

Abstract
Appropriate tyrosine kinase signaling depends on coordinated sequential coupling of protein-protein interactions with catalytic activation. Focal adhesion kinase (FAK) integrates signals from integrin and growth factor receptors to regulate cellular responses including cell adhesion, migration and survival. Here, we describe crystal structures representing both autoinhibited and active states of FAK. The inactive structure reveals a mechanism of inhibition in which the N-terminal FERM domain directly binds the kinase domain, blocking access to the catalytic cleft and protecting the FAK activation loop from Src phosphorylation. Additionally the FERM domain sequesters the Tyr397 autophosphorylation and Src recruitment site, which lies in the linker connecting the FERM and kinase domains. The active phosphorylated FAK kinase adopts a conformation that is immune to FERM inhibition. Our biochemical and structural analysis shows how the architecture of autoinhibited FAK orchestrates an activation sequence of FERM domain displacement, linker autophosphorylation, Src recruitment and full catalytic activation.
INTRODUCTION
Focal adhesions are integrin-mediated points of contact between the cell surface and the extracellular matrix. Coordinated assembly and disassembly of focal adhesions as well as intracellular signaling from these sites modulate cell adhesion, migration, proliferation, differentiation and survival in response to cues in the extracellular milieu (Bershadsky et al., 2003; Ridley et al., 2003). Focal adhesion kinase (FAK, gene name PTK2) is a central node in the signaling network emanating from focal adhesions (Mitra et al., 2005; Parsons, 2003). FAK is at once a signaling switch and a signaling scaffold – engagement of integrins and growth factor receptors on the cell surface induces catalytic activation of FAK, and also promotes its recruitment and activation of Src kinases and other signaling proteins. FAK is ubiquitously expressed, and is required for diverse developmental processes including neuronal pathfinding and epithelial and vascular morphogenesis (Nikolopoulos and Giancotti, 2005; Schock and Perrimon, 2002). FAK deficient mouse embryos are not viable due to a poorly developed vascular system (Ilic et al., 1995; Ilic et al., 2003). FAK overexpression in invasive tumors and its probable role in metastasis have made it a target for the development of anti-cancer drugs (Cohen and Guan, 2005a; McLean et al., 2005).
FAK and the closely related proline-rich tyrosine kinase 2 (Pyk2, also known as CAKβ) share a domain structure that includes an N-terminal FERM (band 4.1, ezrin, radixin, moesin homology) domain, followed by a ∼40 residue linker region, a central kinase domain, a ∼220 residue proline rich low complexity region and a C-terminal focal adhesion targeting (FAT) domain (Figure 1A). The FAT domain consists of a four helix bundle (Arold et al., 2002; Hayashi et al., 2002) and is critical for targeting FAK to focal adhesions via binding to paxillin, but it is not thought to play a direct role in catalytic regulation of FAK. The FERM domain is a 3-lobed protein interaction domain (Pearson et al., 2000) found in a number of cytoskeletal proteins (Chishti et al., 1998). In FAK, it has been shown to bind and inhibit the kinase domain (Cooper et al., 2003), and may mediate interaction of FAK with the cytoplasmic regions of activating receptors including β-integrins (Guan and Shalloway, 1992; Schaller et al., 1992) and the PDGFR (Sieg et al., 2000), EGFR (Sieg et al., 2000), EphA2 (Carter et al., 2002; Miao et al., 2000) and c-Met (Chen and Chen, 2006) growth factor receptors. The precise mechanisms by which cell-surface receptors initiate FAK activation are not clear, but integrin or growth factor activation induces FAK to autophosphorylate on Tyr 397 within the linker (Schaller et al., 1994). The phosphorylated Tyr 397 site and a nearby “PxxP”- motif recruit and activate Src via binding to its SH2 and SH3 domains, and Src in turn phoshorylates Tyr 576 and Tyr 577 in the activation loop of the FAK kinase (Calalb et al., 1995). The activated FAK/Src complex phosphorylates substrates including paxillin (Schaller and Parsons, 1995) and p130cas (Tachibana et al., 1997) and effects reorganization of the actin cytoskeleton via Rho-family GTPases (Mitra et al., 2005).
Figure 1. Structure of autoinhibited FAK.

(A) Domain structure of FAK. Key tyrosine phosphorylation sites are indicated. (B) Overall structure of autoinhibited FAK including the FERM, linker, and kinase regions. In the autoinhibited state, the FERM domain (blue ribbon representation) binds the kinase domain (red), primarily through an interaction between the FERM F2 lobe and the kinase C-lobe. A section of the linker that contains the autophosphorylation site Tyr 397 (yellow) is located between the FERM F1 lobe and the kinase N-lobe. The FERM domain also blocks access to the active site cleft and to the kinase activation loop (A-loop, green). Disordered segments are indicated as dashed lines. The staurosporine analogue AFN941 is bound to the active site of the kinase and is shown in stick representation. (C) Sequence alignment of the FERM, linker and kinase regions of avian FAK (cFAK1), human FAK (hFAK1) and human Pyk2 (hPyk2). cFAK1 shares 94% sequence identity with hFAK1, and hFAK1 43% with hPyk2. Secondary structure elements are indicated and the sequence is shaded to correspond to the colors in panel b. Residues involved in the FERM F2 lobe/kinase C-lobe interaction are indicated by an asterisk and regulatory tyrosines are colored magenta.
Among the major classes of non-receptor tyrosine kinases, which are all thought to be regulated by intramolecular domain interactions, a structural level understanding is only available for Src (Sicheri and Kuriyan, 1997; Xu et al., 1997) and Abl (Nagar et al., 2003) kinases, which share a related SH3-SH2-Kinase regulatory apparatus (Harrison, 2003). Other major classes with divergent domain architectures, including ZAP-70/Syk (SH2-SH2-Kinase domains) and Jak-family kinases (FERM-SH2-Pseudokinase-Kinase domains) in addition to FAK, have not been previously described. In order to understand the structural basis for FAK regulation and to elucidate general principles underlying regulation of non-receptor kinases, we determined the structure of a large fragment of FAK, containing the FERM and kinase domains, in an autoinhibited state. The structure reveals that the autoinhibited assembly is stabilized through an interaction between the FERM domain and the kinase C-lobe and we show that disruption of this interaction activates the kinase. Strikingly, a section of the linker region that contains the autophosphorylation site Tyr397 is positioned far from the active site between the FERM domain and the kinase N-lobe. We demonstrate that the autoinhibited assembly indeed prevents autophosphorylation of Tyr 397, and also blocks Src phosphorylation of the activation loop. Our structural and biochemical analysis of the FAK kinase in an active, Src-phosphorylated state shows that once the activation loop is phosphorylated, the kinase is no longer subject to inhibition by the FERM domain.
RESULTS
Overall structure of autoinhibited FAK
We crystallized a large fragment of avian FAK containing the FERM, linker, and kinase domains in an inactive, non-phosphorylated conformation. The 2.8Å resolution structure reveals a compact assembly with extensive interactions among the FERM, kinase and linker regions (Figure 1B). In this autoinhibited state, the three-lobed FERM domain bridges between the N- and C-lobes of the kinase domain, extending across the active site cleft. The major contact between the two domains is formed between the F2 lobe of the FERM domain and the C-lobe of the kinase domain. A portion of the linker segment containing the Tyr 397 autophosphorylation site binds to a groove on the F1 lobe of the FERM domain, and also makes a glancing contact with the N-terminal lobe of the kinase domain. We find the same overall conformation in two additional crystal forms of autoinhibited FAK, confirming that the observed domain organization is not a happenstance of crystal packing (see supplemental Table S1 for crystallographic details).
The FERM domain exhibits the characteristic 3-lobed architecture (lobes F1, F2 and F3, Figure 1B), and its structure is essentially the same as observed previously in our studies of the isolated FERM domain (Ceccarelli et al., 2006) (RMSD ∼ 1.24 Å). The Tyr 397 autophosphorylation site and flanking residues in the linker form an antiparallel β-strand interaction with the FERM F1 lobe. Strikingly, this contact positions the sidechain of Tyr 397 some 35Å away from the catalytic cleft of the kinase. The linker then extends into the kinase N-lobe, where it is in van der Waals contact with the N-terminal end of the C-helix (αC in Figure 1B). The amino terminal portion of the linker -- including the Src SH3 docking site -- is disordered in the present structure (residues 363−393). Interestingly, mutation of Lys 38 in the F1 lobe has been shown to increase phosphorylation of Tyr 397 (Cohen and Guan, 2005b). This residue is near (but does not contact) the linker suggesting the possibility that it could destabilize the linker/FERM interaction.
The kinase domain in the autoinhibited structure adopts a conformation similar to that of the isolated kinase domain in the unphosphorylated state (Nowakowski et al., 2002) (RMSD ∼ 1.49), but with a small difference in the relative orientation between the N- and C-lobes (a counterclockwise rotation of the N-lobe of approximately 12° when viewed from above). The staurosporine compound AFN941, a broad-spectrum protein kinase inhibitor that was included to improve crystal quality, is bound in the active site cleft in a manner analogous to that observed in other tyrosine kinases including Jak3 (Boggon et al., 2005) and Lck (Zhu et al., 1999). The kinase activation loop adopts an inactive conformation in which it extends toward the FERM domain (near Ser46 in the F1 lobe) and is then unstructured between residues Asp 573 and Leu 584. Although the Tyr 576 and Tyr 577 phosphorylation sites in the activation loop are not visible, they must lie in the space between the active site cleft and the F1 and F2 lobes of the FERM domain.
Why is the autoinhibited structure downregulated, and how will release of the FERM/kinase interface activate the kinase? The overall structure of the FERM-linker-kinase assembly suggests multiple inhibitory mechanisms: i) the position of the FERM domain sterically blocks access of the kinase domain to potential substrates, ii) the partially unstructured, inactive conformation of the activation loop is not expected to support binding of peptide substrates, and the FERM domain blocks the catalytically competent conformation of the unphosphorylated activation loop, iii) autophosphorylation of Tyr 397 is inhibited as this segment of the linker is incorporated into a β-sheet in the FERM domain ∼ 35Å from the active site, and iv) Tyrosines 576 and 577 within the activation loop are “sequestered” by the FERM domain, preventing their efficient phosphorylation by Src. We confirm the relevance of these autoinhibitory mechanisms via comparisons with the active structure of the FAK kinase and via functional dissection of the FERM/kinase interface as described in the following sections.
Structure of the active FAK kinase domain
In order to better understand FAK autoinhibition and activation, we determined the structure of the isolated kinase domain in an active conformation with both Tyr 576 and Tyr 577 phosphorylated. The structure was determined in complex with Mg2+ and the non-hydrolysable ATP analog AMP-PNP at 2.3 Å resolution (see Methods and supplemental Table S1). In this structure, the activation loop adopts a conformation very different from that in the autoinhibited structure (Figure 2A). The doubly-phosphorylated loop exhibits a β-hairpin-like conformation similar to that seen in other active tyrosine kinases including the insulin receptor (Hubbard, 1997), Lck (Yamaguchi and Hendrickson, 1996) and Jak3 (Boggon et al., 2005). The observed conformation of the activation loop appears to be stabilized primarily by hydrogen bond and electrostatic interactions with the phosphate group of pTyr 577 (Figure 2B). The sidechain and phosphate group of pTyr 576 extends into solution. Superposition of the C-lobes of the active FAK kinase on the autoinhibited structure reveals a similar overall structure for the kinase domain (RMSD= 1.44 Å), but a different conformation of the activation loop past the DFG motif (Figure 2C, supplemental Figure S1). Whereas in the autoinhibited structure residues 567−573 in the activation loop extend toward the FERM domain and partially occlude the catalytic cleft, the activation loop in the active kinase allows free access to the active site.
Figure 2. Structure of the active kinase domain of FAK.
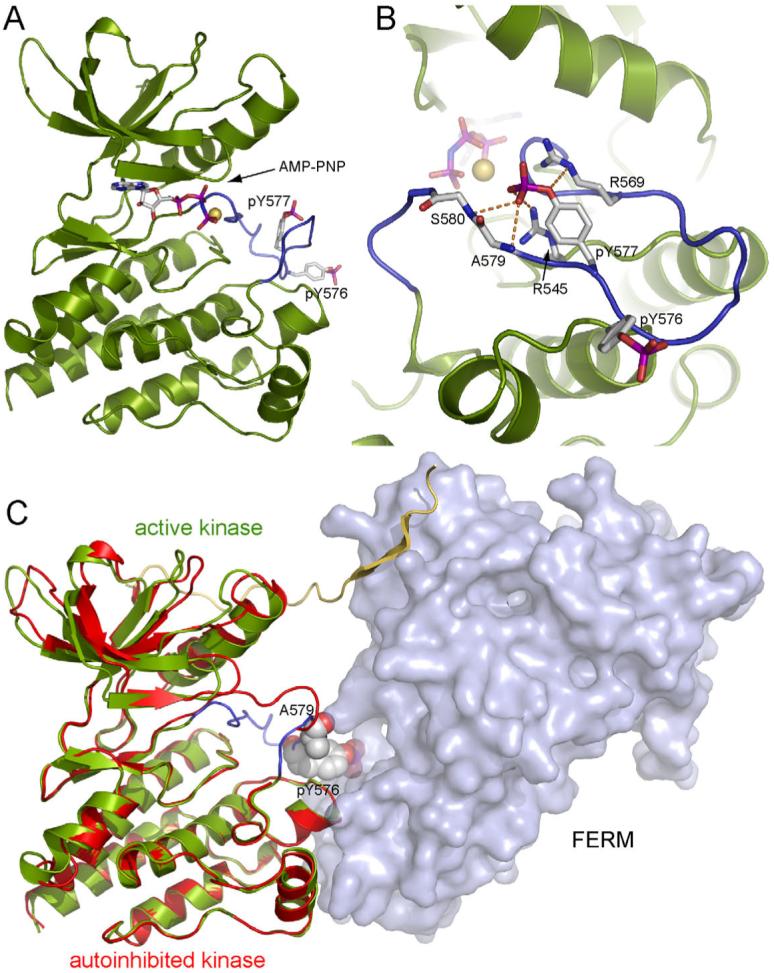
(A) The structure of the FAK kinase domain phosphorylated by Src is shown in ribbon representation (green) with the activation loop in blue. The side chains of phosphotyrosines 576 and 577 and AMP-PNP, which is bound to the active site, are shown in stick representation. A Mg2+ ion at the active site is shown as a yellow sphere. (B) Close up view of the activation loop with the side chains of pY576, pY577, R569, R545 and main chains of A579 and S580 shown in stick representation. A network of hydrogen bonds (orange dashed lines) involving the phosphate group of pY577 stabilizes the conformation of the activation loop. (C) Superposition of active and inactive FAK kinases. The autoinhibited structure is shown with the FERM domain as a surface representation (light blue), and the linker and kinase domains in a ribbon representation colored yellow and red, respectively. The structure of the active kinase domain (green ribbon and blue activation loop) is superimposed based on the kinase C-lobes. The side chain of pY576 and the main chain carbonyl of A579 in the active kinase (both residues are shown in space filling representations) clash with the FERM domain in the autoinhibited structure.
In many kinases, the C-helix is an integral regulatory component -- it pivots out of the active site cleft in the inactive conformation, and is restored to the active position by activation loop phosphorylation and/or appropriate protein-protein interactions (Huse and Kuriyan, 2002). In FAK, comparison of the active and autoinhibited kinases shows that the position of the C-helix (relative to the rest of the N-lobe) does not change upon activation – it is in a position typical of active kinases in both states (Figure 2C). Thus “C-helix displacement” is not an element of FAK regulation. Additionally, a disulfide bond that was observed in the previously reported structure of the unphosphorylated kinase domain (Nowakowski et al., 2002) is not present in any of our structures, active or inactive, and is therefore unlikely to play a role in FAK regulation.
Superposition of the active kinase domain on the inactive structure also shows that pTyr576 and Ala579 in the phosphorylated activation loop collide with the FERM domain in the autoinhibited structure, suggesting that that an active conformation of the activation loop and FERM domain inhibition would be mutually exclusive (Figure 2C).
Phosphorylation of the activation loop overrides FERM domain inhibition
In order to dissect the inter-related roles of FERM domain engagement and activation loop conformation, we compared the in vitro catalytic activity of the crystallized FERM+kinase construct with that of the isolated kinase domain. Both proteins were prepared in defined, homogeneous phosphorylation states in which Tyrosines 576 and 577 in the activation loop were either both unphosphorylated or both phosphorylated. This experiment was performed using a Y397F mutant of the FERM+kinase protein to preclude any confounding effects due to Tyr 397 phosphorylation. We find that in the absence of activation loop phosphorylation, the FERM-containing protein is approximately five-fold less active than the isolated kinase (Figure 3). Strikingly, phosphorylation of the activation loop by Src increases the activity of the FERM+kinase protein more than twenty-fold, activating it to a level equivalent to that of the isolated kinase domain in the phosphorylated state. Considering this result and the steric incompatibility of the phosphorylated activation loop and FERM engagement, we conclude that once the activation loop is phosphorylated, the FERM domain is no longer able to downregulate FAK activity in vitro. These findings suggest that in a cellular context, dephosphorylation of the FAK activation loop will be a necessary step in restoration of the autoinhibited state.
Figure 3. Phosphorylation of the activation loop overrides FERM domain inhibition.
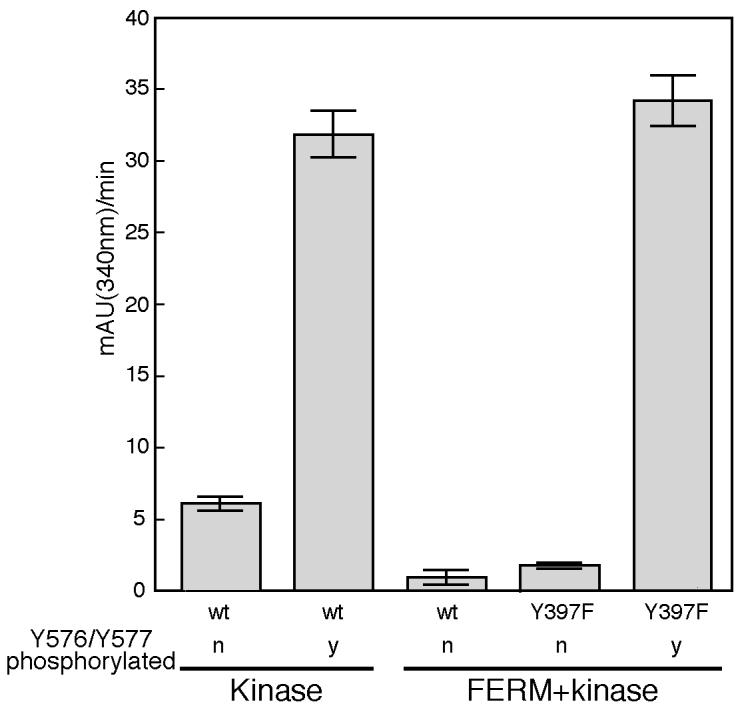
The in vitro kinase activity of the FAK kinase (FAK411−686) and single-chain FERM+kinase (FAK31−686) proteins is plotted for proteins that are unphosphorylated (n) or phosphorylated on Y576 and Y577 (y). Phosphorylated proteins were obtained by treatment with Src and subsequent repurification of phosphorylated FAK proteins. Note the reduced activity of the FERM+kinase protein compared to that of the isolated kinase domain prior to Src phosphorylation. Phosphorylation of the FERM+kinase protein by Src on Tyr 576 and Tyr 577 (shown for Y397F mutant, see text) yields an activity equal to that of the isolated kinase domain in the phosphorylated state.
The conserved FERM/kinase interface inhibits FAK
There are two points of contact between the FERM and kinase domains – a glancing, indirect contact that connects the FERM F1 lobe via the linker segment to the kinase N-lobe and a more extensive and direct contact between the F2 lobe and the kinase C-lobe (Figures 1B, 4A and 4B). The latter buries a surface area of 649 Å2, and appears to be the major source of stabilization for the autoinhibited assembly (Figure 4A). It is also highly conserved across species in both FAK and the related Pyk2 families (Figures 1C, 4C).
Figure 4. The FERM/kinase interface inhibits FAK.

(A) Detailed view of the interaction between the FERM F2 lobe and the kinase C-lobe. Sidechains that participate in the interaction are shown in stick form and are labeled. At the center of the interface F596 on the kinase domain inserts into a hydrophobic pocket on the FERM domain formed by Y180, M183, V196, L197. The periphery of the interaction is predominantly polar. Interdomain hydrogen bonds are shown as yellow dashed lines. (B) Detailed view of the linker region, which forms a b-sheet interaction with the FERM F1 lobe and contacts the kinase N-lobe. Side chains of the autophosphorylation site Y397 and residues at the linker/kinase contact point are shown. (C) Surface representation of separated FERM (right), linker (center) and kinase (left) domains colored according to sequence conservation. Viewing angles are indicated relative to Figure 1B. Conservation is indicated with a color scale from red (most highly conserved) to green (most variable). Note that the interacting surfaces of the FERM F2 lobe and kinase domain are highly conserved. Additionally, a patch of residues at the bottom of the F2 lobe (a potential binding site for a competitive activator, see text) and Trp266 and surrounding residues on the FERM F3 lobe are also highly conserved. Interestingly, Trp 266 and adjacent residues on the FERM domain form the same two-fold symmetric “dimer” interaction in every crystal structure containing the FERM domain, even though these FAK proteins appear to be monomeric in solution. The analysis was performed with ClustalW (Chenna et al., 2003) and ConSurf (Glaser et al., 2003) servers, and the figure was generated with Pymol (DeLano, 2002). (D) In vitro kinase activity of purified wt and mutant FERM+kinase proteins are plotted as negative slopes of NADH depletion (see methods). Disruption of the FERM F2 lobe/kinase C-lobe interface (Y180A, M183A and F596D mutations) activates the autoinhibited form of FAK. (E) Size exclusion chromatography of purified wild-type and mutant single-chain FERM+kinase proteins. Stokes radii were calculated by comparison with standards (Catalase, Aldolase, BSA, Ovalbumin, Chymotrypsinogen and Ribonuclease)(Siegel and Monty, 1966). Mutations that activate FAK result in a larger stokes radius, indicating an ‘open’ conformation, compared to the ‘closed’ autoinhibited form.
In order to test the importance of these interactions in FAK regulation, we introduced mutations at the FERM/kinase interface and tested their effects on FAK activation and phosphorylation in vitro and in vivo. To disrupt the contact with the kinase C-lobe, we mutated either the FERM F2 lobe (Y180A, M183A double mutant) or Phe 596 in the kinase domain (F596D). In the N-lobe interface, we mutated Ser 463 in the kinase C-helix (S463Y) (Figure 4B). We find that both the Y180A, M183A and F596D mutants have dramatically increased kinase activity as compared with the wild-type protein in vitro (Figure 4D). Additionally, both proteins have a larger hydrodynamic radius than the wild-type protein as measured by size-exclusion chromatography (Figure 4E). Not unexpectedly, mutation of Ser 463 in the very modest N-lobe interface only slightly increased FAK activity (Figure 4D), and this mutant eluted from the sizing column at a volume very close to that of the wild-type protein (Figure 4E). We expect that the larger hydrodynamic radius of the activated mutants reflects an “open” conformation in which the FERM domain is released entirely from the kinase domain, but we have not ruled out the possibility of dimerization or an alternative mode of FERM domain interaction that results in a larger hydrodynamic radius.
Mutations in the FERM/kinase interface also activate full-length FAK both in vitro and in vivo. We studied the same mutants described above, and also an additional activated FERM domain variant (a V196D, L197D double mutant). We transiently expressed wild-type or mutant FAK in 293 cells and found that activating mutations lead to increased levels of autophosphorylation on Tyr397 and Src phosphorylation on Tyr 576, 577, 861 and 925 (Figure 5A). We further assayed phosphorylation of recombinant paxillin by immunoprecipitated FAK proteins. All of the activated mutants readily phosphorylated paxillin, whereas the wild-type and S463Y proteins did so far less efficiently (Figure 5B). Importantly, the activated mutants induced higher levels of cellular tyrosine phosphorylation, and also more efficiently phosphorylated co-expressed paxillin in 293 cells (Figures 5C and 5D). Finally, the activating mutants also abrogated the usual adhesion-dependence of FAK activation (Figures 5E and supplemental Figure S2A). We confirmed that FAK and paxillin phosphorylation was indeed mediated by FAK and not Src by employing a FAK kinase dead mutant or a Src-specific kinase inhibitor (supplemental Figures S2B and S2C). Further, we confirmed that disruption of the FERM/kinase interface does not affect FAK localization to focal adhesions (supplemental Figure S3). These results demonstrate that the regulatory interactions we describe here are relevant to the full-length kinase in a cellular context.
Figure 5. Analysis of wild type and mutant FAK in HEK 293 cells.

Mutations were introduced into full-length FAK to disrupt the interaction of the FERM F2 lobe with the kinase C-lobe (Y180A, M183A and V196D, L197D on the FERM domain and F596D on the kinase domain) or to disrupt the contact of the linker with the kinase N-lobe (S463Y). (A) Full-length wild-type or mutant FAK proteins were transiently expressed in HEK 293 cells and cell lysates were analyzed by Western blotting with the indicated phospho-specific antibodies or with the 4.47anti-FAK antibody. (B) In vitro kinase assay for paxillin phosphorylation. Wild type and mutant full-length FAK proteins were immunoprecipitated from HEK 293 lysates using BC4. The immune complex was incubated in a kinase assay utilizing recombinant GST-paxillin-N-C3 as an exogenous substrate (Lyons et al., 2001). Phosphorylation of paxillin was detected using the phosphospecific antibodies pY31 and pY118, as well as the 4G10 phosphotyrosine antibody. Equal amounts of substrate were verified by blotting for paxillin using a polyclonal antiserum. Equal recovery of FAK proteins in the immune complex was verified by blotting for FAK. (C) HEK 293 cells transiently expressing FAK proteins were lysed and analyzed for cellular phosphotyrosine by Western blotting cell lysates with 4G10. (D) Paxillin and FAK were transiently co-expressed in HEK 293 cells and paxillin phosphorylation analyzed by Western blotting lysates using the pY31 antibody (upper panel). Lysates were blotted for paxillin using a monoclonal antibody as a loading control (middle panel) and for FAK to verify comparable expression of the wild type and mutant FAK proteins (bottom panel). (E) Wild type or mutant full-length FAK was transiently expressed in FAK null cells and adherent cells (Ad) or cells incubated in suspension at 37°C for 1 hour (sus) were lysed. FAK was immunoprecipitated using BC4 and the immune complexes analyzed by Western blotting for phosphotyrosine using 4G10 (top panel). Equal amounts of FAK in the immune complexes were verified by blotting for FAK (bottom panel).
Activating phosphorylation sites are sequestered
Because the Tyr 397 autophosphorylation site is incorporated into a β-sheet of the FERM domain and because the FERM domain apparently blocks access of Src to the activation loop, we sought to test whether protection of these sites from phosphorylation indeed contributes to FAK inhibition. We compared Tyr 397 autophosphorylation of the autoinhibited, wild-type protein with that of the mutants that release the FERM/kinase interface (Figure 6A). In the activated mutants we expect an increase in autophosphorylation for two reasons – loss of repression of the kinase domain and also exposure of the Tyr 397 phosphorylation site. To distinguish between these effects, we included a substrate in which the Tyr 397 site is fully exposed as an internal control (F-L, which contains the FERM and linker regions of FAK, residues 31−405). We find that both the F596D and Y180A, M183A FERM+kinase mutants autophosphorylate their Tyr 397 site as efficiently as they phosphorylate the exposed Y397 in the control substrate (Figure 6A). In contrast, in the inhibited FERM+kinase proteins (wt and S463Y) autophosphorylation was markedly less efficient than Tyr 397 phosphorylation of the control substrate. Hence, we conclude that Tyr 397, as positioned in autoinhibited FAK, is indeed protected from autophosphorylation, and that this protection is lost when the FERM domain is released from the kinase. It remains unclear whether physiologically relevant autophosphorylation of Tyr 397 occurs in cis, in trans, or by both mechanisms, but kinase-inactive FAK can clearly be transphosphorylated on Tyr 397 in vivo (Leu et al., 2002; Toutant et al., 2002; Sieg et al., 2000). In vitro, we find that Tyr 397 phosphorylation can occur both in cis and trans (Supplemental Figure S4). In any case, our structural and biochemical results show that incorporation of the Tyr 397 site as a β-strand in the FERM F1 lobe does indeed protect it from autophosphorylation, whether it occurs in cis or trans.
Figure 6. Tyrosine phosphorylation sites are protected in autoinhibited FAK.

(A) Time course of autophosphorylation at Tyr 397 was carried out for wild type and mutant purified single-chain FERM+kinase proteins (upper bands, labeled F-L-K for FERM-Linker-Kinase). FERM+linker protein (Y180A, M183A mutant), which has Y397 exposed, was included in the reactions as a control substrate (lower bands, labeled F-L for FERM-Linker). Tyr 397 phosphorylation was monitored by Western blotting using a specific anti-pY397 antibody (left panels). As loading controls equivalent gels were stained by Coomassie (right panels). Note that autoinhibited FERM+kinase protein (wt and S463Y mutant) autophosphorylate on Y397 much less efficiently than they phosphorylate Y397 of the control substrate, but that this discrimination is lost on the ‘open’ mutants (Y180A, M183A and F596D), which autophosphorylate as rapidly as they phosphorylate the control substrate. Also, note the overall increase in phosphorylation in the open mutants, consistent with their increased catalytic activity (as shown in Figure 4C). (B) and (C) Time course of Src phosphorylation of wt and mutant FERM+kinase on Tyr 576 and Tyr 577. Phosphorylation was monitored by Western blotting using specific anitibodies against pY576 (b, left panels) or pY577 (c, left panels) and equivalent gels were Coomassie stained as loading controls (right panels). Note that Src phosphorylation of the activation loop tyrosines is diminished in the intact, autoinhibited protein.
We also compared the ability of Src to phosphorylate the activation loop of wild-type versus mutant FAK. Both the F596D and Y180A, M183A proteins were more rapidly phosphorylated on tyrosines 576 and 577 by Src in vitro (Figures 6B and 6C), although the in vitro effect is not as striking as that observed in 293 cells (Figures 5A). The increased levels of FAK phosphorylation on Tyr 576, 577, 861 and 925 observed in 293 cells (Figure 5A) is likely due both to exposure of the activation loop in the active state (as demonstrated above in vitro), and to enhanced recruitment and activation of Src kinases to the Tyr 397 autophosphorylation site (supplemental Figure S2D).
DISCUSSION
To a remarkable extent, the logic of FAK signaling is encoded in the structure of the autoinhibited enzyme. The architecture of autoinhibited FAK appears to have evolved to enforce orderly, sequential activation in response to receptor engagement (Figure 7). In the autoinhibited state, the kinase is locked in an inactive conformation, and the activating phosphorylation sites in both the linker and the activation loop are sequestered. Release of the FERM domain would appear to be required to allow autophosphorylation of Tyr 397 and subsequent steps in activation. In analogy with Src-family kinases, which are activated by displacement of their SH3 and/or SH2 domains (Moarefi et al., 1997), we propose that FERM domain displacement is a key initial step in FAK activation. In this regard, a conserved basic patch on the F2 lobe of the FERM domain that is immediately adjacent to the site of contact with the kinase domain has been shown to be important for FAK activation (Chen and Chen, 2006; Dunty et al., 2004). It is tempting to speculate that this site could represent an initial site of docking for an activating protein, which might then directly disrupt the FERM/kinase interface to activate FAK. Precisely how integrin or growth factor engagement couples to FAK activation, and whether the coupling is direct (for example, via integrin cytoplasmic tails) or involves additional proteins, will require further investigation. Irrespective of the mechanism of its displacement, once the FERM domain is released, Tyr 397 is rapidly autophosphorylated and the activation loop is exposed for Src phosphorylation, indicative of a cooperative “disassembly” of interactions among the FERM, linker and kinase regions. Subsequent Src recruitment to the Tyr 397 site and phosphorylation of the activation loop yield the fully active FAK enzyme.
Figure 7. Schematic of autoinhibited FAK and a sequential model of activation.

In the inactive state (left), the FERM domain blocks the kinase active site and sequesters the Tyr 397 and activation loop phosphorylation sites. Although not observed in the present structure, we indicate the docking of the PxxP motif in the linker to the F3 lobe of the FERM domain based on previous work (Ceccarelli et al., 2006). We propose that FAK activation will be initiated by displacement of the FERM domain by competitive binding of an activating protein (orange star) to the FERM F2 surface. Such interactions have not been structurally characterized, but candidate activating proteins include the cytoplasmic regions of b-integrins or growth factor receptors EGFR, PDGFR, c-Met or EphA2. Disassembly of the autoinhibited conformation allows rapid autophosphorylation of the linker residue Tyr 397, and exposes the Src docking sites in the linker (center panel). In a subsequent step, Src is recruited and activated via SH2 binding to pTyr 397 and SH3 binding to the PxxP sequence in the linker region. Localized Src then phosphorylates the activation loop residues Tyr 576 and Tyr 577 of FAK (right panel). Phosphorylation of the activation loop yields full catalytic activity, and even in the event of activator dissociation will not allow kinase inhibition by the FERM domain. Note that for simplicity we have depicted FAK autophosphorylation in cis, it may, however, also occur in trans.
Comparison of FAK and Src reveals marked differences in their autoinhibitory mechanisms, but also fundamental similarities. In Src kinases, the SH3 and SH2 domains assemble on the “back” of the kinase domain, opposite the catalytic cleft, and inactivate the kinase domain indirectly by stabilizing an inactive conformation in which the C-helix is displaced from the active site (see Boggon and Eck, 2004; Sicheri and Kuriyan, 1997 for review). In contrast, the FAK FERM domain directly blocks the catalytic cleft and the overall conformation of the FAK kinase is remarkably similar in the autoinhibited and active conformations. Although Src is fully activated by autophosphorylation, FAK requires partnership with Src to achieve full catalytic activity. Despite their very different domain architectures and mechanisms of kinase inhibition, Src and FAK do share similar features that are fundamental to their function. In both, linker segments that connect the regulatory and kinase domains are used to stabilize the autoinhibited assembly, in addition to direct contacts between the regulatory and catalytic domains. Furthermore, in both kinases the activating phosphorylation sites are sequestered in the inactive conformations, and appropriate interactions with the regulatory domains are expected to disassemble the autoinhibitory interactions to expose these sites for phosphorylation. These similarities in Src and FAK may represent underlying “design principles” in tyrosine kinase signal transduction, as it is likely that they evolved independently in both kinases.
EXPERIMENTAL PROCEDURES
Protein expression and crystallization
Avian FAK containing the FERM and kinase domains (wt and mutant FAK31−686) or only the kinase domain (FAK411−686) were expressed in insect Hifive™ cells (Invitrogen) using the baculovirus expression system. The FERM domain was expressed and purified as described in (Ceccarelli et al., 2006). FERM+kinase proteins (expressed as GST fusions), were initially purified by affinity purification on glutathione sepharose beads (GE Healthcare). After TEV cleavage (to remove GST) proteins were further purified by anion exchange (MonoQ, GE Healthcare) and size exclusion (Superdex200, GE Healthcare) chromatography. Purified autoinhibited FERM+kinase was concentrated to approximately 10 mg/ml in 20 mM Tris pH 8.0, 200 mM NaCl, 5% Glycerol and 2 mM TCEP for crystallization. Optimal crystallization was performed in hanging drops by mixing the protein with an equal volume of 13% PEG10K, 250 mM NaCl, 100 mM Tris pH 8.5, 10 mM TCEP and 0.2 volume of a 1mg/μl aqueous suspension of AFN941 (Novartis). Crystals were flash-frozen in liquid nitrogen after brief soaking in precipitant solution containing additionally 25% ethylene glycol (EG) as cryoprotectant. The FAK kinase domain (expressed with 6xhis-tag) was initially purified on Ni sepharose beads (GE Healthcare). Subsequently, the his-tag was removed with TEV protease. Further purification was carried out by cation exchange (MonoS, GE Healthcare) and size exclusion (Superdex200) chromatography. The purified FAK kinase domain was incubated for 6h at RT with human Src (containing SH3, SH2 and kinase domains and a phosphorylated Tyr 416) at a molar Src:FAK ratio of 1:50. Phosphorylation on Tyr 576 and Tyr 577 was monitored with specific anti-pTyr antibodies (Biosource) and unphosphorylated protein was removed by cation exchange chromatography (MonoS). Phosphorylated kinase was further purified and buffer exchanged by size exclusion and concentrated to 4 mg/ml in 20 mM HEPES pH 7.0, 150 mM NaCl, 5% Glycerol and 2 mM TCEP. Crystallization was performed by pre-incubating the protein with 2 mM AMP-PNP and 4 mM MgCl2 for 1h at 4°C before setting up hanging drops with equal volume of 26% PEG4K, 200 mM LiSO4, 100 mM Tris pH 8.5, 10 mM TCEP. Crystals were flash-frozen in precipitant containing 18% EG.
Data collection and structure determination
Data was collected at beamline 24-ID (NE-CAT) at the Advanced Photon Source (Argonne National Laboratory) and beamline X29A at the National Synchrotron Light Source (Brookhaven National Laboratory). Data was processed with HKL2000 (Otwinowski and Minor, 1997) and yielded the following space groups and cell parameters: C212121, a=67.5, b=91.3, c=250.7 Å for FERM+kinase crystals and P212121, a=44.3, b=71.8, c=86.9 Å for active kinase crystals. Phases were calculated using the molecular replacement program PHASER (Storoni et al., 2004), using the FAK FERM (2AEH) (Ceccarelli et al., 2006) and/or the kinase domain (1MP8) (Nowakowski et al., 2002) as molecular search probes. Both structures contained one molecule in the asymmetric unit cell (52.1 % solvent, VM=2.57 Å3/Da, (Matthews, 1968) for the FERM+kinase structure and 44.9 % solvent, VM=2.23 Å3/Da for the active kinase structure). Refinement was performed using the program Refmac (Murshudov et al., 1997), manual rebuilding was carried out with the programs O (Jones et al., 1991) and Coot (Emsley and Cowtan, 2004). Final R-factors obtained were 21.7/27.4 (Rwork/Rfree) for the FERM+kinase structure and 21.4/26.3 for the active kinase structure. Due to disorder residues 31−34, 363−393, 574−583 were not included in the final FERM+kinase structure. For a complete summary of data collection and refinement parameters see supplemental Table S1.
In vitro kinase assays
Kinase assays were essentially performed as described in (LaFevre-Bernt et al., 1998). In brief, 1 μM kinase was added to a reaction mixture containing 20 mM HEPES pH 7.0, 150 mM NaCl, 1mM TCEP, 10mM MgCl2, 100 mM phosphoenolpyruvate, 0.28 mM NADH, 0.08 units/μl pyruvate kinase, 0.1 units/μl lactate dehydrogenase, 100 μM E4Y (as polyGlu-Tyr, 4:1 Glu:Tyr, Sigma) and reactions were initiated by addition of 0.5 mM ATP. NADH depletion (which is coupled to the rate of the kinase reaction) was monitored by absorbance at 340 nm using a Spectra MAX Plus spectrometer (Molecular Devices). For assaying activation loop-phosphorylated states of the FAK kinase or FERM+kinase, proteins were incubated for 6h at RT with human Src (containing SH3, SH2 and kinase domains) at a molar Src:FAK ratio of 1:50. Phosphorylation on Tyr 576 and Tyr 577 was monitored with specific anti-pTyr antibodies (Biosource) and unphosphorylated protein and Src was removed by subsequent ion exchange and size exclusion chromatography.
Size exclusion chromatography
75 μg of wt or mutant FERM+kinase proteins were injected onto a PC3.2/30 Superdex 200 (column volume = 2.4 ml) and eluting proteins were detected by absorption at 280 nm. Stokes radii were calculated via elution volumes of Catalase, Aldolase, BSA, Ovalbumin, Chymotrypsinogen and Ribonuclease (Siegel and Monty, 1966).
Auto and Src phosphorylation
FERM+kinase proteins as expressed in insect cells were partially phosphorylated on Y397. Therefore, proteins were first dephosphorylated for 4 h at RT with CD45 phosphatase at molar CD45:FAK ratio of 1:20 molar. Dephosphorylation was monitored by Western blotting using a specific anti-pTyr 397 antibody (Biosource). Autophosphorylation reactions were performed at RT with 1 μM dephosphorylated FERM+kinase protein (FAK31−686 wt or mutant) and 1 μM mutant FERM+linker protein (FAK31−405 Y180A, M183A) in 20 mM Tris pH 8.0, 150 mM NaCl, 5% Glycerol, 1 mM TCEP, 1 mM sodium vanadate, 2 mM ATP and 4 mM MgCl2. For Src phosphorylation reactions 2 μM FERM+kinase protein, 0.2 μM human Src (SH3+SH2+kinase) and otherwise identical conditions as for autophosphorylation reactions were used. Equal amounts were removed at time points indicated in Figure 6 and analyzed by Western blotting using a specific anti-pTyr 397 antibody for autophosphorylation or anti-pTyr 576 and anti-pTyr 577 antibodies for Src phosphorylation reactions (Biosource).
Analysis of FAK in HEK293 cells
For expression of full-length FAK in cells, mutations were engineered into pcDNA3.1-FAK constructs. The avian paxillin cDNA was subcloned into pcDNA3−1 for transient expression. HEK 293 cells were maintained in Dulbecco’s modified Eagle’s medium/Ham’s F-12 medium containing 10% fetal bovine serum. Cells were transfected using Lipofectamine Plus according to the manufacturer’s recommended protocol (Invitrogen). Cells were lysed 24 hours post transfection using modified RIPA buffer (Reynolds et al., 1989). Phosphorylation was examined by Western blotting cell lysates using phosho-specific antibodies (anti pY925 antibody was obtained from Santa Cruz, all others from Biosource). For immune complex kinase assays, FAK was immunoprecipitated using the BC4 polyclonal antiserum and the immune complex incubated with 2 μg of GST-paxillin-N-C3 for 10 minutes using previously described kinase reaction conditions (Lyons et al., 2001). The reaction was terminated by the addition of sample buffer and phosphorylation of paxillin examined by Western blotting with phospho-specific antibodies (Biosource) or the 4G10 phosphotyrosine antibody (UBI). FAK was detected by Western blotting using the 4.47 antibody (UBI). A polyclonal antibody (Thomas et al., 1999) or commercially available monoclonal antibody (BD Transduction Labs) was used for paxillin blotting. For detection of adhesion dependent FAK phosphorylation wt or mutant full-length FAK was transiently expressed in FAK null fibroblasts. In some experiments, cells were taken into suspension by trypsinization and incubated in suspension at 37°C for 1 hour. The cells were collected by centrifugation and lysed in modified RIPA buffer. FAK was immunoprecipitated using BC4 and analyzed by Western blotting using the 4G10 phosphotyrosine antibody (UBI).
ACKNOWLEDGEMENTS
We thank staff at the APS beamline 24ID and NSLS beamline X29A for assistance, F. Poy for technical assistance and members of the Springer laboratory for helpful discussions. The research was supported in part by NIH grants HL048675 and CA080942 (MJE) and HL45100 (MDS). MJE is the recipient of a scholar award from the leukaemia and lymphoma society. DL was supported by an institutional training grant from the NCI (5T32CA09361). We thank Paul Manley (Novartis) for the kind gift of AFN941.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Supplemental Data include four figures and one table and can be found with this article online at www.cell.com
Accession Numbers
The atomic coordinates of autoinhibited FERM+kinase and the phosphorylated kinase domain of FAK have been deposited in the Protein Data Bank with the accession codes 2J0J and 2J0L respectively.
Supplementary Material
Figure S1. 2Fo-Fc electron density maps near the DFG motive of the activation loop 2Fo-Fc electron density maps countered at 1σ are shown for the autoinhibited (A) and active (B) FAK kinase. Note that past the DFG sequence the backbones of the inhibited and active structures follow very different paths.
Figure S2. Analysis of wt and mutant full-length FAK in 293 cells (A) Wild type FAK or FAK mutants were transiently expressed in HEK293 cells and adherent cells (cul) or cells incubated in suspension at 37°C for 1 hour (sus) were lysed. FAK was immunoprecipitated using BC4 and the immune complexes analyzed by Western blotting for phosphotyrosine using 4G10. (B) Wild type FAK or FAK mutants were transiently expressed in HEK293 cells. FAK was immunoprecipitated from lysates using BC4 and the immune complexes analyzed by Western blotting for phosphotyrosine using 4G10 (top panel). Equal amounts of FAK in each immune complex was verified by blotting for FAK (bottom panel). (C) In vitro kinase assay for paxillin phosphorylation in the presence or absence of Src inhibitor PP2. Wild type and mutant FAK proteins were immunoprecipitated from HEK293 lysates using BC4. The immune complexes were incubated in a kinase assay utilizing recombinant GST-paxillin-N-C3 as an exogenous substrate in the presence of 0.5 μM PP2 (+) or vehicle alone (DMSO) (−). Phosphorylation of paxillin was detected using the 4G10 phosphotyrosine antibody (two exposures shown in the middle panels). Equal amounts of substrate was verified by blotting for paxillin using a polyclonal antiserum (bottom panel). Equal recovery of FAK in the immune complexes was verified by blotting for FAK (top panel). (D) HEK293 cells were transiently transfected with Flag-FAK and GFP-Lyn constructs. The cells were lysed and exogenous GFP-Lyn was immunoprecipitated using a GFP monoclonal antibody (Roche) (top two panels). The immune complexes were blotted for FAK using monoclonal antibody 4.47 (top panel). Equal recovery of Lyn was verified by blotting for GFP (second panel). Exogenous FAK was immunoprecipitated using the Flag monoclonal antibody (bottom two panels) and the immune complexes blotted for GFP to detect GFP-Lyn (bottom panel). As a loading control the immune complexes were also blotted for FAK using monoclonal antibody 4.47 (third panel). (E) HEK293 cells transiently expressing wild type FAK or FAK mutants were lysed. FAK was immunoprecipitated using 2A7 (top two panels) and the immune complexes analyzed by Western blotting for paxillin (second panel). Equal amount of FAK in each immune complex was verified by blotting for FAK (top panel). Paxillin was directly immunoprecipitated from cell lysates using a commercially available paxillin antibody (bottom two panels). FAK was detected by blotting with monoclonal antibody 4.47 (third panel). Equal recovery of paxillin was validated by blotting for paxillin (bottom panel).
Figure S3. Localization of wt and mutant FAK in HeLa cells HeLa cells transiently expressing YFP (A), YFP-FAK (B) or YFP-FAK-Y180A/M183A (C) were plated on fibronectin and the localization of the proteins determined by fluorescent microscopy. Methods: The Y180A/M183A mutations were subcloned into YFP-FAK for localization studies. HeLa cells were transiently transfected with the YFP constructs. After 16 hours the cells were trypsinized and plated onto fibronectin coated coverslips for 3 hours at 37°C. Cells were imaged using a Zeiss Axiovert microscope with a 100× lens.
Figure S4. Autophosphorylation of Tyr 397 occurs in cis and trans. Phosphorylation reactions containing purified single chain wt FERM+kinase (middle band, labeled F-L-K for FERM-Linker-Kinase), GST-FERM+kinase K454R (upper band, labeled GST-F-L-Kd for GST-tagged FERM-linker-kinase dead) and FERM+linker (lower band, labeled F-L for FERM-Linker) were incubated for 8 minutes at room temperature (lanes 3−8). As controls, one reaction was not incubated (lane 1) and one was incubated without active wt FERM+kinase (lane 2). Tyr 397 phosphorylation was monitored by Western blotting using a specific anti-pY397 antibody (left panel). As loading control an equivalent gel was stained by Coomassie (right panel). Phosphorylation of the GST-F-L-Kd and F-L proteins must occur in trans, as these constructs lack catalytic activity. Phosphorylation of the active F-L-K protein may in principle occur in cis or trans. If phosphorylation occurred only in trans, one would expect equal phosphorylation of the active F-L-K and inactive GST-F-L-Kd species (as they are present in equimolar concentrations), and also equal competition by the excess F-L substrate. Instead, note i) that the GST-FERM+Kinase dead protein is phosphorylated at a lower level than the active FERM+kinase, and ii) that excess amounts of FERM+linker protein diminish phosphorylation of GST-FERM+kinase dead protein more than the active FERM+kinase. These results indicate that autophosphorylation in vitro occurs both in cis and trans.
Supplemental Table S1. Data collection and refinement statistics
REFERENCES
- Arold ST, Hoellerer MK, Noble ME. The structural basis of localization and signaling by the focal adhesion targeting domain. Structure (Camb) 2002;10:319–327. doi: 10.1016/s0969-2126(02)00717-7. [DOI] [PubMed] [Google Scholar]
- Bershadsky AD, Balaban NQ, Geiger B. Adhesion-dependent cell mechanosensitivity. Annu Rev Cell Dev Biol. 2003;19:677–695. doi: 10.1146/annurev.cellbio.19.111301.153011. [DOI] [PubMed] [Google Scholar]
- Boggon TJ, Eck MJ. Structure and regulation of Src family kinases. Oncogene. 2004;23:7918–7927. doi: 10.1038/sj.onc.1208081. [DOI] [PubMed] [Google Scholar]
- Boggon TJ, Li Y, Manley PW, Eck MJ. Crystal structure of the Jak3 kinase domain in complex with a staurosporine analog. Blood. 2005;106:996–1002. doi: 10.1182/blood-2005-02-0707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calalb MB, Polte TR, Hanks SK. Tyrosine phosphorylation of focal adhesion kinase at sites in the catalytic domain regulates kinase activity: a role for Src family kinases. Mol Cell Biol. 1995;15:954–963. doi: 10.1128/mcb.15.2.954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter N, Nakamoto T, Hirai H, Hunter T. EphrinA1-induced cytoskeletal re-organization requires FAK and p130(cas) Nat Cell Biol. 2002;4:565–573. doi: 10.1038/ncb823. [DOI] [PubMed] [Google Scholar]
- Ceccarelli DF, Song HK, Poy F, Schaller MD, Eck MJ. Crystal structure of the FERM domain of focal adhesion kinase. J Biol Chem. 2006;281:252–259. doi: 10.1074/jbc.M509188200. [DOI] [PubMed] [Google Scholar]
- Chen SY, Chen HC. Direct interaction of focal adhesion kinase (FAK) with Met is required for FAK to promote hepatocyte growth factor-induced cell invasion. Mol Cell Biol. 2006;26:5155–5167. doi: 10.1128/MCB.02186-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chenna R, Sugawara H, Koike T, Lopez R, Gibson TJ, Higgins DG, Thompson JD. Multiple sequence alignment with the Clustal series of programs. Nucleic Acids Res. 2003;31:3497–3500. doi: 10.1093/nar/gkg500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chishti AH, Kim AC, Marfatia SM, Lutchman M, Hanspal M, Jindal H, Liu SC, Low PS, Rouleau GA, Mohandas N, et al. The FERM domain: a unique module involved in the linkage of cytoplasmic proteins to the membrane. Trends Biochem Sci. 1998;23:281–282. doi: 10.1016/s0968-0004(98)01237-7. [DOI] [PubMed] [Google Scholar]
- Cohen LA, Guan JL. Mechanisms of focal adhesion kinase regulation. Curr Cancer Drug Targets. 2005a;5:629–643. doi: 10.2174/156800905774932798. [DOI] [PubMed] [Google Scholar]
- Cohen LA, Guan JL. Residues within the first subdomain of the FERM-like domain in focal adhesion kinase are important in its regulation. J Biol Chem. 2005b;280:8197–8207. doi: 10.1074/jbc.M412021200. [DOI] [PubMed] [Google Scholar]
- Cooper LA, Shen TL, Guan JL. Regulation of focal adhesion kinase by its amino-terminal domain through an autoinhibitory interaction. Mol Cell Biol. 2003;23:8030–8041. doi: 10.1128/MCB.23.22.8030-8041.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLano WL. The PyMOL Molecular Graphics System. 2002.
- Dunty JM, Gabarra-Niecko V, King ML, Ceccarelli DF, Eck MJ, Schaller MD. FERM domain interaction promotes FAK signaling. Mol Cell Biol. 2004;24:5353–5368. doi: 10.1128/MCB.24.12.5353-5368.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- Glaser F, Pupko T, Paz I, Bell RE, Bechor-Shental D, Martz E, Ben-Tal N. ConSurf: identification of functional regions in proteins by surface-mapping of phylogenetic information. Bioinformatics. 2003;19:163–164. doi: 10.1093/bioinformatics/19.1.163. [DOI] [PubMed] [Google Scholar]
- Guan JL, Shalloway D. Regulation of focal adhesion-associated protein tyrosine kinase by both cellular adhesion and oncogenic transformation. Nature. 1992;358:690–692. doi: 10.1038/358690a0. [DOI] [PubMed] [Google Scholar]
- Harrison SC. Variation on an Src-like theme. Cell. 2003;112:737–740. doi: 10.1016/s0092-8674(03)00196-x. [DOI] [PubMed] [Google Scholar]
- Hayashi I, Vuori K, Liddington RC. The focal adhesion targeting (FAT) region of focal adhesion kinase is a four-helix bundle that binds paxillin. Nat Struct Biol. 2002;9:101–106. doi: 10.1038/nsb755. [DOI] [PubMed] [Google Scholar]
- Hubbard SR. Crystal structure of the activated insulin receptor tyrosine kinase in complex with peptide substrate and ATP analog. Embo J. 1997;16:5572–5581. doi: 10.1093/emboj/16.18.5572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huse M, Kuriyan J. The conformational plasticity of protein kinases. Cell. 2002;109:275–282. doi: 10.1016/s0092-8674(02)00741-9. [DOI] [PubMed] [Google Scholar]
- Ilic D, Furuta Y, Kanazawa S, Takeda N, Sobue K, Nakatsuji N, Nomura S, Fujimoto J, Okada M, Yamamoto T. Reduced cell motility and enhanced focal adhesion contact formation in cells from FAK-deficient mice. Nature. 1995;377:539–544. doi: 10.1038/377539a0. [DOI] [PubMed] [Google Scholar]
- Ilic D, Kovacic B, McDonagh S, Jin F, Baumbusch C, Gardner DG, Damsky CH. Focal adhesion kinase is required for blood vessel morphogenesis. Circ Res. 2003;92:300–307. doi: 10.1161/01.res.0000055016.36679.23. [DOI] [PubMed] [Google Scholar]
- Jones TA, Zou J-Y, Cowan SW, Kjeldgaard M. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Cryst. 1991;A47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- LaFevre-Bernt M, Sicheri F, Pico A, Porter M, Kuriyan J, Miller WT. Intramolecular regulatory interactions in the Src family kinase Hck probed by mutagenesis of a conserved tryptophan residue. J Biol Chem. 1998;273:32129–32134. doi: 10.1074/jbc.273.48.32129. [DOI] [PubMed] [Google Scholar]
- Leu TH, Maa MC. Tyr-863 phosphorylation enhances focal adhesion kinase autophosphorylation at Tyr-397. Oncogene. 2002;21:6992–7000. doi: 10.1038/sj.onc.1205904. [DOI] [PubMed] [Google Scholar]
- Lyons PD, Dunty JM, Schaefer EM, Schaller MD. Inhibition of the catalytic activity of cell adhesion kinase beta by protein-tyrosine phosphatase-PEST-mediated dephosphorylation. J Biol Chem. 2001;276:24422–24431. doi: 10.1074/jbc.M011080200. [DOI] [PubMed] [Google Scholar]
- Matthews BW. Solvent content of protein crystals. J Mol Biol. 1968;33:491–497. doi: 10.1016/0022-2836(68)90205-2. [DOI] [PubMed] [Google Scholar]
- McLean GW, Carragher NO, Avizienyte E, Evans J, Brunton VG, Frame MC. The role of focal-adhesion kinase in cancer - a new therapeutic opportunity. Nat Rev Cancer. 2005;5:505–515. doi: 10.1038/nrc1647. [DOI] [PubMed] [Google Scholar]
- Miao H, Burnett E, Kinch M, Simon E, Wang B. Activation of EphA2 kinase suppresses integrin function and causes focal-adhesion-kinase dephosphorylation. Nat Cell Biol. 2000;2:62–69. doi: 10.1038/35000008. [DOI] [PubMed] [Google Scholar]
- Mitra SK, Hanson DA, Schlaepfer DD. Focal adhesion kinase: in command and control of cell motility. Nat Rev Mol Cell Biol. 2005;6:56–68. doi: 10.1038/nrm1549. [DOI] [PubMed] [Google Scholar]
- Moarefi I, LaFevre-Bernt M, Sicheri F, Huse M, Lee CH, Kuriyan J, Miller WT. Activation of the Src-family tyrosine kinase Hck by SH3 domain displacement. Nature. 1997;385:650–653. doi: 10.1038/385650a0. [DOI] [PubMed] [Google Scholar]
- Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- Nagar B, Hantschel O, Young MA, Scheffzek K, Veach D, Bornmann W, Clarkson B, Superti-Furga G, Kuriyan J. Structural basis for the autoinhibition of c-Abl tyrosine kinase. Cell. 2003;112:859–871. doi: 10.1016/s0092-8674(03)00194-6. [DOI] [PubMed] [Google Scholar]
- Nikolopoulos SN, Giancotti FG. Netrin-integrin signaling in epithelial morphogenesis, axon guidance and vascular patterning. Cell Cycle. 2005;4:e131–135. [PubMed] [Google Scholar]
- Nowakowski J, Cronin CN, McRee DE, Knuth MW, Nelson CG, Pavletich NP, Rogers J, Sang BC, Scheibe DN, Swanson RV, Thompson DA. Structures of the cancer-related Aurora-A, FAK, and EphA2 protein kinases from nanovolume crystallography. Structure (Camb) 2002;10:1659–1667. doi: 10.1016/s0969-2126(02)00907-3. [DOI] [PubMed] [Google Scholar]
- Otwinowski Z, Minor W. Processing X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- Parsons JT. Focal adhesion kinase: the first ten years. J Cell Sci. 2003;116:1409–1416. doi: 10.1242/jcs.00373. [DOI] [PubMed] [Google Scholar]
- Pearson MA, Reczek D, Bretscher A, Karplus PA. Structure of the ERM protein moesin reveals the FERM domain fold masked by an extended actin binding tail domain. Cell. 2000;101:259–270. doi: 10.1016/s0092-8674(00)80836-3. [DOI] [PubMed] [Google Scholar]
- Reynolds AB, Roesel DJ, Kanner SB, Parsons JT. Transformation-specific tyrosine phosphorylation of a novel cellular protein in chicken cells expressing oncogenic variants of the avian cellular src gene. Mol Cell Biol. 1989;9:629–638. doi: 10.1128/mcb.9.2.629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridley AJ, Schwartz MA, Burridge K, Firtel RA, Ginsberg MH, Borisy G, Parsons JT, Horwitz AR. Cell migration: integrating signals from front to back. Science. 2003;302:1704–1709. doi: 10.1126/science.1092053. [DOI] [PubMed] [Google Scholar]
- Schaller MD, Borgman CA, Cobb BS, Vines RR, Reynolds AB, Parsons JT. pp125FAK a structurally distinctive protein-tyrosine kinase associated with focal adhesions. Proc Natl Acad Sci U S A. 1992;89:5192–5196. doi: 10.1073/pnas.89.11.5192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaller MD, Hildebrand JD, Shannon JD, Fox JW, Vines RR, Parsons JT. Autophosphorylation of the focal adhesion kinase, pp125FAK, directs SH2-dependent binding of pp60src. Mol Cell Biol. 1994;14:1680–1688. doi: 10.1128/mcb.14.3.1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaller MD, Parsons JT. pp125FAK-dependent tyrosine phosphorylation of paxillin creates a high-affinity binding site for Crk. Mol Cell Biol. 1995;15:2635–2645. doi: 10.1128/mcb.15.5.2635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schock F, Perrimon N. Molecular mechanisms of epithelial morphogenesis. Annu Rev Cell Dev Biol. 2002;18:463–493. doi: 10.1146/annurev.cellbio.18.022602.131838. [DOI] [PubMed] [Google Scholar]
- Sicheri F, Kuriyan J. Structures of Src-family tyrosine kinases. Curr Opin Struct Biol. 1997;7:777–785. doi: 10.1016/s0959-440x(97)80146-7. [DOI] [PubMed] [Google Scholar]
- Sieg DJ, Hauck CR, Ilic D, Klingbeil CK, Schaefer E, Damsky CH, Schlaepfer DD. FAK integrates growth-factor and integrin signals to promote cell migration. Nat Cell Biol. 2000;2:249–256. doi: 10.1038/35010517. [DOI] [PubMed] [Google Scholar]
- Siegel LM, Monty KJ. Determination of molecular weights and frictional ratios of proteins in impure systems by use of gel filtration and density gradient centrifugation. Application to crude preparations of sulfite and hydroxylamine reductases. Biochim Biophys Acta. 1966;112:346–362. doi: 10.1016/0926-6585(66)90333-5. [DOI] [PubMed] [Google Scholar]
- Storoni LC, McCoy AJ, Read RJ. Likelihood-enhanced fast rotation functions. Acta Crystallogr D Biol Crystallogr. 2004;60:432–438. doi: 10.1107/S0907444903028956. [DOI] [PubMed] [Google Scholar]
- Tachibana K, Urano T, Fujita H, Ohashi Y, Kamiguchi K, Iwata S, Hirai H, Morimoto C. Tyrosine phosphorylation of Crk-associated substrates by focal adhesion kinase. A putative mechanism for the integrin-mediated tyrosine phosphorylation of Crk-associated substrates. J Biol Chem. 1997;272:29083–29090. doi: 10.1074/jbc.272.46.29083. [DOI] [PubMed] [Google Scholar]
- Thomas JW, Cooley MA, Broome JM, Salgia R, Griffin JD, Lombardo CR, Schaller MD. The role of focal adhesion kinase binding in the regulation of tyrosine phosphorylation of paxillin. J Biol Chem. 1999;274:36684–36692. doi: 10.1074/jbc.274.51.36684. [DOI] [PubMed] [Google Scholar]
- Toutant M, Costa A, Studler JM, Kadare G, Carnaud M, Girault JA. Alternative splicing controls the mechanisms of FAK autophosphorylation. Mol Cell Biol. 2002;22:7731–7743. doi: 10.1128/MCB.22.22.7731-7743.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu W, Harrison SC, Eck MJ. Three-dimensional structure of the tyrosine kinase c-Src. Nature. 1997;385:595–602. doi: 10.1038/385595a0. [DOI] [PubMed] [Google Scholar]
- Yamaguchi H, Hendrickson WA. Structural basis for activation of human lymphocyte kinase Lck upon tyrosine phosphorylation. Nature. 1996;384:484–489. doi: 10.1038/384484a0. [DOI] [PubMed] [Google Scholar]
- Zhu X, Kim JL, Newcomb JR, Rose PE, Stover DR, Toledo LM, Zhao H, Morgenstern KA. Structural analysis of the lymphocyte-specific kinase Lck in complex with non-selective and Src family selective kinase inhibitors. Structure. 1999;7:651–661. doi: 10.1016/s0969-2126(99)80086-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. 2Fo-Fc electron density maps near the DFG motive of the activation loop 2Fo-Fc electron density maps countered at 1σ are shown for the autoinhibited (A) and active (B) FAK kinase. Note that past the DFG sequence the backbones of the inhibited and active structures follow very different paths.
Figure S2. Analysis of wt and mutant full-length FAK in 293 cells (A) Wild type FAK or FAK mutants were transiently expressed in HEK293 cells and adherent cells (cul) or cells incubated in suspension at 37°C for 1 hour (sus) were lysed. FAK was immunoprecipitated using BC4 and the immune complexes analyzed by Western blotting for phosphotyrosine using 4G10. (B) Wild type FAK or FAK mutants were transiently expressed in HEK293 cells. FAK was immunoprecipitated from lysates using BC4 and the immune complexes analyzed by Western blotting for phosphotyrosine using 4G10 (top panel). Equal amounts of FAK in each immune complex was verified by blotting for FAK (bottom panel). (C) In vitro kinase assay for paxillin phosphorylation in the presence or absence of Src inhibitor PP2. Wild type and mutant FAK proteins were immunoprecipitated from HEK293 lysates using BC4. The immune complexes were incubated in a kinase assay utilizing recombinant GST-paxillin-N-C3 as an exogenous substrate in the presence of 0.5 μM PP2 (+) or vehicle alone (DMSO) (−). Phosphorylation of paxillin was detected using the 4G10 phosphotyrosine antibody (two exposures shown in the middle panels). Equal amounts of substrate was verified by blotting for paxillin using a polyclonal antiserum (bottom panel). Equal recovery of FAK in the immune complexes was verified by blotting for FAK (top panel). (D) HEK293 cells were transiently transfected with Flag-FAK and GFP-Lyn constructs. The cells were lysed and exogenous GFP-Lyn was immunoprecipitated using a GFP monoclonal antibody (Roche) (top two panels). The immune complexes were blotted for FAK using monoclonal antibody 4.47 (top panel). Equal recovery of Lyn was verified by blotting for GFP (second panel). Exogenous FAK was immunoprecipitated using the Flag monoclonal antibody (bottom two panels) and the immune complexes blotted for GFP to detect GFP-Lyn (bottom panel). As a loading control the immune complexes were also blotted for FAK using monoclonal antibody 4.47 (third panel). (E) HEK293 cells transiently expressing wild type FAK or FAK mutants were lysed. FAK was immunoprecipitated using 2A7 (top two panels) and the immune complexes analyzed by Western blotting for paxillin (second panel). Equal amount of FAK in each immune complex was verified by blotting for FAK (top panel). Paxillin was directly immunoprecipitated from cell lysates using a commercially available paxillin antibody (bottom two panels). FAK was detected by blotting with monoclonal antibody 4.47 (third panel). Equal recovery of paxillin was validated by blotting for paxillin (bottom panel).
Figure S3. Localization of wt and mutant FAK in HeLa cells HeLa cells transiently expressing YFP (A), YFP-FAK (B) or YFP-FAK-Y180A/M183A (C) were plated on fibronectin and the localization of the proteins determined by fluorescent microscopy. Methods: The Y180A/M183A mutations were subcloned into YFP-FAK for localization studies. HeLa cells were transiently transfected with the YFP constructs. After 16 hours the cells were trypsinized and plated onto fibronectin coated coverslips for 3 hours at 37°C. Cells were imaged using a Zeiss Axiovert microscope with a 100× lens.
Figure S4. Autophosphorylation of Tyr 397 occurs in cis and trans. Phosphorylation reactions containing purified single chain wt FERM+kinase (middle band, labeled F-L-K for FERM-Linker-Kinase), GST-FERM+kinase K454R (upper band, labeled GST-F-L-Kd for GST-tagged FERM-linker-kinase dead) and FERM+linker (lower band, labeled F-L for FERM-Linker) were incubated for 8 minutes at room temperature (lanes 3−8). As controls, one reaction was not incubated (lane 1) and one was incubated without active wt FERM+kinase (lane 2). Tyr 397 phosphorylation was monitored by Western blotting using a specific anti-pY397 antibody (left panel). As loading control an equivalent gel was stained by Coomassie (right panel). Phosphorylation of the GST-F-L-Kd and F-L proteins must occur in trans, as these constructs lack catalytic activity. Phosphorylation of the active F-L-K protein may in principle occur in cis or trans. If phosphorylation occurred only in trans, one would expect equal phosphorylation of the active F-L-K and inactive GST-F-L-Kd species (as they are present in equimolar concentrations), and also equal competition by the excess F-L substrate. Instead, note i) that the GST-FERM+Kinase dead protein is phosphorylated at a lower level than the active FERM+kinase, and ii) that excess amounts of FERM+linker protein diminish phosphorylation of GST-FERM+kinase dead protein more than the active FERM+kinase. These results indicate that autophosphorylation in vitro occurs both in cis and trans.
Supplemental Table S1. Data collection and refinement statistics