development of vascular defects in endoglin heterozygous mice with hereditary hemorrhagic telangiectasia. Am J Pathol. 2001;158:2011–2020. doi: 10.1016/s0002-9440(10)64673-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvalho RL, Jonker L, Goumans MJ, Larsson J, Bouwman P, Karlsson S, Dijke PT, Arthur HM, Mummery CL. Defective paracrine signalling by TGFβ in yolk sac vasculature of endoglin mutant mice: a paradigm for hereditary haemorrhagic telangiectasia. Development. 2004;131:6237–6247. doi: 10.1242/dev.01529. [DOI] [PubMed] [Google Scholar]
- David L, Mallet C, Mazerbourg S, Feige JJ, Bailly S. Identification of BMP9 and BMP10 as functional activators of the orphan activin receptor-like kinase 1 (ALK1) endothelial cells. Blood. 2007;109(5):1953–1961. doi: 10.1182/blood-2006-07-034124. [DOI] [PubMed] [Google Scholar]
- Duff SE, Li C, Garland JM, Kumar S. CD105 is important for angiogenesis: Evidence and potential applications. FASEB J. 2003;17:984–992. doi: 10.1096/fj.02-0634rev. [DOI] [PubMed] [Google Scholar]
- Guttmacher AE, Marchuk DA, White RI., Jr Hereditary hemorrhagic telangiectasia. N Engl J Med. 1995;333:918–924. doi: 10.1056/NEJM199510053331407. [DOI] [PubMed] [Google Scholar]
- Jerkic M, Rivas-Elena JV, Santibanez JF, Prieto M, Rodriguez-Barbero A, Perez-Barriocanal F, Pericacho M, Arevalo M, Vary CP, Letarte M, Bernabeu C, Lopez-Novoa JM. Endoglin regulates cyclooxygenase-2 expression and activity. Circ Res. 2006;99:248–256. doi: 10.1161/01.RES.0000236755.98627.69. [DOI] [PubMed] [Google Scholar]
- Johnson DW, Berg JN, Baldwin MA, Gallione CJ, Marondel I, Yoon SJ, Stenzel TT, Speer M, Pericak-Vance MA, Diamond A, Guttmacher AE, Jackson CE, Attisano L, Kucherlapati R, Porteous ME, Marchuk DA. Mutations in the activin receptor-like kinase 1 gene in hereditary haemorrhagic telangiectasia type 2. Nat Genet. 1996;13:189–195. doi: 10.1038/ng0696-189. [DOI] [PubMed] [Google Scholar]
- Jonker L, Arthur HM. Endoglin expression in early development is associated with vasculogenesis and angiogenesis. Mech Dev. 2002;110:193–196. doi: 10.1016/s0925-4773(01)00562-7. [DOI] [PubMed] [Google Scholar]
- Lallemand Y, Luria V, Haffner-Krausz R, Lonai P. Maternally expressed PGK-Cre transgene as a tool for early and uniform activation of the Cre site-specific recombinase. Transgenic Res. 1998;7:105–112. doi: 10.1023/a:1008868325009. [DOI] [PubMed] [Google Scholar]
- Lebrin F, Goumans MJ, Jonker L, Carvalho RL, Valdimarsdottir G, Thorikay M, Mummery C, Arthur HM, ten Dijke P. Endoglin promotes endothelial cell proliferation and TGF-β/ALK1 signal transduction. EMBO J. 2004;23:4018–4028. doi: 10.1038/sj.emboj.7600386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leneuve P, Colnot S, Hamard G, Francis F, Niwa-Kawakita M, Giovannini M, Holzenberger M. Cre-mediated germline mosaicism: A new transgenic mouse for the selective removal of residual markers from tri-lox conditional alleles. Nucleic Acids Res. 2003;31:e21. doi: 10.1093/nar/gng021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li DY, Sorensen LK, Brooke BS, Urness LD, Davis EC, Taylor DG, Boak BB, Wendel DP. Defective angiogenesis in mice lacking endoglin. Science. 1999;284:1534–1537. doi: 10.1126/science.284.5419.1534. [DOI] [PubMed] [Google Scholar]
- McAllister KA, Baldwin MA, Thukkani AK, Gallione CJ, Berg JN, Porteous ME, Guttmacher AE, Marchuk DA. Six novel mutations in the endoglin gene in hereditary hemorrhagic telangiectasia type 1 suggest a dominant-negative effect of receptor function. Hum Mol Genet. 1995;4:1983–1985. doi: 10.1093/hmg/4.10.1983. [DOI] [PubMed] [Google Scholar]
- Nagy A. Cre recombinase: The universal reagent for genome tailoring. Genesis. 2000;26:99–109. [PubMed] [Google Scholar]
- Raab U, Velasco B, Lastres P, Letamendia A, Cales C, Langa C, Tapia E, Lopez-Bote JP, Paez E, Bernabeu C. Expression of normal and truncated forms of human endoglin. Biochem J. 1999;339(Part 3):579–588. [PMC free article] [PubMed] [Google Scholar]
- Toporsian M, Gros R, Kabir MG, Vera S, Govindaraju K, Eidelman DH, Husain M, Letarte M. A role for endoglin in coupling eNOS activity and regulating vascular tone revealed in hereditary hemorrhagic telangiectasia. Circ Res. 2005;96:684–692. doi: 10.1161/01.RES.0000159936.38601.22. [DOI] [PubMed] [Google Scholar]
- Torsney E, Charlton R, Diamond AG, Burn J, Soames JV, Arthur HM. Mouse model for hereditary hemorrhagic telangiectasia has a generalized vascular abnormality. Circulation. 2003;107:1653–1657. doi: 10.1161/01.CIR.0000058170.92267.00. [DOI] [PubMed] [Google Scholar]
- Torsney E, Charlton R, Parums D, Collis M, Arthur HM. Inducible expression of human endoglin during inflammation and wound healing in vivo. Inflamm Res. 2002;1:464–470. doi: 10.1007/pl00012413. [DOI] [PubMed] [Google Scholar]
- van Laake LW, van den Driesche S, Post S, Feijen A, Jansen MA, Driessens MH, Mager JJ, Snijder RJ, Westermann CJ, Doevendans PA, van Echteld CJ, Ten Dijke P, Arthur HM, Goumans MJ, Lebrin F, Mummery CL. Endoglin has a crucial role in blood cell-mediated vascular repair. Circulation. 2006;114:2288–2297. doi: 10.1161/CIRCULATIONAHA.106.639161. [DOI] [PubMed] [Google Scholar]