

Abstract
Cardiomyocyte injury following ischemia–reperfusion can lead to cell death and result in cardiac dysfunction. A wide range of cardioprotective factors have been studied to date, but only recently has the cardioprotective role of fatty acids, specifically arachidonic acid (AA), been investigated. This fatty acid can be found in the membranes of cells in an inactive state and can be released by phospholipases in response to several stimuli, such as ischemia. The metabolism of AA involves the cycloxygenase (COX) and lipoxygenase (LOX) pathways, as well as the less well characterized cytochrome P450 (CYP) monooxygenase pathway. Current research suggests important differences with respect to the cardiovascular actions of specific CYP mediated arachidonic acid metabolites. For example, CYP mediated hydroxylation of AA produces 20-hydroxyeicosatetraenoic acid (20-HETE) which has detrimental effects in the heart during ischemia, pro-inflammatory effects during reperfusion and potent vasoconstrictor effects in the coronary circulation. Conversely, epoxidation of AA by CYP enzymes generates 5,6-, 8,9-, 11,12- and 14,15-epoxyeicosatrienoic acids (EETs) that have been shown to reduce ischemia–reperfusion injury, have potent anti-inflammatory effects within the vasculature, and are potent vasodilators in the coronary circulation. This review aims to provide an overview of current data on the role of these CYP pathways in the heart with an emphasis on their involvement as mediators of ischemia–reperfusion injury. A better understanding of these relationships will facilitate identification of novel targets for the prevention and/or treatment of ischemic heart disease, a major worldwide public health problem.
Keywords: Arachidonic acid, Cytochrome P450, Eicosanoids, Ischemia reperfusion injury, Cardioprotection
1. Introduction
Heart disease and stroke are major causes of illness, disability and death in Western societies, and impose a great burden to national health care systems [1-6]. For example, cardiovascular disease (CVD) accounted for the death of approximately 76,500 Canadians in 2002 [2,4] and over 910,000 individuals in the US [6]. As the population ages and co-morbidities, such as obesity and diabetes become more prevalent, both the human cost and economic burden of CVD will likely increase. Acute myocardial infarction (AMI) continues to be a leading cause of death worldwide [7,8]. Myocardial infarction occurs when ischemia exceeds a critical threshold and overwhelms cellular repair mechanisms that are designed to maintain normal operating function and homeostasis. Ischemia at this critical threshold level results in irreversible myocardial cell damage or death. Such injury contributes to the pathogenesis of heart failure (HF), AMI and sudden death [9]. Advances in early reperfusion therapy, such as thromobolytic drugs, coronary angioplasty or bypass graft surgery, have reduced morbidity, HF and infarct-associated ventricular arrhythmias. Preconditioning (PC) is another powerful cardioprotective strategy that renders the heart resistant to injury [10]. Brief, non-detrimental episodes of ischemia or pharmacological mimetics given prior to a prolonged ischemic event can initiate signaling events that protect the myocardium [10]. Unfortunately, both early reperfusion therapy and cardioprotective drugs given prior to ischemia have limited clinical utility as patients typically present after the onset of ischemia and/or are unable to reach medical facilities [11,12]. In light of the increasing incidence and prevalence of HF after AMI [1,3,4], there is a profound need for a better understanding of the underlying pathophysiology and a need for development of strategies to protect the myocardium from ischemic-reperfusion injury. Thus, novel therapeutic strategies are required in order to prevent the adverse consequences and impact of CVD.
2. Arachidonic acid, CYP epoxygenases and soluble epoxide hydrolase
Arachidonic acid (AA), a polyunsaturated fatty acid normally found esterified to cell membrane glycerophospholipids, can be released by phospholipases in response to several stimuli, such as ischemia [13]. Free AA is then available for metabolism by prostaglandin H2 synthases, lipoxygenases and cytochrome P450 monooxygenases to generate numerous metabolites, collectively termed eicosanoids [14,15]. CYP epoxygenases metabolize AA to four regioisomeric epoxyeicosatrienoic acids (5,6-, 8,9-, 11,12- and 14,15-EETs), all of which are biologically active (Fig. 1) [16,17]. The actions of EETs are terminated by conversion to the corresponding and less biologically active dihydroxyeicosatrienoic acids (DHETs) by epoxide hydrolases [13]. Two major epoxide hydrolases are found in mammalian tissues, the microsomal epoxide hydrolase (mEH) and the soluble epoxide hydrolase (sEH or EPHX2) [14]. Previous work has demonstrated that sEH is the main enzyme involved in the in vivo hydrolysis of the EETs [15,17]. The majority of endogenous EETs (>85%) are esterified to membrane glycerophospholipids, particularly phosphatidylcholine and phosphatidylinositol, where they are generally considered inactive until their release [17]. In this regard, ischemia has been shown to activate cytosolic phospholipase A2 leading to the release of bioactive eicosanoids from glycerophospholipids [18].
Fig. 1.

CYP epoxygenase and hydroxylase mediated metabolism of arachidonic acid.
EETs are important components of many intracellular signaling pathways in both cardiac and extracardiac tissues. For example, EETs activate Ca2+-sensitive K+ channels (BKCa) in vascular smooth muscle cells resulting in hyperpolarization of the resting membrane potential and vasodilation of the coronary circulation [19-21]. This effect is diminished upon hydrolysis of EETs to DHETs by sEH [22]. Other studies have shown that EETs display anti-inflammatory, thrombolytic and angiogenic properties within the vasculature [23-25]. In endothelial cells, EETs activate mitogen activated protein kinase (MAPK) and phosphatidylinositol-3 kinase (PI3K)-Akt signaling pathways [25], increase intracellular cAMP levels [24], upregulate expression of nitric oxide synthase [26] and protect against hypoxia-reoxygenation injury [27]. In general, the effects of DHETs on these pathways are less pronounced [23,24]. Within the heart, EETs activate cardiac ATP-sensitive K+ (KATP) channels [28-31], enhance L-type calcium currents [32,33] and improve postischemic recovery of left ventricular function [31,34-36]. Thus, alteration in the production and/or elimination of EETs may affect steady-state cellular levels of these bioactive eicosanoids in vivo and could potentially influence cardiac function (Fig. 2).
Fig. 2.
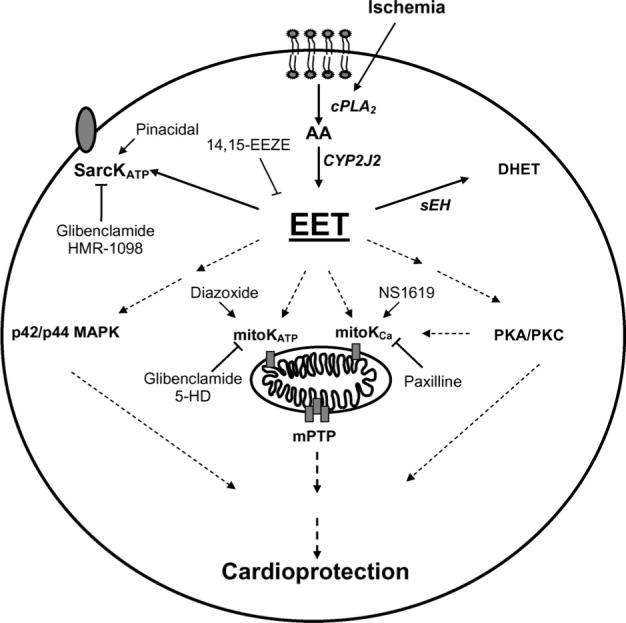
Schematic of proposed mechanisms of EET-mediated cardioprotection. CYP2J2-derived eicosanoids, readily removed by sEH, activate K+ channels, p42/p44-MAPK, PKA and/or PKC pathways leading to cardioprotection.
Arachidonic acid can be metabolized by CYP epoxygenases and CYP ω-hydroxylases to products that have vastly different physiologic effects. For example, EETs have potent vasodilatory properties [37] and 20-hydroxyeicosatetraenoic acid (20-HETE) has potent vasoconstrictive effects [38]. Therefore, changes in the expression and/or activity of specific CYP epoxygenase and hydroxylase enzymes can alter the delicate balance between EETs and 20-HETE. For instance, recent data has demonstrated that inhibition of CYP ω-hydroxylases results in reduction of infarct size in rats and dogs following ischemic injury, which suggests that 20-HETE has detrimental effects in the heart [39,40]. These investigators originally showed that 20-HETE was released into coronary venous blood in high concentrations at the end of 60 min of ischemia and throughout 3 h of reperfusion. Furthermore, the non-selective CYP inhibitor, miconazole (MIC) and the more selective inhibitors, 17-octadecanoic acid (17-ODYA) and N-methylsulfonyl-12,12-dibromododec-11-enamide (DDMS), all reduced 20-HETE release from the ischemic/reperfused heart and produced marked reductions in myocardial infarct size (expressed as a percent of the area at risk) from 19.6 ± 1.7% to 8.4 ± 2.5% (MIC), 5.9 ± 2.2% (17-ODYA) and 10.8 ± 1.8% (DDMS), respectively [39,40]. Conversely, exogenous administration of 20-HETE significantly increased infarct size to 26.9 ± 1.9%. These investigators also showed that at least three isoforms of the ω-hydroxylases were present in canine heart tissue – CYP4A1, CYP4A2 and CYP4F – all of which were markedly inhibited by incubation with 17-ODYA. Reductions in infarct size were also observed by this same group in rat hearts in which MIC,17-ODYA and DDMS were adminstered prior to ischemia or 5 min prior to reperfusion [40] These results suggest that the major effect of inhibiting ω-hydroxylase in rats occurs during reperfusion and is associated with in a decrease in “reperfusion injury” More recently, this same group demonstrated that inhibition of ω-hydroxylases in dog hearts with DDMS reduced infarct size to a degree equal to that of ischemic PC, the “gold standard” in cardioprotection research. These same investigators found that the administration of the 20-HETE antagonist, 20-HEDE, reduced infarct size similar to that observed with ischemic PC and DDMS. Interestingly, the combination of ischemic PC and DDMS produced a significantly greater reduction in infarct size than either intervention alone. Together, the results suggest that these two treatments may be acting via different signaling pathways and may be a potent combination in alleviating the sequelae of ischemia and/or reperfusion injury in a clinical setting, such as coronary artery bypass graft surgery.
Transgenic hearts from mice overexpressing human CYP2J2 have improved postischemic functional recovery as evidenced by studies using Langendorff perfused hearts subjected to ischemia/reperfusion injury [31]. Perfusion with the selective P450 epoxygenase inhibitor N-methylsulphonyl-6-(2-proparglyloxyphenyl)hexanamide (MS-PPOH) for 20 min prior to ischemia resulted in reduction of postischemic LVDP recovery in wild type hearts and abolished the improved postischemic LVDP recovery in CYP2J2 transgenic hearts. These data provided evidence that the cardioprotective effects of CYP2J2 overexpression involved P450 epoxygenase metabolites. Moreover, these data suggest an important role for endogenous P450 epoxygenases in postischemic functional recovery. Mechanistic studies by these investigators [31] suggested that enhanced activation of ATP-sensitive K+ channels (KATP) were involved in the improved recovery observed in the CYP2J2 transgenic mice. In addition, CYP2J2 overexpressing mice exhibited increased expression of phospho-p42/p44 mitogen-activated protein kinase (MAPK) following ischemia. The addition of the p42/p44 MAPK kinase (MEK) inhibitor PD98059 during reperfusion abolished the cardioprotective effects observed in the CYP2J2 mice. Together, these data suggest that CYP2J2-derived metabolites are cardioprotective following ischemia and the mechanism for this cardioprotection involves activation of KATP channels and p42/p44 MAPK.
Many CYP inhibitors lack isoform specificity and also may have effects on other signaling pathways. Reduction in infarct size observed in rat and rabbit hearts following treatment with non-specific CYP inhibitors has been attributed to suppression of CYP-dependent ROS production [41]. CYP2C and CYP2J isozymes are the predominant AA epoxygenases in the cardiovascular system. While CYP2C9 has been shown to generate ROS in coronary arteries [42], CYP2J2 is not a relevant source of ROS [27,42]. These studies highlight the complexity of the CYP enzyme system, emphasize the role of different CYP metabolites in cardioprotection and suggest caution in the interpretation of results when using non-selective CYP inhibitors. Recent epidemiologic data suggests an association between single nucleotide polymorphisms in the genes encoding CYP2J2, CYP2C8, CYP2C9 and EPHX2 and cardiovascular disease risk in humans supporting the functional relevance of the CYP epoxygenase pathway in the heart [43-47].
3. Ischemic injury and cardioprotection
Ischemic heart disease is an underlying cause of most AMIs, congestive HF, arrhythmias and sudden cardiac death. Myocardial ischemia is characterized by inadequate blood flow to the heart resulting in limited glucose, oxygen and delayed metabolic by-product removal. Ultimately, ischemic events result in cellular death and myocyte loss which is the primary pathology behind many CVDs. Myocytes are not easily replaced, although stem cell therapy shows great initial promise in overcoming this problem. Nevertheless, preserving the viability of ischemic myocardium is the major goal for cardioprotection. Cardioprotective mechanisms modulate cell metabolism or signaling pathways directly or via posttranslational modification of key proteins, as well initiate new transcription and translation [10,12]. A diverse spectrum of signals converge onto a few common end effectors that maintain membrane integrity and inhibit cell death [48]. Mitochondria are one of these important convergence points, due to their critical function in cell survival and death; notably, ATP production and apoptosis [10,12,48]. Key mitochondrial proteins, such as several potassium (K+) channels [49-53] and the mitochondrial permeability transition pore (mPTP) [48,54-56], act as effectors integrating these upstream signals into cardioprotective responses. The mechanisms by which CYP metabolites alter these important effectors are not well defined and require further experimentation.
3.1. K+ channels and cardioprotection
K+ channels are membrane proteins involved in many physiological processes, such as regulation of heart rate, muscle contraction and cell volume [57]. Two pharmacologically distinct ATP-sensitive potassium channels (KATP) have been identified in cardiomyocytes, sarcolemmal KATP (Sarc KATP) and mitochondrial KATP (mito KATP) [58]. sarc KATP is activated during cardiac ischemia when cytoplasmic ATP is depleted and this affects membrane excitability. Activation leads to shortening of the cardiac action potential and reduced intracellular calcium overload [59,60]. Although there are no selective sarc KATP openers, a number of drugs known to open this channel have been shown to produce beneficial effects in the myocardium in animal models of ischemia/reperfusion injury, and several non-selective inhibitors of sarc KATP (such as glibenclamide) block ischemic PC [59,60]. In spite of these findings, the strongest evidence for a role of sarc KATP channels in ischemic PC has been demonstrated in the KiR 6.2 knockout mouse where ischemic PC could not be produced [60]. The EETs have been shown to be potent openers of sarc KATP by reducing channel sensitivity to ATP in isolated rat myocytes using the inside-out patch clamp technique; however, the exact site on the channel that interacts with EETs remains unknown [30,61]. These authors also showed that membrane hyperpolarization occurred in isolated rat myocytes by 11,12-EET addition, an effect blocked by glibenclamide, the non-selective KATP channel inhibitor. A role for the sarc KATP channel in mediating the cardioprotective effect of inhibiting ω-hydroxylase at the time of reperfusion in the rat heart was recently described [34]. These investigators showed that the cardioprotective effect resulting from administering MIC and 17-ODYA just prior to reperfusion in rats was completely abolished by HMR 1098, a selective inhibitor of the sarc KATP channel. In contrast, the selective mito KATP channel inhibitor, 5-hydroxydecanoic acid (5-HD), had no effect. Although it can be assumed that a shift in the 20-HETE/EET balance in favor of the EETs is responsible for the cardioprotective effect of inhibiting 20-HETE synthesis, it remains unknown if the cardioprotective effect of the ω-hydroxylase inhibitors is the result of the action of the EETs on the sarc KATP channel in the myocardium.
In contrast to the sarc KATP channel, the precise molecular composition of the mito KATP channel remains elusive. However, recent studies suggest it is part of a multiprotein complex including the adenine nucleotide transporter (ANT) and succinate dehydrogenase (SDH) [62]. Importantly, pharmacologic data indicate that selective activation of mito KATP confers cardioprotection following ischemia and that this channel is the major one mediating ischemic PC [59,60,63,64]. While the precise pathways by which mito KATP activation confers cardioprotection remain unknown, potentially beneficial consequences of opening mito KATP include partial depolarization of the intramitochondrial membrane, transient swelling of the intramitochondrial space, enhanced respiration via the electron transport chain, reduced mitochondrial calcium overload, and altered production of reactive oxygen species [59,60,64]. Cardioprotective effects of CYP2J2 overexpression involve activation of mito KATP channels [31]. Increased flavoprotein fluorescence (a marker of mitochondrial redox status) [63] in CYP2J2 Tr cardiomyocytes is consistent with enhanced mito KATP activation in the presence of CYP2J2 overexpression [31]. Moreover, treatment of wild type cardiomyocytes with physiologically relevant concenrations of EETs increased flavoprotein fluorescence [31].
Ca2+-activated K+ (KCa) channels include large conductance (BKCa), voltage-sensitive K+ selective proteins expressed in various tissues including heart mitochondria [53]. KCa channels can be activated by elevations in intracellular Ca2+ and membrane depolarization [65,66]. CYP epoxygenase derived EETs are known activators of BKCa channels in vascular smooth muscle [20,67], whereas, CYP ω-hydroxylases derived HETEs are known inhibitors in vascular smooth muscle [38,68,69]. Recent evidence suggests that newly identified KCa channels in cardiac mitochondria (mito KCa) [53,70] are important mediators of cardioprotection. It is proposed that these mitochondrial K+ channels work in concert with mito KATP and other mitochondrial proteins in response to ischemia [49]. Activation of K+ channels by kinases, such as PKC or PKA, and other unknown signals is predicted to increase mitochondrial K+ uptake and in turn reduce Ca2+ overload in cardiomyocytes. The cardioprotective mechanisms associated with opening of these channels include a mild uncoupling, depolarization of the intramitochondrial membrane, transient swelling of the intramitochondrial space, enhanced respiration via the electron transport chain and altered production of reactive oxygen species [49,53,59,60,64,70].
3.2. Mitochondrial permeability transition pore
mPTP is a protein complex on the inner mitochondrial membrane which includes ANT, cyclophilin D and the voltage-dependent anion channel (VDAC) [48,49,71]. mPTP remains closed under normal physiological conditions but opens under cellular stress, such as during reperfusion following an ischemic event [48,49,54]. Opening allows free passage of molecules >1.5 kDa which initiate adverse effects, like large osmotic pressure changes and uncoupling of oxidative phosphorylation, that ultimately result in cell death [48,72]. Mitochondrial Ca2+ overload, oxidative stress, adenine nucleotide depletion and mitochondrial depolarization contribute to the pore opening. Inhibition of prolonged mPTP opening (high-conductance) during reperfusion with cyclosporine-A (CsA) or sanglifehrin-A (SfA) can reduce cardiomyocyte injury [73-75]. Conversely, evidence suggests that the transient (low-conductance) opening of mPTP during ischemic PC plays a role in cell survival [54]. Cell culture experiments suggest that mito KATP openers, such as diazoxide, trigger mPTP opening which can be blocked by CsA or 5HD [54]. Recent evidence demonstrates that multiple cardioprotective kinases, PKA, PKB/Akt and PKC can converge upon glycogen synthase kinase-3β (GSK-3β), which will induce inhibition of the mPTP [76]. While it is very likely that signaling pathways involved in EET-mediated cardioprotection target the mitochondria, it is unknown whether the response involves transient opening of mPTP prior to ischemia or prevents prolonged opening during reperfusion. EETs have been shown to activate sarc KATP [30,61] and mito KATP channels [31] in the heart, and KCa channels [20,67], in vascular smooth muscle cells [38,68,69]. Increased flavoprotein fluorescence in CYP2J2 Tr cardiomyocytes and wild type cardiomyocytes treated with EETs [31], strongly suggest convergence of a cardioprotective signal onto the mitochondria. It is expected that EETs will alter the flavin nucleotide fluorescence and intracellular [Ca2+] within the myocytes and most likely alter mPTP opening. It is unknown whether EETs work solely via mito KATP channels; however, it is likely they work together with other proteins, such as mito KCa channels. Future experiments will help determine whether EETs influence the transient opening or closing of mPTP, as well how they influence intramitochondrial [Ca2+] levels.
4. Cardioprotective signaling pathways
There is considerable controversy regarding the role of cytochrome P450s in the heart, notably the beneficial versus the detrimental effects of arachidonic acid metabolites [31,34,35,39,40,77,78]. We have demonstrated that EETs play a significant role in the improved postischemic functional recovery in isolated mouse hearts overexpressing CYP2J2 [31,34,35,57,78]. Recent results suggest potential cardioprotective mechanisms and indicate that KATP channels, p42/p44-MAPK and PI3K are involved. However, as previously discussed EETs are known to activate a number of signaling elements and ion channels, therefore, further work needs to be done to determine the important protective mediators involved in this novel cardioprotective pathway. Preliminary data suggested that EETs might only partially activate [31] the mito KATP channel and could be working synergistically with other cardioprotective sites, such as the sarc KATP channel or the mito KCa channel. Additional evidence has demonstrated an important role for PKA and PKC in the cardioprotective pathway involving K+ channels [79,80,81]. Further experiments will help determine if EET-mediated cardioprotection involves early activation or co-localization of PKA or PKC to the mitochondria.
5. Conclusion
There are numerous reasons for investigating the role of this novel endogenous pathway in myocardial function and ischemic injury. First, CYP-derived metabolites of arachidonic acid play critical roles in modulating fundamental biological processes [68,82]. Second, environmental or genetic factors that alter P450 expression and/or function lead to changes in the production of bioactive eicosanoids [68,82]. Such effects can influence cell and organ function in either an adverse or beneficial manner. Third, CYP isozymes expressed in the heart, notably CYP2J2 [36,83], generate EETs, which have been shown to be cardioprotective [31,36,83]. In contrast, other CYP isozymes (CYP2C and CYP4A) generate products, which are thought to be detrimental to the heart, such as ROS and 20-HETE [39-42]. Traditionally, investigation into the role of CYP isozymes has focused on hepatic and renal drug metabolism and function. There is little known about the importance of this endogenous system within the heart even though the heart contains significant levels of functionally active CYP. Finally, and most importantly, recent human epidemiological evidence has identified associations between CYP2J2 polymorphisms and CAD, and EPHX2 polymorphisms and CHD [43,45,46,84]. These findings provide strong evidence supporting the notion that EETs play an important role in postischemic functional recovery and perhaps infarct size reduction [31,33,36,83]. Recent studies have only begun to address the underlying mechanisms of EET-mediated cardioprotection and highlight the potential for this novel endogenous system as a therapeutic target for CVD. Ultimately, understanding the basic cellular mechanisms of EET-mediated cardioprotection will enhance our knowledge of this important phenomenon and will lead to the development of novel therapeutics for the treatment of cardiovascular diseases. Targeting CYP2J2, ω-hydroxylases or sEH, either through pharmacological or gene therapy methods, represents a novel therapeutic approach to the management of ischemic heart disease in humans.
Abbreviations
- AA
arachidonic acid
- AMI
acute myocardial infarction
- ANT
adenine nucleotide transporter
- BKCa
large conductance calcium activated potassium channels
- CYP
cytochrome P450 monooxygenase
- CVD
cardiovascular disease
- DHET
dihydroepoxyeicosatrienoic acid
- EETs
epoxyeicosatrienoic acid
- GSK-3β
glycogen synthase kinase-3β
- 20-HETE
20-hydroxyeicosatetraenoic acid
- HF
heart failure
- IPC
ischemic preconditioning
- IR
ischemic-reperfusion
- KATP
ATP-sensitive potassium channel
- KCa
calcium-activated potassium channel
- LVDP
left ventricular developed pressure
- MAPK
mitogen activated protein kinase
- mEH
microsomal epoxide hydrolase
- mito KATP
mitochondrial ATP-sensitive potassium channel
- mPTP
mitochondrial permeability transition pore
- MS-PPOH
N-methylsulphonyl-6-(2-proparglyloxyphenyl) hexanamide
- PKA
protein kinase A
- PKC
protein kinase C
- PI3K
phosphatidylinositol-3 kinase
- sarc KATP
sarcolemmal ATP-sensitive potassium channel
- sEH/Ephx2
soluble epoxide hydrolase
- SDH
succinate dehydrogenase
- VDAC
voltage-dependent anion channel
References
- 1.Health Canada Economic burden of illness in Canada. 1998.
- 2.Heart and Stroke Foundation of Canada The growing burden of heart disease and stroke in Canada. 2003.
- 3.Manuel DG, Leung M, Nguyen K, Tanuseputro P, Johansen H. Burden of cardiovascular disease in Canada. Can J Cardiol. 2003;19:997–1004. [PubMed] [Google Scholar]
- 4.Manuel DG, Luo W, Ugnat AM, Mao Y. Cause-deleted health-adjusted life expectancy of Canadians with selected chronic conditions. Chronic Dis Can. 2003;24:108–15. [PubMed] [Google Scholar]
- 5.Statistics Canada Causes of death, 2002. 2004.
- 6.Thom T, Haase N, Rosamond W, et al. Heart disease and stroke statistics—2006 update: a report from the American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Circulation. 2006;113:e85–e151. doi: 10.1161/CIRCULATIONAHA.105.171600. [DOI] [PubMed] [Google Scholar]
- 7.Tu JV, Austin PC, Filate WA, Johansen HL, et al. Outcomes of acute myocardial infarction in Canada. Can J Cardiol. 2003;19:893–901. [PubMed] [Google Scholar]
- 8.Tu JV, Cameron C. Impact of an acute myocardial infarction report card in Ontario, Canada. Int J Qual Health Care. 2003;15:131–7. doi: 10.1093/intqhc/mzg015. [DOI] [PubMed] [Google Scholar]
- 9.Bolli R, Becker L, Gross G, Mentzer R, Jr, Balshaw D, Lathrop DA. Myocardial protection at a crossroads: the need for translation into clinical therapy. Circ Res. 2004;95:125–34. doi: 10.1161/01.RES.0000137171.97172.d7. [DOI] [PubMed] [Google Scholar]
- 10.Yellon DM, Downey JM. Preconditioning the myocardium: from cellular physiology to clinical cardiology. Physiol Rev. 2003;83:1113–51. doi: 10.1152/physrev.00009.2003. [DOI] [PubMed] [Google Scholar]
- 11.Cannon R., III Mechanisms, management and future directions for reperfusion injury after myocardial infarction. Nat Clin Pract. 2005;2:88–94. doi: 10.1038/ncpcardio0096. [DOI] [PubMed] [Google Scholar]
- 12.Murphy E. Primary and secondary signaling pathways in early preconditioning that converge on the mitochondria to produce cardioprotection. Circ Res. 2004;94:7–16. doi: 10.1161/01.RES.0000108082.76667.F4. [DOI] [PubMed] [Google Scholar]
- 13.Morisseau C, Hammock BD. Epoxide hydrolases: mechanisms, inhibitor designs, and biological roles. Annu Rev Pharmacol Toxicol. 2004 doi: 10.1146/annurev.pharmtox.45.120403.095920. [DOI] [PubMed] [Google Scholar]
- 14.Fang X, Weintraub NL, McCaw RB, et al. Effect of soluble epoxide hydrolase inhibition on epoxyeicosatrienoic acid metabolism in human blood vessels. Am J Physiol Heart Circ Physiol. 2004;287:H2412–20. doi: 10.1152/ajpheart.00527.2004. [DOI] [PubMed] [Google Scholar]
- 15.Zeldin DC, Kobayashi J, Falck JR, et al. Regio- and enantiofacial selectivity of epoxyeicosatrienoic acid hydration by cytosolic epoxide hydrolase. J Biol Chem. 1993;268:6402–7. [PubMed] [Google Scholar]
- 16.Spector AA, Fang X, Snyder GD, Weintraub NL. Epoxyeicosatrienoic acids (EETs): metabolism and biochemical function. Prog Lipid Res. 2004;43:55–90. doi: 10.1016/s0163-7827(03)00049-3. [DOI] [PubMed] [Google Scholar]
- 17.Zeldin DC, DuBois RN, Falck JR, Capdevila JH. Molecular cloning, expression and characterization of an endogenous human cytochrome p450 arachidonic acid epoxygenase isoform. Arch Biochem Biophys. 1995;322:76–86. doi: 10.1006/abbi.1995.1438. [DOI] [PubMed] [Google Scholar]
- 18.Saluja I, Song D, O'Regan MH, Phillis JW. Role of phospholipase a2 in the release of free fatty acids during ischemia–reperfusion in the rat cerebral cortex. Neurosci Lett. 1997;233:97–100. doi: 10.1016/s0304-3940(97)00646-0. [DOI] [PubMed] [Google Scholar]
- 19.Campbell WB, Gebremedhin D, Pratt PF, Harder DR. Identification of epoxyeicosatrienoic acids as endothelium-derived hyperpolarizing factors. Circ Res. 1996;78:415–23. doi: 10.1161/01.res.78.3.415. [DOI] [PubMed] [Google Scholar]
- 20.Fisslthaler B, Popp R, Kiss L, et al. Cytochrome p450 2c is an edhf synthase in coronary arteries. Nature. 1999;401:493–7. doi: 10.1038/46816. [DOI] [PubMed] [Google Scholar]
- 21.Fleming I. Cytochrome p450 epoxygenases as edhf synthase(s). Pharmacol Res. 2004;49:525–33. doi: 10.1016/j.phrs.2003.11.016. [DOI] [PubMed] [Google Scholar]
- 22.Fang X, Kaduce TL, Weintraub NL, et al. Pathways of epoxyeicosatrienoic acid metabolism in endothelial cells. Implications for the vascular effects of soluble epoxide hydrolase inhibition. J Biol Chem. 2001;276:14867–74. doi: 10.1074/jbc.M011761200. [DOI] [PubMed] [Google Scholar]
- 23.Node K, Huo Y, Ruan X, et al. Anti-inflammatory properties of cytochrome p450 epoxygenase-derived eicosanoids. Science. 1999;285:1276–9. doi: 10.1126/science.285.5431.1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Node K, Ruan XL, Dai J, et al. Activation of gas mediates induction of tissue-type plasminogen activator gene transcription by epoxyeicosatrienoic acids. J Biol Chem. 2001;276:15983–9. doi: 10.1074/jbc.M100439200. [DOI] [PubMed] [Google Scholar]
- 25.Wang Y, Wei X, Xiao X, et al. Arachidonic acid epoxygenase metabolites stimulate endothelial cell growth and angiogenesis via mitogen-activated protein kinase and phosphatidylinositol 3-kinase/Akt signaling pathways. J Pharmacol Exp Ther. 2005;314:522–32. doi: 10.1124/jpet.105.083477. [DOI] [PubMed] [Google Scholar]
- 26.Wang H, Lin L, Jiang J, et al. Up-regulation of endothelial nitric-oxide synthase by endothelium-derived hyperpolarizing factor involves mitogen-activated protein kinase and protein kinase c signaling pathways. J Pharmacol Exp Ther. 2003;307:753–64. doi: 10.1124/jpet.103.052787. [DOI] [PubMed] [Google Scholar]
- 27.Yang B, Graham L, Dikalov S, et al. Overexpression of cytochrome p450 cyp2j2 protects against hypoxia-reoxygenation injury in cultured bovine aortic endothelial cells. Mol Pharmacol. 2001;60:310–20. doi: 10.1124/mol.60.2.310. [DOI] [PubMed] [Google Scholar]
- 28.Lee HC, Lu T, Weintraub NL, VanRollins M, Spector AA, Shibata EF. Effects of epoxyeicosatrienoic acids on the cardiac sodium channels in isolated rat ventricular myocytes. J Physiol. 1999;519(Pt 1):153–68. doi: 10.1111/j.1469-7793.1999.0153o.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lu T, Hoshi T, Weintraub NL, Spector AA, Lee HC. Activation of ATP-sensitive k(+) channels by epoxyeicosatrienoic acids in rat cardiac ventricular myocytes. J Physiol. 2001;537:811–27. doi: 10.1111/j.1469-7793.2001.00811.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lu T, VanRollins M, Lee HC. Stereospecific activation of cardiac ATP-sensitive k(+) channels by epoxyeicosatrienoic acids: a structural determinant study. Mol Pharmacol. 2002;62:1076–83. doi: 10.1124/mol.62.5.1076. [DOI] [PubMed] [Google Scholar]
- 31.Seubert J, Yang B, Bradbury JA, et al. Enhanced postischemic functional recovery in cyp2j2 transgenic hearts involves mitochondrial ATP-sensitive K+ channels and p42/p44 mapk pathway. Circ Res. 2004;95:506–14. doi: 10.1161/01.RES.0000139436.89654.c8. [DOI] [PubMed] [Google Scholar]
- 32.Xiao YF, Huang L, Morgan JP. Cytochrome p450: a novel system modulating Ca2+ channels and contraction in mammalian heart cells. J Physiol. 1998;508:777–92. doi: 10.1111/j.1469-7793.1998.777bp.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Xiao YF, Ke Q, Seubert JM, et al. Enhancement of cardiac l-type Ca2+ currents in transgenic mice with cardiac-specific overexpression of cyp2j2. Mol Pharmacol. 2004;66:1607–16. doi: 10.1124/mol.104.004150. [DOI] [PubMed] [Google Scholar]
- 34.Nithipatikom K, Endsley MP, Moore JM, et al. Effects of selective inhibition of cytochrome p-450 omega-hydroxylases and ischemic preconditioning in myocardial protection. Am J Physiol Heart Circ Physiol. 2006;290:H500–5. doi: 10.1152/ajpheart.00918.2005. [DOI] [PubMed] [Google Scholar]
- 35.Nithipatikom K, Moore JM, Isbell MA, Falck JR, Gross GJ. Epoxyeicosatrienoic acids (EETs) in cardioprotection: ischemic versus reperfusion injury. Am J Physiol Heart Circ Physiol. 2006 doi: 10.1152/ajpheart.00071.2006. [DOI] [PubMed] [Google Scholar]
- 36.Wu S, Chen W, Murphy E, et al. Molecular cloning, expression, and functional significance of a cytochrome p450 highly expressed in rat heart myocytes. J Biol Chem. 1997;272:12551–9. doi: 10.1074/jbc.272.19.12551. [DOI] [PubMed] [Google Scholar]
- 37.Medhora M, Narayanan J, Harder D. Dual regulation of the cerebral microvasculature by epoxyeicosatrienoic acids. Trends Cardiovasc Med. 2001;11:38–42. doi: 10.1016/s1050-1738(01)00082-2. [DOI] [PubMed] [Google Scholar]
- 38.Roman RJ, Maier KG, Sun CW, Harder DR, Alonso-Galicia M. Renal and cardiovascular actions of 20-hydroxyeicosatetraenoic acid and epoxyeicosatrienoic acids. Clin Exp Pharmacol Physiol. 2000;27:855–65. doi: 10.1046/j.1440-1681.2000.03349.x. [DOI] [PubMed] [Google Scholar]
- 39.Gross ER, Nithipatikom K, Hsu AK, et al. Cytochrome p450 omega-hydroxylase inhibition reduces infarct size during reperfusion via the sarcolemmal kATP channel. J Mol Cell Cardiol. 2004;37:1245–9. doi: 10.1016/j.yjmcc.2004.10.008. [DOI] [PubMed] [Google Scholar]
- 40.Nithipatikom K, Gross ER, Endsley MP, et al. Inhibition of cytochrome p450omega-hydroxylase: a novel endogenous cardioprotective pathway. Circ Res. 2004;95:e65–71. doi: 10.1161/01.RES.0000146277.62128.6f. [DOI] [PubMed] [Google Scholar]
- 41.Granville DJ, Tashakkor B, Takeuchi C, et al. Reduction of ischemia and reperfusion-induced myocardial damage by cytochrome p450 inhibitors. Proc Natl Acad Sci USA. 2004;101:1321–6. doi: 10.1073/pnas.0308185100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fleming I, Michaelis UR, Bredenkotter D, et al. Endothelium-derived hyperpolarizing factor synthase (cytochrome p450 2c9) is a functionally significant source of reactive oxygen species in coronary arteries. Circ Res. 2001;88:44–51. doi: 10.1161/01.res.88.1.44. [DOI] [PubMed] [Google Scholar]
- 43.Fornage M, Boerwinkle E, Doris PA, Jacobs D, Liu K, Wong ND. Polymorphism of the soluble epoxide hydrolase is associated with coronary artery calcification in African–American subjects. The coronary artery risk development in young adults (cardia) study. Circulation. 2004 doi: 10.1161/01.CIR.0000109487.46725.02. [DOI] [PubMed] [Google Scholar]
- 44.Fornage M, Lee CR, Doris PA, et al. The soluble epoxide hydrolase gene harbors sequence variation associated with susceptibility to and protection from incident ischemic stroke. Hum Mol Genet. 2005;14:2829–37. doi: 10.1093/hmg/ddi315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Przybyla-Zawislak BD, Srivastava PK, Vazquez-Matias J, et al. Polymorphisms in human soluble epoxide hydrolase. Mol Pharmacol. 2003;64:482–90. doi: 10.1124/mol.64.2.482. [DOI] [PubMed] [Google Scholar]
- 46.Spiecker M, Darius H, Hankeln T, et al. Risk of coronary artery disease associated with polymorphism of the cytochrome p450 epoxygenase cyp2j2. Circulation. 2004;110:2132–6. doi: 10.1161/01.CIR.0000143832.91812.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yasar U, Bennet AM, Eliasson E, et al. Allelic variants of cytochromes p450 2c modify the risk for acute myocardial infarction. Pharmacogenetics. 2003;13:715–20. doi: 10.1097/00008571-200312000-00002. [DOI] [PubMed] [Google Scholar]
- 48.Halestrap AP, Clarke SJ, Javadov SA. Mitochondrial permeability transition pore opening during myocardial reperfusion—a target for cardioprotection. Cardiovasc Res. 2004;61:372–85. doi: 10.1016/S0008-6363(03)00533-9. [DOI] [PubMed] [Google Scholar]
- 49.Hanley PJ, Daut J. K(ATP) channels and preconditioning: a re-examination of the role of mitochondrial k(ATP) channels and an overview of alternative mechanisms. J Mol Cell Cardiol. 2005;39:17–50. doi: 10.1016/j.yjmcc.2005.04.002. [DOI] [PubMed] [Google Scholar]
- 50.Liu Y, Sato T, Seharaseyon J, Szewczyk A, O'Rourke B, Marban E. Mitochondrial ATP-dependent potassium channels. Viable candidate effectors of ischemic preconditioning. Ann NY Acad Sci. 1999;874:27–37. doi: 10.1111/j.1749-6632.1999.tb09222.x. [DOI] [PubMed] [Google Scholar]
- 51.O'Rourke B. Evidence for mitochondrial K+ channels and their role in cardioprotection. Circ Res. 2004;94:420–32. doi: 10.1161/01.RES.0000117583.66950.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wang X, Yin C, Xi L, Kukreja RC. Opening of Ca2+-activated K+ channels triggers early and delayed preconditioning against i/r injury independent of nos in mice. Am J Physiol Heart Circ Physiol. 2004;287:H2070–7. doi: 10.1152/ajpheart.00431.2004. [DOI] [PubMed] [Google Scholar]
- 53.Xu W, Liu Y, Wang S, et al. Cytoprotective role of Ca2+-activated K+ channels in the cardiac inner mitochondrial membrane. Science. 2002;298:1029–33. doi: 10.1126/science.1074360. [DOI] [PubMed] [Google Scholar]
- 54.Hausenloy D, Wynne A, Duchen M, Yellon D. Transient mitochondrial permeability transition pore opening mediates preconditioning-induced protection. Circulation. 2004;109:1714–7. doi: 10.1161/01.CIR.0000126294.81407.7D. [DOI] [PubMed] [Google Scholar]
- 55.Hausenloy DJ, Duchen MR, Yellon DM. Inhibiting mitochondrial permeability transition pore opening at reperfusion protects against ischaemia-reperfusion injury. Cardiovasc Res. 2003;60:617–25. doi: 10.1016/j.cardiores.2003.09.025. [DOI] [PubMed] [Google Scholar]
- 56.Weiss JN, Korge P, Honda HM, Ping P. Role of the mitochondrial permeability transition in myocardial disease. Circ Res. 2003;93:292–301. doi: 10.1161/01.RES.0000087542.26971.D4. [DOI] [PubMed] [Google Scholar]
- 57.Wickenden A. K(+) channels as therapeutic drug targets. Pharmacol Ther. 2002;94:157–82. doi: 10.1016/s0163-7258(02)00201-2. [DOI] [PubMed] [Google Scholar]
- 58.Hu H, Sato T, Seharaseyon J, et al. Pharmacological and histochemical distinctions between molecularly defined sarcolemmal kATP channels and native cardiac mitochondrial kATP channels. Mol Pharmacol. 1999;55:1000–5. [PubMed] [Google Scholar]
- 59.Gross GJ, Peart JN. KATP channels and myocardial preconditioning: an update. Am J Physiol Heart Circ Physiol. 2003;285:H921–30. doi: 10.1152/ajpheart.00421.2003. [DOI] [PubMed] [Google Scholar]
- 60.Gross GJ, Fryer RM. Sarcolemmal versus mitochondrial ATP-sensitive K+ channels and myocardial preconditioning. Circ Res. 1999;84:973–9. doi: 10.1161/01.res.84.9.973. [DOI] [PubMed] [Google Scholar]
- 61.Lu T, Hoshi T, Weintraub NL, Spector AA, Lee HC. Activation of ATP-sensitive k(+) channels by epoxyeicosatrienoic acids in rat cardiac ventricular myocytes. J Physiol. 2001;537:811–27. doi: 10.1111/j.1469-7793.2001.00811.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ardehali H, Chen Z, Ko Y, Mejia-Alvarez R, Marban E. Multiprotein complex containing succinate dehydrogenase confers mitochondrial ATP-sensitive K+ channel activity. Proc Natl Acad Sci USA. 2004;101:11880–5. doi: 10.1073/pnas.0401703101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Liu Y, Sato T, O'Rourke B, Marban E. Mitochondrial ATP-dependent potassium channels: novel effectors of cardioprotection? Circulation. 1998;97:2463–9. doi: 10.1161/01.cir.97.24.2463. [DOI] [PubMed] [Google Scholar]
- 64.O'Rourke B. Evidence for mitochondrial K+ channels and their role in cardioprotection. Circ Res. 2004;94:420–32. doi: 10.1161/01.RES.0000117583.66950.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Brayden JE, Nelson MT. Regulation of arterial tone by activation of calcium-dependent potassium channels. Science. 1992;256:532–5. doi: 10.1126/science.1373909. [DOI] [PubMed] [Google Scholar]
- 66.Calderone V. Large-conductance, ca(2+)-activated k(+) channels: function, pharmacology and drugs. Curr Med Chem. 2002;9:1385–95. doi: 10.2174/0929867023369871. [DOI] [PubMed] [Google Scholar]
- 67.Campbell WB, Harder DR. Endothelium-derived hyperpolarizing factors and vascular cytochrome p450 metabolites of arachidonic acid in the regulation of tone. Circ Res. 1999;84:484–8. doi: 10.1161/01.res.84.4.484. [DOI] [PubMed] [Google Scholar]
- 68.Roman RJ. p-450 metabolites of arachidonic acid in the control of cardiovascular function. Physiol Rev. 2002;82:131–85. doi: 10.1152/physrev.00021.2001. [DOI] [PubMed] [Google Scholar]
- 69.Kroetz DL, Xu F. Regulation and inhibition of arachidonic acid omega-hydroxylases and 20-hete formation. Annu Rev Pharmacol Toxicol. 2005;45:413–38. doi: 10.1146/annurev.pharmtox.45.120403.100045. [DOI] [PubMed] [Google Scholar]
- 70.Siemen D, Loupatatzis C, Borecky J, Gulbins E, Lang F. Ca2+-activated k channel of the bk-type in the inner mitochondrial membrane of a human glioma cell line. Biochem Biophys Res Commun. 1999;257:549–54. doi: 10.1006/bbrc.1999.0496. [DOI] [PubMed] [Google Scholar]
- 71.Halestrap AP. Mitochondrial permeability: dual role for the ADP/ATP translocator? Nature. 2004;430:983–4. doi: 10.1038/nature02816. [DOI] [PubMed] [Google Scholar]
- 72.Crompton M. The mitochondrial permeability transition pore and its role in cell death. Biochem J. 1999;341(Pt 2):233–49. [PMC free article] [PubMed] [Google Scholar]
- 73.Nazareth W, Yafei N, Crompton M. Inhibition of anoxia-induced injury in heart myocytes by cyclosporin a. J Mol Cell Cardiol. 1991;23:1351–4. doi: 10.1016/0022-2828(91)90181-k. [DOI] [PubMed] [Google Scholar]
- 74.Griffiths EJ, Halestrap AP. Protection by cyclosporin a of ischemia/reperfusion-induced damage in isolated rat hearts. J Mol Cell Cardiol. 1993;25:1461–9. doi: 10.1006/jmcc.1993.1162. [DOI] [PubMed] [Google Scholar]
- 75.Griffiths EJ, Ocampo CJ, Savage JS, Stern MD, Silverman HS. Protective effects of low and high doses of cyclosporin a against reoxygenation injury in isolated rat cardiomyocytes are associated with differential effects on mitochondrial calcium levels. Cell Calcium. 2000;27:87–95. doi: 10.1054/ceca.1999.0094. [DOI] [PubMed] [Google Scholar]
- 76.Juhaszova M, Zorov DB, Kim SH, et al. Glycogen synthase kinase-3beta mediates convergence of protection signaling to inhibit the mitochondrial permeability transition pore. J Clin Invest. 2004;113:1535–49. doi: 10.1172/JCI19906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Moffat MP, Ward CA, Bend JR, Mock T, Farhangkhoee P, Karmazyn M. Effects of epoxyeicosatrienoic acids on isolated hearts and ventricular myocytes. Am J Physiol. 1993;264:H1154–60. doi: 10.1152/ajpheart.1993.264.4.H1154. [DOI] [PubMed] [Google Scholar]
- 78.Seubert JM, Goralski K, Sinal CJ, Murphy E, Zeldin DC. Role of soluble epoxide hydrolase in postischemic recovery of heart contractile function. Circulation. 2004;110(Suppl):808. doi: 10.1161/01.RES.0000237390.92932.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Liu GS, Cohen MV, Mochly-Rosen D, Downey JM. Protein kinase c-epsilon is responsible for the protection of preconditioning in rabbit cardiomyocytes. J Mol Cell Cardiol. 1999;31:1937–48. doi: 10.1006/jmcc.1999.1026. [DOI] [PubMed] [Google Scholar]
- 80.Cross HR, Murphy E, Bolli R, Ping P, Steenbergen C. Expression of activated pkc epsilon (pkc epsilon) protects the ischemic heart, without attenuating ischemic h(+) production. J Mol Cell Cardiol. 2002;34:361–7. doi: 10.1006/jmcc.2001.1518. [DOI] [PubMed] [Google Scholar]
- 81.Sato T, Saito T, Saegusa N, Nakaya H. Mitochondrial Ca2+-activated K+ channels in cardiac myocytes: a mechanism of the cardioprotective effect and modulation by protein kinase a. Circulation. 2005;111:198–203. doi: 10.1161/01.CIR.0000151099.15706.B1. [DOI] [PubMed] [Google Scholar]
- 82.Zeldin DC. Epoxygenase pathways of arachidonic acid metabolism. J Biol Chem. 2001;276:36059–62. doi: 10.1074/jbc.R100030200. [DOI] [PubMed] [Google Scholar]
- 83.Wu S, Moomaw CR, Tomer KB, Falck JR, Zeldin DC. Molecular cloning and expression of cyp2j2, a human cytochrome p450 arachidonic acid epoxygenase highly expressed in heart. J Biol Chem. 1996;271:3460–8. doi: 10.1074/jbc.271.7.3460. [DOI] [PubMed] [Google Scholar]
- 84.Lee CR, Bray NK, Fornage MS, et al. Genetic variation in soluble epoxide hydrolase (EPHX2) and risk of coronary heart disease: the atherosclerosis risk in communities (ARIC) study. Hum Mol Genet. 2006 doi: 10.1093/hmg/ddl085. [Epub] [DOI] [PMC free article] [PubMed] [Google Scholar]