

Abstract
Imaging agents capable of assessing amyloid-beta (Aβ) content in vivo in the brains of Alzheimer’s disease (AD) subjects likely will be important as diagnostic agents to detect Aβ plaques in the brain, to help test the amyloid cascade hypothesis of AD, and as an aid to assess the efficacy of anti-amyloid therapeutics currently under development and in clinical trials. Positron emission tomography (PET) imaging studies of amyloid deposition in human subjects with several Aβ imaging agents are currently underway. We reported the first PET studies of the carbon-11-labeled thioflavin-T derivative Pittsburgh Compound B ([11C]PiB) in 2004, and this work has subsequently been extended to include a variety of subject groups including AD, mild cognitive impairment (MCI), and healthy controls. The ability to quantify regional Aβ plaque load in the brains of living human subjects has provided a means to begin to apply this technology as a diagnostic agent to detect regional concentrations of Aβ plaques and as a surrogate marker of therapeutic efficacy in anti-amyloid drug trials.
Keywords: Amyloid imaging, amyloid-beta, Aβ, PiB, Alzheimer’s disease, anti-amyloid therapy
Introduction
Alzheimer’s disease (AD) is the most prevalent cause of dementia, accounting for 60–70% of all dementia cases, and there were about 4.5 million people afflicted with AD in the United States in 2000 [1]. The most significant risk factor for developing AD is age, with about 30% prevalence by 85 years [2], and increased longevity is expected raise the number of cases in the US to more than 13 million by 2050 [1]. These numbers highlight the need for more effective therapeutic interventions for AD beyond presently used drugs. Currently used drugs, such as cholinesterase inhibitors and NMDA receptor antagonists, treat the symptoms of AD, but do not halt or reverse the pathophysiological causes of AD. An imaging agent that could provide direct evidence of the delay or reversal of the root cause(s) of AD could hasten drug development and intervention efforts and assist in delivering therapies that are truly effective in delaying the onset or modifying the progression of AD.
AD is characterized histopathologically by the presence and abundance of two different abnormal aggregated proteins, amyloid plaques and neurofibrillary tangles, in brain tissue (Fig 1) [3]. Amyloid plaques are predominantly comprised of insoluble amyloid-beta (Aβ) peptides, mostly 40 or 42 amino acids in length, with Aβ42 being the most prevalent component [4]. Neurofibrillary tangles (NFTs) are composed mainly of hyperphosphorylated forms of the microtubule-associated protein, tau [5]. Most investigators referring to “amyloid” in AD equate the term to Aβ, but amyloid is a more general term and refers to many types of beta-pleated sheet conformation proteins found both systemically and in the central nervous system [6,7]. A recent report by the Nomenclature Committee of the International Society of Amyloidosis defined amyloid as “extracellular depositions of protein fibrils with characteristic appearance in electron microscope, typical X-ray diffraction pattern, and affinity for Congo red with concomitant green birefringence” [8]. By this definition, Aβ plaques, as well as prion proteins associated with spongioform encephalopathies (e.g., mad cow disease and scrapies), are amyloid proteins, but NFTs and Lewy bodies (comprised mainly of α-synuclein protein) are not amyloids. Instead, they are defined as “close relatives” of amyloid because of their predominantly intracellular or intraneuronal locations. In AD, the Aβ component far outweighs the other amyloid-related protein components, such as NFTs, on a total mass basis in most brain regions [9]. This presents more binding sites (higher Bmax) for aggregated Aβ binding relative to NFT binding sites for ligands with high binding affinities for these protein deposits. It is interesting and important that some thioflavin-T derivatives, such as Pittsburgh Compound B (PiB) (2-(4′-methylaminophenyl)-6-hydroxybenzothiazole) (Fig 2), bind fibrillar Aβ deposits, with no detectable binding to soluble Aβ forms nor to NFTs or Lewy bodies [10] under conditions relevant to positron emission tomography (PET) studies (at ligand concentrations ~1 nM).
Figure 1.
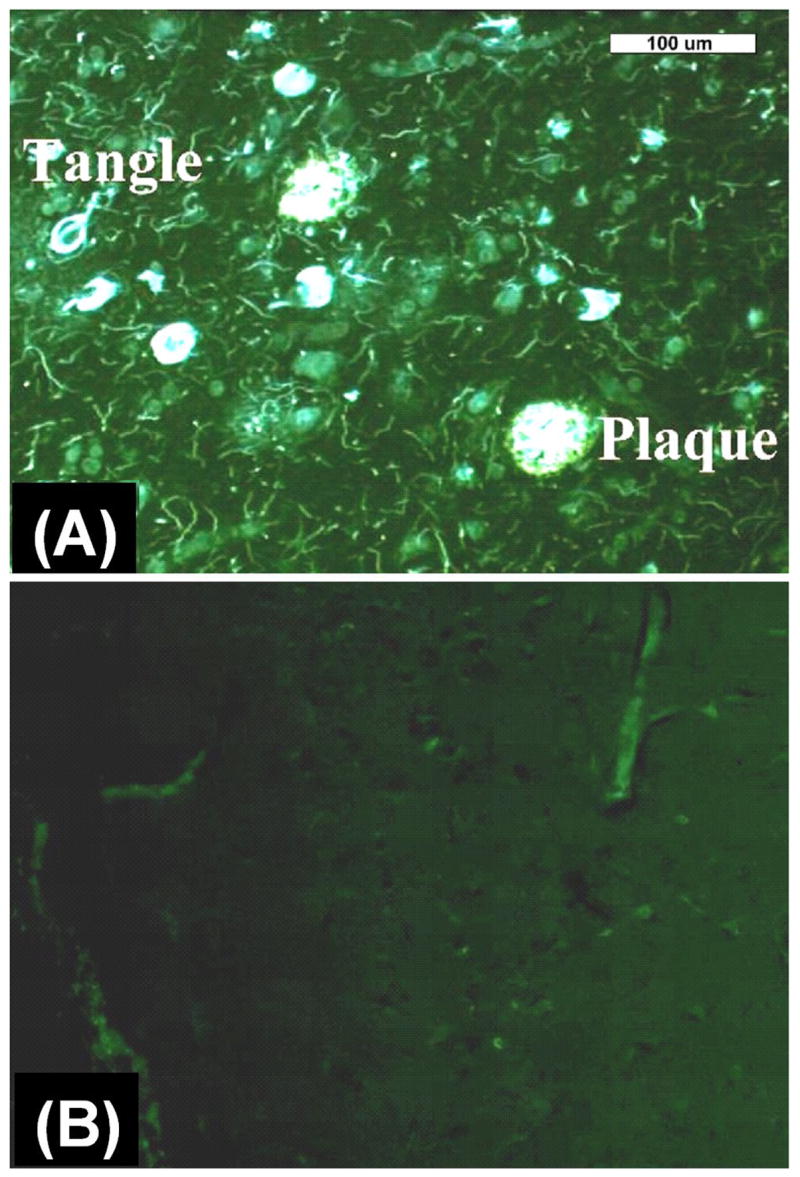
(A) Post mortem fluorescent microscopy image of an 8 μm thick section from AD brain frontal cortex stained with X-34 showing amyloid plaques and neurofibrillary tangles; (B) Post mortem fluorescent microscopy image of an 8 μm thick section from an aged, non-demented control brain stained with X-34 showing lack of pathology.
Figure 2.

Structures of thioflavin-T and a neutral, lipophilic derivative of thioflavin-T, PiB. The site of radiolabelling with 11C is indicated with an asterisk (*).
Potential Uses of an Aβ Imaging Agent
Several potential uses of Aβ radioligands labeled with positron or single photon emitting radionuclides are evident: the first is their use as imaging agents to diagnose AD. While Aβ plaques are one of the characteristic features of AD confirmed by post mortem evaluation (Fig 1), Aβ deposition in the brain is not unique to clinically apparent AD and also has been found in normal aging, prompting the suggestion that there is a presymptomatic stage of AD [11,12]. Although the time course of Aβ deposition in normal subjects who go on to develop AD has not been fully elucidated, evidence gained through post mortem study of Down syndrome (a condition in which Aβ deposition is always present by age 40 and dementia is very common) suggests that Aβ deposition begins over a decade prior to the clinical symptoms of dementia [13]. Aβ deposition is believed to begin in normal elderly subjects, who subsequently may develop signs of mild cognitive impairment (MCI), and then may finally develop AD (Figs 3 and 4), in whom post mortem analysis demonstrates the characteristic abundance of Aβ plaques in specific brain areas. Recent PET studies using [11C]PiB in elderly normal control subjects support the existence of a preclinical AD stage in which Aβ plaques are found in discrete brain regions by demonstrating the presence of significant radioligand retention, approaching levels seen in AD subjects, in about 10% of the elderly control subjects [14]. In addition, Aβ plaques are often found in Parkinson’s disease subjects with co-morbid dementia and in diffuse Lewy body (DLB) disease patients [15]. Thus, significant Aβ deposits may be found in subjects who do not have clinically apparent dementia, or who have dementia symptoms that suggest a clinical diagnosis other than AD. This has lead Klunk and coworkers to suggest that Aβ imaging agents might most definitively be used to demonstrate the presence of cerebral beta-amyloidosis, rather than as highly specific diagnostic markers of clinical AD [16]. This does not mean that Aβ imaging agents will not be useful in assisting with the diagnosis of AD, but rather that the presence of cerebral Aβ deposits are not unique or specific to clinically apparent AD.
Figure 3.

PET images in transaxial and sagittal planes of parametric Logan distribution volume ratios (DVR, relative to cerebellum) of [11C]PIB (370–555 MBq, 90 min scans) in a normal control subject (NC), [11C]PIB-positive normal control (NC+), [11C]PIB-negative mild cognitive impairment subject (MCI-), [11C]PIB-positive MCI subject (MCI+), highly [11C]PIB-positive MCI subject (MCI++), and a [11C]PIB-positive Alzheimer’s disease (AD) subject. The small amount of signal seen in the NC- and MCI- scans in white matter areas (including brain stem) reflects slowly clearing, non-specifically bound tracer. The low level signal in the NC+ subject indicates specifically bound tracer in frontal cortex, a brain region often among the first to show [11C]PIB binding in cognitively normal control subjects. The three MCI subjects show highly variable amounts of [11C]PIB binding, with the MCI- subject nearly identical to the NC- subject and the MCI++ nearly identical to the AD subject.
Figure 4.

Schematic of the hypothetical progression of Aβ deposition over time from the early initiation (ei) phase, to the continuously progressive (p) phase, and finally the late equilibrium (eq) phase. Subjects may experience either a long (p1/t1) or brief (p2/t2) progressive phase of Aβ deposition, which snapshots such as shown in Figure 3 can capture at a single point in time. Cognitive symptoms may not be evident until the equilibrium (eq) phase (MCI+, MCI++, and AD in Figure 3), but the cascade of pathological events that may lead to these symptoms (i.e. neurofibrillary pathology, synapse and neuron loss) may be initiated during the progressive phase (p) (figure from [94])
It is known that Aβ plaques occur earliest in neocortex in cognitively normal subjects where they are relatively evenly distributed [17]. Neurofibrillary tangles appear first in limbic areas such as the transentorhinal cortex and progress in a predictable pattern of regional distribution to the neocortex [18]. Arnold and coworkers mapped the distribution of NFTs and neuritic plaques (Aβ plaques surrounded by dystrophic neurites) in the brains of patients with AD [19]. Compared to NFTs, neuritic plaques were, in general, more evenly distributed throughout the cortex, with the exceptions of notably fewer neuritic plaques in limbic periallocortex and allocortex (the areas with greatest NFT density such as the hippocampus). This is supported by the findings of Price and Morris who found little Aβ pathology in the hippocampus of elderly controls or very mild AD patients [12]. Thus, while limbic areas have early and severe NFT pathology, the mesial temporal lobe has relatively little neuritic plaque pathology early in the course of AD. The cerebellum is notably free of neuritic plaques in AD (Fig 3), although diffuse Aβ deposits which do not label with fibrillar dyes, such as Congo red and PiB, are commonly observed [10,20,21]. Thus from an AD diagnostic perspective, one would look for the presence of Aβ deposits or the binding of an Aβ imaging agent in specific brain regions, and the absence of binding in other regions. However, the presence of significant Aβ deposits in specific brain regions does not necessarily indicate the presence or severity of clinical AD, as some control and MCI subjects contain concentrations of Aβ plaques in their brains as high as contained in the brains of AD subjects (Fig 5) [14,22,23]. The coincidence of high Aβ loads and the absence or lack of extensive cognitive deficits in some subjects could be interpreted as high resistance to Aβ damage (i.e., a high cognitive reserve) or could indicate that excessive Aβ has not been present for a sufficiently long time in some subjects to cause as much damage as in AD subjects. Comprehensive longitudinal studies using Aβ imaging agents likely will help sort out these possibilities that single time point cross-sectional studies are not able to distinguish.
Figure 5.

Distribution volume ratio (DVR) outcome measures of [11C]PiB for individual control (n=8), MCI (n=10), and AD (n=6) subjects for posterior cingulate gyrus (PCG) and frontal cortex (FRC) regions of interest. The regional DVR outcome measures were determined using the non-invasive Logan graphical analysis method with cerebellum as the reference region over 90 min post-injection. The numbered circles represent the individual subjects, and subjects with overlapping values are placed adjacent to one another. Note that the MCI group displays DVR values ranging from control-like (<1.4) to AD-like (>2.0) with a few MCI subjects displaying intermediate DVR values (between 1.5 and 2.0). Adapted from [22].
A second and closely related potential use of Aβ imaging agents is to help test the “amyloid cascade hypothesis” of AD [24,25,26]. A growing consensus points to the overabundance of extracellular Aβ protein in the human brain as the central event in the pathogenesis of AD [27]. The amyloid cascade hypothesis holds that the overproduction of Aβ, or the failure to clear this protein from the brain, leads to AD (Fig 6). Proponents of this hypothesis posit that high levels of Aβ subsequently lead to a series of downstream neuropathological events, including the production of extensive intracellular NFT deposits, inflammation, oxidative damage, excitotoxicity, loss of synaptic connections, neuronal cell death, and, eventually, clinical symptoms such as memory impairment and AD [28]. The single, most important piece of evidence supporting the amyloid cascade hypothesis of AD is the demonstration that many different mutations in the Aβ precursor protein (APP) gene on chromosome 21, all lying in or near the Aβ peptide region, cause early-onset or familial forms of AD [28,29,30]. Further genetic support for the amyloid cascade hypothesis comes from the finding that the most common form of early-onset, autosomal dominant familial AD – the chromosome 14 mutations – is caused by mutations in the presenilin-1 (PS1) gene, which codes for a protein that is strongly implicated to be an essential component of the “γ-secretase” enzyme complex responsible for C-terminal cleavage of Aβ from its precursor, APP (Fig 6) [31].
Figure 6.

Schematic of the production of Aβ (1–40) and Aβ (1–42) peptides from amyloid precursor protein (APP) via sequential β-and γ-secretase cleavages. The Aβ (1–40) and Aβ (1–42) monomers are believed to aggregate in the excellular space to form soluble Aβ species (oligomers). Some Aβ oligomers are believed to form proto-fibrils, which then form slightly soluble Aβ fibrils and plaques. Oligomers and Aβ-containing fibrils and plaques are believed to set in motion subsequent damaging processes, such as tau hyperphosphorylation and neurofibrillary tangle formation, generation of excitotoxic species, oxidative damage, neuroinflammation, enlargement of axons and dendrites with deposits of hyperphosphorylated tau filaments (dystrophic neurites), loss of synaptic junctions, and neuronal cell death.
Studies in triple transgenic mice overexpressing both APP and tau have provided insights into the mechanistic relationship between Aβ and tau pathology in AD. These so-called 3xTg-AD mice bear both the Swedish (KM670/671NL) APP mutation and the PS1 (M146V) mutation leading to overproduction and deposition of Aβ as well as the human four-repeat tau (P301L) mutation leading to overproduction and deposition of tau [32,33]. Billings and coworkers [34] have summarized the neuropathological aspects of these mice that are reminiscent of AD: (1) Aβ plaques and neurofibrillary pathology develop in a hierarchical manner in AD-relevant brain regions, mainly the hippocampus, cortex, and amygdala; (2) Aβ plaque pathology precedes tangle formation, and plaques consist of the longer, more amyloidogenic Aβ42; (3) the pattern of conformational and phosphorylation changes that the tau protein undergoes parallels the sequence in the human AD brain; and (4) the 3xTg-AD mice show selective loss of nicotinic α7 receptors in the hippocampus and cortex. It also has been shown that both Aβ immunotherapy and γ-secretase treatment lead to clearance not only of Aβ, but also early-stage tau lesions [35]. Importantly, Aβ immunotherapy did not reverse late, well-established tau aggregation, pointing out the importance of preventing this pathology very early before synaptic and neuronal losses have occurred (see below). These and other studies [36,37] suggest that, at least in the mouse model, Aβ deposition precedes and exacerbates NFT formation.
While there is considerable agreement that Aβ plays a key causative role in at least early-onset, autosomal dominant forms of familial AD, there is disagreement about the molecular and cellular mechanisms through which Aβ exerts its pathophysiological effects. Butterfield and Bush have reviewed the evidence that factors such as oxygen, the single methionine-35 residue of Aβ42, and redox metal ions (Zn+2 and Cu+2) are important for the oxidative stress and neurotoxic properties of Aβ (Butterfield 2004). Others have pointed out that the assembly of Aβ42 into toxic species is intrinsic to the primary structure of Aβ42 and does not require chemical modification of the peptide or the invocation of peptide-associated enzymatic activity [38]. This latter discussion has focused increasing attention on the important, and perhaps unique, role of small, soluble, oligomeric assemblies of Aβ42 in the cascade of synaptic dysfunction and neurotoxicity (Fig 6). That is, it may be the soluble Aβ42 oligomers, more than insoluble fibrillar deposits, that most contribute to the neurotoxic effects of high extracellular concentrations of Aβ. Oligomeric Aβ42 has been shown to be 10- to 100-fold more toxic than fibrillar and monomeric Aβ42 when incubated with Neuro-2A cells [38,39]. However, at 10 μM concentrations, all “starting” aggregation states of Aβ42 (i.e., monomeric, oligomeric, and fibrillar) produce equivalent toxicity suggesting either that aggregation state-dependent toxicity is a dose-related and not an absolute phenomenon or, more likely, that at higher concentrations an equilibrium is established under physiologic conditions that produces a spectrum of toxic species independent of the “starting” aggregation state.
Regardless of the equilibrium state under any given set of physiological conditions, all of the protein forms (monomer, soluble oligomer, and fibril) are separated by energy barriers that can be crossed in both directions. Studies of immunotherapies in transgenic mice [40,41] and autopsy studies of humans treated with active immunization in the AN-1792 trial [42,43,44], strongly suggest that Aβ in fibrillar deposits can be mobilized and cleared. The equilibrium-based nature of this clearance is perhaps best exemplified by the observation that immunization of Aβ-depositing transgenic mice in a manner that produces antibodies specific for oligomeric Aβ led to marked reduction of not only oligomeric forms of Aβ, but also resulted in clearance of thioflavin-S positive insoluble plaque forms of Aβ as well [45,46]. This most likely occurred by shifting the equilibrium away from the fibrillar species toward the oligomeric forms that could be cleared by the antibodies.
One should also consider that insoluble Aβ exceeds soluble forms of Aβ by a factor of about 100-fold in AD brain [47], so even a 100-fold greater toxic potency of the oligomeric form leaves questions about the relative toxicities of various aggregation states in vivo. Kuo and coworkers reported that the soluble pool of Aβ displays a continuous distribution of monomeric and oligomeric Aβ [47]. Centricon membrane molecular weight fractionation of the soluble pool showed that approximately 75% of the soluble Aβ pool in human brain was oligomeric. It also is notable that Kuo found the parenchymal levels of soluble oligomeric Aβ to be significantly higher (~50-fold) than CSF Aβ levels (and higher still than blood levels), suggesting that there is some barrier to free diffusion from the brain parenchyma to CSF (and blood). In summary, the evidence that oligomeric Aβ42 is the most toxic species of Aβ is convincing. Studies strongly support the following: 1) all else being equal, enriched oligomeric preparations of Aβ42 appear to be the most toxic species [38,39]; 2) oligomeric species of Aβ42 can inhibit long term potentiation (LTP) and lead to synaptic dysfunction [48,49]; 3) memory loss occurs prior to extensive deposition of fibrillar Aβ42 in transgenic mice and can be reversed by targeting soluble species of Aβ42 [50,51,52,53]; and 4) oligomeric Aβ42 species exist in AD brain [54]. However, it is important to appreciate that the removal or reduction of the soluble, toxic oligomeric forms of Aβ42 from brain parenchyma will require the concomitant reduction and removal of the insoluble (fibrillar) Aβ42 pool as well. Chemists can readily appreciate the analogous situation in the case of a mass of a slightly soluble precipitate at the bottom of a beaker covered by a saturated aqueous solution of the precipitated compound. One can remove the saturated solution and replace it with a fresh supply of water, but the water solution above the precipitate will become saturated again in short order (when equilibrium is reached). Only when the precipitate is completely removed, or dissolved, will the aqueous solution be able to contain lower quantities of the precipitate than contained in a saturated solution above the precipitate. Likewise if the concentration of the solute exceeds its solubility limit, new precipitate will form. The relationship between soluble and insoluble forms of Aβ in the extracellular space of parenchymal tissues may not be this direct, but likely is analogous; the permanent lowering of soluble Aβ in the extracellular solution will depend upon a proportional lowering of insoluble (or more correctly, slightly soluble) forms of Aβ (i.e., Aβ plaques) as well. These Aβ deposits provide for the storage of the vast majority of Aβ in AD brain (>99%), and only when they are removed will the concentration of the more toxic, soluble oligomeric forms of Aβ be permanently reduced. Therefore an implication of the amyloid cascade hypothesis is that an anti-amyloid therapy directed at removing insoluble Aβ stores will likely be effective in reducing soluble Aβ levels as well.
The third potential use of Aβ imaging agents would be to assist with the development of AD therapeutic agents, specifically with drugs aimed at halting or reversing the increased concentration of toxic soluble Aβ oligomers in brain and their subsequent deposition and storage in the forms of Aβ fibrils and plaques. Aβ imaging agents could be used to identify subjects who would benefit from anti-Aβ therapies, i.e., subjects with Aβ deposits in their brains, as well as to assess the efficacy of various treatment approaches in halting or reversing Aβ deposition in these subjects. It is not surprising that the metabolism of Aβ has become an important therapeutic target in AD research. A corollary of the amyloid cascade hypothesis is that prevention of Aβ accumulation in oligomers or plaques should prevent AD. Approaches to “anti-amyloid” therapy have focused on decreasing synthesis, increasing clearance, or decreasing the aggregation/toxicity of Aβ.
Because soluble forms of Aβ in AD brain comprise ~1% of the total Aβ (on a molar basis) [47] imaging agents targeting soluble Aβ will have a much lower target density (Bmax) than agents targeting Aβ plaques. In addition, the development of imaging agents specific for soluble, oligomeric Aβ binding sites relative to insoluble Aβ binding sites could prove to be problematic, not only because of the much higher density of insoluble Aβ binding sites in AD brain, but also because of the likely difficulty of developing a small molecule imaging agent specific for binding only soluble Aβ oligomers in the presence of high concentrations of fibrillar, insoluble Aβ deposits. Fortunately, PiB (Fig 2) has been shown to bind specifically to Aβ40 and Aβ42 synthetic fibrils and insoluble Aβ plaques containing Aβ42 and Aβ40 found in AD brain. In contrast, PiB does not bind appreciably to soluble Aβ and probably does not bind to oligomeric forms of Aβ until they reach some critical size (yet to be determined). PiB binding requires an extended β-pleated sheet structure found in Aβ fibrils and plaques in order to bind with high affinity. Hence if one wishes to follow the amount of insoluble Aβ in AD brain tissue, a radiotracer such as [11C]PiB with a high selectivity for insoluble Aβ likely would prove useful in vivo in human brain [16,22].
Studies in transgenic mice that overexpress mutant human APP have been interpreted to suggest that small soluble forms of Aβ are a critical toxic species. In Tg2576 mice, no obvious correspondence between memory and insoluble Aβ levels was apparent [53]. Furthermore, passive immunization studies of this same Tg2576 strain of mice showed that memory deficits and disruption of LTP can be reversed without affecting the levels of soluble or insoluble Aβ, suggesting “neutralization” of soluble Aβ by antibodies may be sufficient to aid in cognitive improvement [51,52]. These results were interpreted to imply that insoluble Aβ is a surrogate marker for “small assemblies” of Aβ that disrupt cognition and occur as intermediates during the formation of insoluble Aβ. This finding was extended to the PDAPP strain of mice by Dodart and colleagues [50]. This group previously reported that chronic treatment of PDAPP mice with the m266 anti-Aβ antibody (every 2 weeks from 4 to 9 months of age) reduced Aβ burden at least in part by increasing peripheral clearance [55]. In the subsequent study in 24 month-old PDAPP mice [50], they found that a “subchronic” six-week course of m266 immunotherapy reversed the cognitive deficits measured, but did not decrease the Aβ immunohistochemical burden (brain Aβ levels were not determined by ELISA). More surprisingly, they found that the cognitive deficits measured in 11 month-old PDAPP mice could be reversed within days of a single dose of the m266 anti-Aβ antibody. Dodart and coworkers did not determine if this acute improvement was transient or long-lasting. This experiment was subsequently performed by Billings et al. in the 3xTg-AD mouse [34]. At four months of age, intraneuronal soluble and insoluble Aβ accumulation appeared to lead to early cognitive deficits prior to extracellular plaque and tangle deposition in the 3xTg-AD mouse. Intracerebroventricular injection of an anti-Aβ antibody into 4 month-old 3xTg-AD mice cleared the intraneuronal Aβ and reversed the early cognitive deficit when tested 1 week later. However, 1 month after this single antibody treatment, intraneuronal Aβ pathology returned along with the early cognitive deficits typical of untreated mice [35]. This important experiment points to the transient nature of acute immunotherapy on early Aβ accumulation and the need to effect and maintain a stable shift in the equilibrium of Aβ accumulation. Taken together, these data suggest that immunotherapy leads to cognitive improvement in these mouse studies via actions primarily directed at soluble oligomeric forms of Aβ.
The impact of current symptomatic treatments, such as cholinesterase inhibitors and NMDA receptor antagonists, likely is not sufficient to make a major impact on the pending public health crisis in AD prevalence in the elderly population as symptomatic treatment strategies are of diminished effectiveness as AD progresses. New, more-effective disease modifying therapies are critically necessary to alter the course of AD, although there is no clear consensus regarding the best targets for new therapeutic approaches [56]. One of the approaches receiving much attention can be generally classified as “anti-amyloid therapy” (e.g., immunotherapies, secretase inhibitors). Anti-amyloid therapies (see below) hold promise for yielding a significant disease-modifying effect based on the hypothesis that the deposition of the Aβ protein in the brain is causative of AD [25,27]. Secretase inhibitors have proven difficult to develop, despite nearly a decade of intense work in this area, but progress in this area continues [56]. Immunotherapies have been shown to have marked anti-amyloid effects in transgenic mice [40,57], and human clinical trials have resulted in modest clinical effects [58]. More intriguing than the clinical effect in these early immunotherapy trials is the fact that three published autopsy reports strongly support the notion that immunotherapy results in significant (albeit focal) Aβ plaque reduction in humans [42,43,44]. Passive immunization is currently in clinical trials, but even if effective, it may be difficult to apply this form of therapy to tens of millions of patients world-wide due to both antibody availability and expense.
Therapeutic Approaches that Decrease Aβ Synthesis
Attempts to decrease Aβ production involve inhibition of two distinct “secretase” enzymes responsible for cleavage of Aβ from its much larger precursor protein (Fig 6) [59]. β-Secretase or β-amyloid cleaving enzyme (BACE) cleaves the N-terminus of Aβ and the γ-secretase enzyme complex cleaves the C-terminus [60]. Studies with transgenic mice with deposit Aβ plaques in their brain have shown that γ-secretase inhibitors can prevent Aβ deposition [61]. Both β- and γ-secretase have proven to be difficult drug targets. γ-Secretase knockout mice are not viable. Inhibition of γ-secretase not only reduces the processing of APP to Aβ, but also reduces the processing of other γ-secretase substrates. The most important of the non-APP substrates appears to be “Notch”, a protein critical in cell proliferation and differentiation. Alteration of Notch metabolism produces marked gastrointestinal toxicity and reduced hematopoiesis in animals. Efforts are underway to develop γ-secretase inhibitors that have selectivity for APP processing over Notch, and some progress has been reported [62,63]. BACE-knockout mice are viable and appear to develop normally, making BACE appear to be the preferred secretase drug target [64]. Ironically, BACE has proven very difficult to potently inhibit with small molecules. This was believed to be a result of the “large catalytic site” responsible for BACE cleavage of APP [65,66]. Despite the problems, progress towards human therapy has been made. Human studies have been begun with γ-secretase inhibitors [67,68].
In addition to drugs specifically designed as β- or γ-secretase inhibitors, drugs such as non-steriodal anti-inflammatory drugs (NSAIDs) and statins may work to reduce Aβ generation through effects on secretases. NSAIDs appear to decrease the proportion of the more aggregation-prone Aβ42 through a non-cyclooxygenase mechanism [69,70], although clinical trials with NSAIDs have yet to show promise [71]. Similarly, statins appear to lower Aβ generation by activating the competitive α-secretase cleavage pathway [72].
Therapeutic Approaches that Increase Aβ Clearance or Decrease Toxicity
Another “anti-amyloid” approach makes use of immunotherapy against Aβ. It is generally regarded that this approach lowers Aβ levels by augmenting clearance of Aβ. Schenk and colleagues first demonstrated that active immunization of Aβ-depositing transgenic mice with Aβ peptides leads to circulating anti-Aβ antibodies and prevention of Aβ deposition in PDAPP mice [40]. One possible mechanism for immunotherapy-induced Aβ clearance is the enhancement of microglial phagocytosis via Fc receptors [57]. Bard and coworkers [73] have extensively studied the characteristics of antibodies that best relate to Aβ clearance and found: 1) only antibodies against the N-terminal regions of Aβ were able to invoke plaque clearance; 2) plaque binding correlated with a clearance response and neuronal protection, whereas the ability of antibodies to capture soluble Aβ was not necessarily correlated with efficacy; 3) the isotype of the antibody dramatically influenced the degree of plaque clearance and neuronal protection; 4) high affinity of the antibody for Fc receptors on microglial cells seemed more important than high affinity for Aβ itself; and 5) complement activation was not required for plaque clearance. These results were interpreted to indicate that antibody Fc-mediated plaque clearance is a highly efficient and effective process for protection against neuropathology in an animal model of Alzheimer’s disease.
Several studies suggest that if anti-Aβ mediated Aβ clearance happens in the brain, it is not solely via a microglial Fc receptor-dependent mechanism. Administration of the F(ab′)2 fragment of an anti-Aβ antibody (i.e., an Aβ binding agent which lacks the Fc region of the antibody) led to a reduction in brain Aβ to the same extent as the full anti-Aβ antibody [74]. This suggests that Aβ binding alone, not microglia-mediated Aβ clearance, is sufficient to reduce Aβ load, although this does not necessarily imply a peripheral effect. Solomon and coworkers have shown that antibodies to Aβ can bind to fibrils and cause disaggregation [75]. More directly, APP transgenic mice crossed with Fc receptor knockout mice responded to vaccination as well as mice with intact Fc receptors [76] showing that Fc receptor mediated phagocytosis was not necessary for Aβ clearance in this mouse. An alternative hypothesis suggests that antibodies present in the peripheral blood alter the central:peripheral Aβ equilibrium [55]. This “peripheral sink hypothesis” is supported by the finding of marked elevations in plasma Aβ levels after passive immunization [55] and active immunization [77].
Anti-Aβ antibodies, in addition to the clearance mechanisms discussed above, can decrease aggregation and induce disaggregation of Aβ in vitro [75]. However, it seems unlikely that the near-stoichiometric amounts of antibody required for inhibition of aggregation can be attained in the brain [75]. Therefore, approaches to decreasing aggregation and toxicity have largely centered on small molecule approaches. It has been pointed out that an anti-aggregation approach that would decrease the aggregation of oligomers and protofibrils into fibrils could actually be counter-productive by increasing the level of the most toxic species [41]. While this is plausible in theory, it should be pointed out that there are no published data to support the hypothesis that a small molecule can affect the intermolecular association of Aβ in such a way as to selectively inhibit aggregation into fibrils without also inhibiting the formation of oligomers. To the contrary, at least one small molecule, curcumin, has been shown to inhibit both oligomer and fibril formation [78]. Numerous other small molecules have been shown to possess anti-aggregation activity in vitro [79]. Another approach to the design of anti-aggregation agents is to start with fragments of Aβ such as 16–20, known to be critical in the aggregation process, and then add prolines, bulky groups or amide N-methyl groups to generate “β-sheet breaker” peptides. The small molecule, 3-amino-1-propanesulfonic acid (3APS; Alzhemed™; Neurochem Inc.) has been reported to prevent Aβ aggregation by competing with endogenous sulfated glycosaminoglycans (GAGs). Kisilevsky et al. have synthesized low-molecular-weight (135–1,000) anionic sulphonate or sulphate compounds. These compounds interfered with heparan sulphate-stimulated Aβ fibril aggregation in vitro [80]. Clioquinol is a metal-protein-attenuating compound that inhibits zinc and copper ions from binding to Aβ, thereby promoting Aβ dissolution and diminishing its toxic properties [81,82]. In a blinded and controlled 9 week study of a mouse model of AD, oral clioquinol decreased brain Aβ by 49% without systemic toxicity [83].
Human Anti-Amyloid Therapy Clinical Trials
The first iteration of the immunotherapeutic approach in clinical trials involved active immunization with Aβ42 itself, along with an immunogenic adjuvant (QS-21). Unfortunately, this “AN-1792” trial was suspended as a result of a 6% incidence of a serious adverse event of meningoencephalitis [58,84] that may be related to the presence of cerebral amyloid angiopathy [85]. A report on a subset of patients suggested that successful immunization to Aβ slows cognitive decline [86], but the results from the larger cohort showed a very modest clinical effect [58]. The effects seemed to be related to the strength of the antibody response [58]. Surprisingly, high antibody titer also was linked to increased atrophy on MRI, a finding that remains unexplained [87]. More promising than the clinical and imaging outcomes were the findings reported in three autopsy cases from this AN-1792 trial. All three cases, one of which had no encephalitis, showed marked focal reduction of Aβ deposition [42,43,44] providing proof-of-concept evidence that Aβ clearance can indeed occur in humans with AD. This has prompted more intense interest in further refinements of the immunotherapeutic anti-amyloid approach such as passive immunization with anti-Aβ antibodies [55,73] which may avoid many untoward effects of active immunization including menigoencephalitis although there has been a caution to the contrary [88].
One of the most encouraging findings from the AN-1792 active immunization trial was that the autopsy data strongly suggested that immunotherapy can have a profound Aβ-clearing effect in humans. Although this finding can be considered a proof-of-concept in many ways, enthusiasm must be tempered not only by the incidence of meningoencephalitis, but also by the relatively modest clinical effect of AN-1792. However, careful consideration of the state of the pathology when AN-1792 treatment was initiated may be instructive as to why this anti-amyloid therapy had such modest clinical effects. Figure 7 shows a small field-of-view from the frontal cortex of the AN-1792 autopsy case reported by Masliah and coworkers (Masliah 2005). The brain has been stained with X-34 (see Fig 1), a highly fluorescent Congo red derivative that stains β-sheet deposits including both fibrillar tau and Aβ pathology [89]. As confirmed by immunohistochemistry with anti-Aβ antibodies (data not shown), this area of the frontal cortex has been cleared of Aβ deposits, but extensive neuritic and neurofibrillary pathology remain untouched. The asterisk points to an area void of Aβ or neuritic pathology and has the appearance of an area that once may have been occupied by an Aβ plaque. This figure graphically points out how Aβ immunotherapy failed to affect tau pathology once it is well-established and the neuronal dysfunction associated with this tau pathology. Evidence from transgenic mouse models of plaque and tangle deposition suggest that plaque deposits precede and enhance tangle pathology at least in these animals [35]. This suggests that effective anti-amyloid therapy should be initiated early in the pathological process of AD in order to be optimally effective. Evidence suggests that Aβ pathology begins a decade or more before clinical symptoms are apparent [13]. Imaging technologies such as [11C]PiB that can detect Aβ deposits are under development by several groups [16,90,91,92]. The exact point in the evolution of Aβ pathology at which these imaging technologies will become useful remains to be determined, but early evidence suggests that detection of extensive Aβ deposition is possible during the mild cognitive impairment (MCI) stage [22] and perhaps prior to the onset of symptoms (Fig 5) [14,23].
Figure 7.
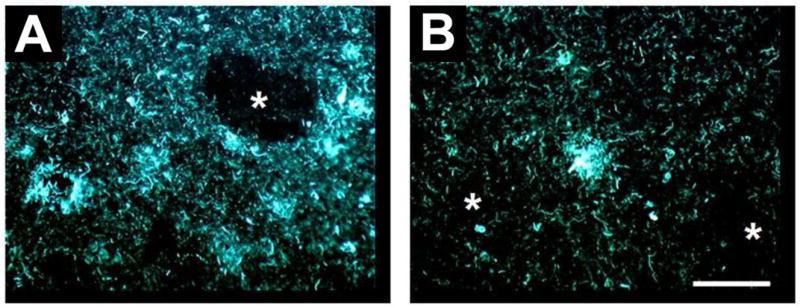
Post mortem fluorescent microscopy images of X-34 staining of 8 μm thick brain sections from an AD patient treated with the anti-amyloid therapy AN-1792 (an active immunization treatment). Sections include tissues frontal cortex (A) and temporal cortex (B) showing mostly neurofibrillary tangles and other tau-containing cellular processes, but the sections are relatively free of Aβ plaques. The asterisks (*) mark “holes” in the brain were Aβ plaques may have previously existed prior to AN-1792 treatment. The white scale bar is 100 μm in length.
There are several implications of Figures 3, 5, and 7 with respect to the application of anti-Aβ therapies and the effective role of Aβ imaging agents in aiding drug development efforts. First, agents such as [11C]PiB can identify subjects who would benefit from anti-Aβ therapies and those who would not. Clearly, AD patients (Figure 3 and 5) with heavy Aβ depositions are candidates for these therapies. However as shown in Figure 7, these treatments may be too late in advanced AD cases that have significant downstream damage from Aβ deposition such as extensive NFT deposits, dystrophic neurites, neuroinflammation, and synaptic and neuronal cell loses. Instead, MCI subjects with moderate to heavy Aβ loads (Fig 5) might be better candidates for these treatments. These subjects may not have the extensive synaptic and neuronal loses suffered by AD patients. In addition, MCI subjects with no evidence of Aβ deposition (Figs 3 and 5) would likely not be candidates for these therapies. These MCI subjects might be among the reported 30–40% of MCI subjects who do not progress on to develop AD [93]. Normal control subjects who have evidence of some Aβ deposition (Figs 3 and 5) also might benefit from anti-Aβ treatments prior to the onset of detectable cognitive symptoms. We have followed an elderly control subject with significant Aβ brain depositions for two years with [11C]PiB imaging (Fig 8). This subject shows increased [11C]PiB binding over two years greater than the test/retest variability in brain regions known to contain high concentrations of Aβ plaques in AD (e.g., precuneus, parietal cortex, frontal cortex, and striatum) and no increase in [11C]PiB binding in brain regions known to contain low concentrations of Aβ plaques in AD (e.g., cerebellum, mesial temporal cortex, sensory motor strip, white matter). This elderly control subject would be an excellent candidate for anti-Aβ therapies at a stage where little neuronal damage is evident.
Figure 8.
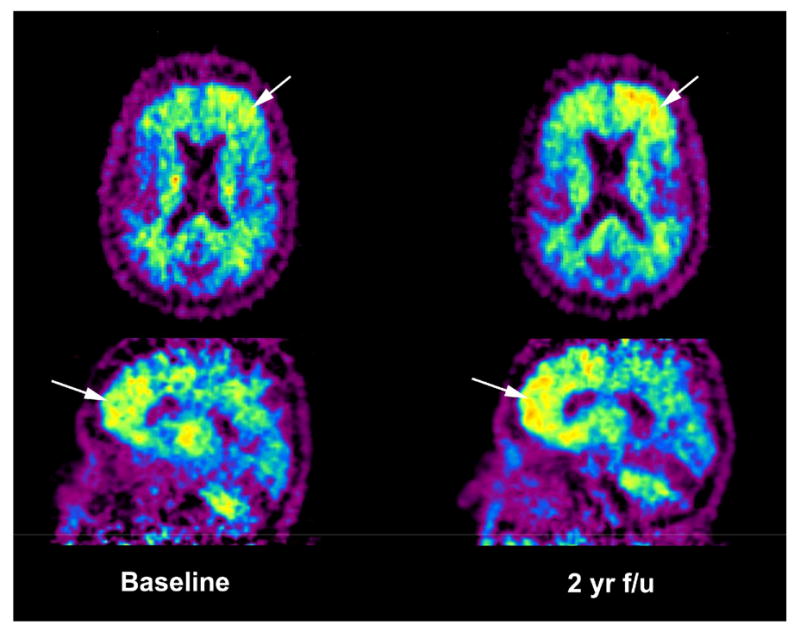
Transaxial and sagittal [11C]PiB parametric images of the reference Logan distribution volume ratio (DVR) using cerebellar tissue as input in an elderly normal control subject at baseline and 2 years later. The subject shows increased, unilateral [11C]PiB binding in the frontal cortex (arrows) over the 2 year time period, perhaps suggesting a future course of cognitive decline.
The second implication of Figures 3, 5, and 7 is that only subjects with detectable Aβ loads will demonstrate decreases in [11C]PiB binding with anti-Aβ therapies. Elderly controls and MCI subjects with no evidence of Aβ deposition are unlikely to show changes in [11C]PiB binding with anti-amyloid therapy. Finally, [11C]PiB and related compounds should be able to detect decreases in insoluble Aβ load about 2-fold greater than the test/retest variability of the imaging method. For [11C]PiB, the test/retest variability depends on the data analysis method and the region of interest [22], but is in the range of 5–10%. Thus 10–20% decreases in Aβ load should prove detectable with [11C]PiB.
The type of anticipated effects of anti-Aβ treatment on Aβ load are shown in Figure 9. This figure shows the result of in vitro [3H]PiB binding assay in post mortem frontal cortex tissue collected from the same patient shown in Figure 7. The amount of [3H]PiB binding in the AD tissue sample is comparable to that found in the frontal cortex of normal control subjects and well below levels found in AD patients. The question is: what level of Aβ plaque deposition did this patient have in frontal cortex before AN-1792 treatments? With a single post mortem sample, one can not address this question. But with amyloid imaging at baseline and after therapy, this question can be addressed. Currently, [11C]PiB is being used in anti-amyloid drug development trials in clinically diagnosed mild to moderate AD subjects to quantitatively assess changes in amyloid plaque densities throughout the brain over the course of the experimental treatments. Accurate pre-treatment, baseline measures of [11C]PiB binding in these subjects will determine a reference point against to judge the effectiveness of the treatments in clearing amyloid plaque burden, as well as confirm the clinical diagnosis of apparent AD.
Figure 9.

In vitro binding assays of [3H]PiB in post mortem homogenized tissues taken from the frontal cortex (Fr) and cerebellum (CB) of normal control subjects (Cntl), untreated Alzheimer’s disease subjects (AD), and the AN-1792-treated AD subject shown in Figure 7. All cerebellar tissues show low levels of specifically bound tracer in accordance with the absence of Aβ plaque pathology in the cerebellum, but the frontal cortex from the untreated AD subjects shows a high level of specifically bound tracer. Frontal cortex from the AN-1792 treated AD subject shows values similar to control subjects, indicating the absence of Aβ plaque pathology. The question is: did this AN-1792 treated subject have Aβ plaque pathology in the frontal cortex prior to treatment? Imaging studies using Aβ imaging agents such [11C]PiB at baseline and after anti-amyloid treatment will be able to answer this question.
Clinical trials of the effectiveness of anti-amyloid therapies must necessarily begin with clinically diagnosed mild to moderate AD subjects, because of the need to clearly demonstrate cognitive improvement and power the drug trials with a reasonable number of subjects over as short a treatment period as possible. Given that 30–40% of MCI subjects do not progress on to AD and the conversion rate of MCI to AD is only 15%/year, use of this group for anti-amyloid clinical trials would require larger subject numbers and longer observational periods than use of AD subject groups would permit. Prevention studies in elderly normal control subjects would be even more time consuming and costly. However, it is likely that therapeutic intervention would benefit [11C]PIB-positive elderly control subjects and MCI subjects with significant Aβ plaque deposits more than it would AD patients. In amyloid-positive elderly control and MCI subjects, extensive irreversible neuronal damage is likely to be considerably lower than in AD patients. This is illustrated graphically in Figure 10.
Figure 10.
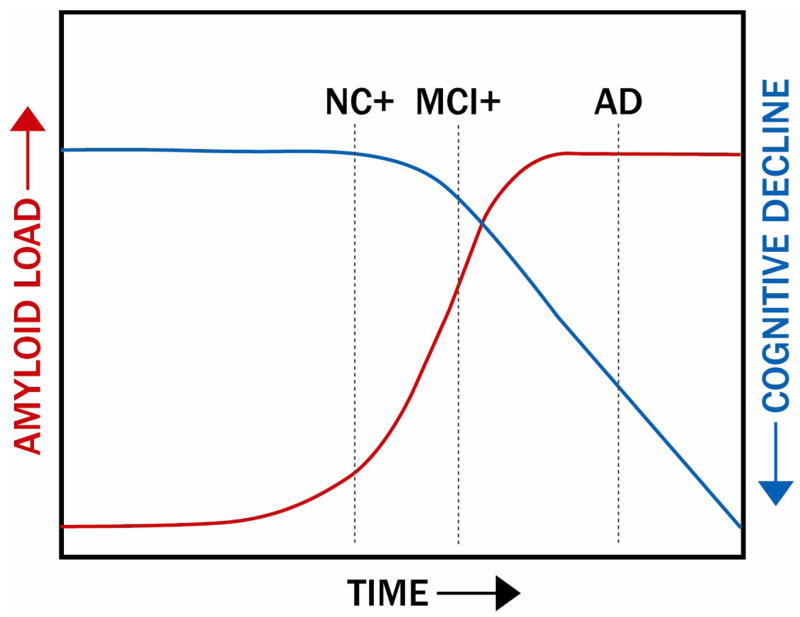
Schematic of the time course of Aβ deposition and the offset in time of subsequent cognitive decline. The dotted line under AD indicates the time point for application of anti-amyloid treatments in clinically apparent AD patients, while the dotted NC+ and MCI+ lines indicate the time points for application of these treatments when cognitive performance is relatively intact, but Aβ deposition is apparent. Treatments with anti-amyloid therapies at these earlier time points could halt the downstream damage caused by excessive cerebral Aβ loads (see Figures 4 and 6).
Conclusions
The potential applications of Aβ imaging include their use as diagnostic agents to detect cerebral beta-amyloidosis, to help test the amyloid cascade hypothesis of AD, and to assist with the development of anti-amyloid therapeutic drugs. While efforts are underway currently in all three areas of research, the most immediate impact of this class of imaging agents will likely be to aid in the in vivo evaluation of new anti-amyloid therapies. The clinical efficacy of these anti-amyloid therapies, aimed at lowering levels of soluble and insoluble Aβ in the brain, remains to be fully demonstrated. But it is likely that Aβ imaging agents will assist with the interpretation of the clinical trial results and hasten the development of these drugs. If anti-amyloid therapies prove efficacious in halting or reversing cognitive decline in AD patients, it is likely that they subsequently will be applied to treat amyloid-positive, pre-AD subjects, who might benefit even more than AD patients from this therapy. Aβ imaging agents likely will help identify pre-AD subjects for these treatments. This, in turn, will more fully employ the first two potential applications of these radioligands: as diagnostic agents to detect cerebral beta-amyloidosis and to help test the amyloid cascade hypothesis of AD.
Acknowledgments
This work was supported by National Institute of Health grants R01 AG018402, P50 AG005133, R01 AG020226, R37 AG025516, and P01 AG025204 and Alzheimer’s Association Grant TLL-01-3381. GE Healthcare holds a license agreement with the University of Pittsburgh based on the [11C]PiB imaging technology described in this manuscript. Drs. Mathis and Klunk are co-inventors of [11C]PiB and, as such, have a financial interest in this license agreement. GE Healthcare provided no financial support for the preparation of this manuscript and had no role in the writing or interpretation of the information contained in this manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hebert LE, Scherr PA, Bienias JL, Bennett DA, Evans DA. Alzheimer disease in the US population: prevalence estimates using the 2000 census. Archives of Neurology. 2003;60(8):1119–1122. doi: 10.1001/archneur.60.8.1119. [DOI] [PubMed] [Google Scholar]
- 2.Jorm AF. Cross-national comparisons of the occurrence of Alzheimer’s and vascular dementias. Eur Arch Psychiatry Clin Neurosci. 1991;240(4–5):218–22. doi: 10.1007/BF02189530. [DOI] [PubMed] [Google Scholar]
- 3.Mirra SS, Heyman A, McKeel D, Sumi SM, Crain BJ, Brownlee LM, et al. The Consortium to Establish a Registry for Alzheimer’s Disease (CERAD). Part II. Standardization of the neuropathologic assessment of Alzheimer’s disease. Neurology. 1991;41:479. doi: 10.1212/wnl.41.4.479. [DOI] [PubMed] [Google Scholar]
- 4.Iwatsubo T, Odaka A, Suzuki N, Mizusawa H, Nukina N, Ihara Y. Visualization of A beta 42(43) and A beta 40 in senile plaques with end-specific A beta monoclonals: evidence that an initially deposited species is A beta 42(43) Neuron. 1994;13:45. doi: 10.1016/0896-6273(94)90458-8. [DOI] [PubMed] [Google Scholar]
- 5.Goedert M. Tau protein and the neurofibrillary pathology of Alzheimer’s disease. Trends in Neurosciences. 1993;16:460–465. doi: 10.1016/0166-2236(93)90078-z. [DOI] [PubMed] [Google Scholar]
- 6.Uversky VNATJRGALF. Protein deposits as the molecular basis of amyloidosis. Part I. Systemic amyloidoses. Med Sci Monit. 1999;5:1001–1012. [Google Scholar]
- 7.Uversky VNATJRGALF. Protein deposits as the molecular basis of amyloidosis. Part II. Localized amyloidosis and neurodegenerative disorders. Med Sci Monit. 1999;5:1238–1254. [Google Scholar]
- 8.Westermark P, Benson MD, Buxbaum JN, Cohen AS, Frangione B, Ikeda S, et al. Amyloid: toward terminology clarification. Report from the Nomenclature Committee of the International Society of Amyloidosis Amyloid. 2005;12(1):1–4. doi: 10.1080/13506120500032196. [DOI] [PubMed] [Google Scholar]
- 9.Forman MS, Mufson EJ, Leurgans S, Pratico D, Joyce S, Leight S, et al. Cortical biochemistry in MCI and Alzheimer disease: lack of correlation with clinical diagnosis. Neurology. 2007;68(10):757–63. doi: 10.1212/01.wnl.0000256373.39415.b1. [DOI] [PubMed] [Google Scholar]
- 10.Klunk WE, Wang Y, Huang GF, Debnath ML, Holt DP, Shao L, et al. The binding of 2-(4′-methylaminophenyl)benzothiazole to postmortem brain homogenates is dominated by the amyloid component. J Neurosci. 2003;23(6):2086–92. doi: 10.1523/JNEUROSCI.23-06-02086.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Morris JC, Storandt M, McKeel DW, Jr, Rubin EH, Price JL, Grant EA, et al. Cerebral amyloid deposition and diffuse plaques in “normal” aging: Evidence for presymptomatic and very mild Alzheimer’s disease. Neurology. 1996;96(3):707. doi: 10.1212/wnl.46.3.707. [DOI] [PubMed] [Google Scholar]
- 12.Price JL, Morris JC. Tangles and plaques in nondemented aging and “preclinical” Alzheimer’s disease. Annals of Neurology. 1999;45(3):358. doi: 10.1002/1531-8249(199903)45:3<358::aid-ana12>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- 13.Hyman BT, West HL, Rebeck GW, Lai F, Mann DM. Neuropathological changes in Down’s syndrome hippocampal formation. Effect of age and apolipoprotein E genotype. Arch Neurol. 1995;52(4):373–8. doi: 10.1001/archneur.1995.00540280059019. [DOI] [PubMed] [Google Scholar]
- 14.Mintun MA, Larossa GN, Sheline YI, Dence CS, Lee SY, Mach RH, et al. [11C]PIB in a nondemented population: potential antecedent marker of Alzheimer disease. Neurology. 2006;67(3):446–52. doi: 10.1212/01.wnl.0000228230.26044.a4. [DOI] [PubMed] [Google Scholar]
- 15.McKeith IG, Galasko D, Kosaka K, Perry EK, Dickson DW, Hansen LA, et al. Consensus guidelines for the clinical and pathologic diagnosis of dementia with Lewy bodies (DLB): report of the consortium on DLB international workshop. Neurology. 1996;47(5):1113–24. doi: 10.1212/wnl.47.5.1113. [DOI] [PubMed] [Google Scholar]
- 16.Klunk WE, Engler H, Nordberg A, Wang Y, Blomqvist G, Holt DP, et al. Imaging brain amyloid in Alzheimer’s disease with Pittsburgh Compound-B. Annals of Neurology. 2004;55:306–319. doi: 10.1002/ana.20009. [DOI] [PubMed] [Google Scholar]
- 17.Thal DR, Rub U, Orantes M, Braak H. Phases of A beta-deposition in the human brain and its relevance for the development of AD. Neurology. 2002;58(12):1791. doi: 10.1212/wnl.58.12.1791. [DOI] [PubMed] [Google Scholar]
- 18.Braak H, Braak E. Neuropathological staging of Alzheimer-related changes. Acta Neuropathologica. 1991;82:239. doi: 10.1007/BF00308809. [DOI] [PubMed] [Google Scholar]
- 19.Arnold SE, Hyman BT, Flory J, Damasio AR, Van Hoesen GW. The topographical and neuroanatomical distribution of neurofibrillary tangles and neuritic plaques in the cerebral cortex of patients with Alzheimer’s disease. Cereb Cortex. 1991;1(1):103–16. doi: 10.1093/cercor/1.1.103. [DOI] [PubMed] [Google Scholar]
- 20.Joachim CL, Morris JH, Selkoe DJ. Diffuse senile plaques occur commonly in the cerebellum in Alzheimer’s disease. American Journal of Pathology. 1989;135:309. [PMC free article] [PubMed] [Google Scholar]
- 21.Yamaguchi H, Hirai S, Morimatsu M, Shoji M, Nakazato Y. Diffuse type of senile plaques in the cerebellum of Alzheimer- type dementia demonstrated by beta protein immunostain. Acta Neuropathologica. 1989;77:314. doi: 10.1007/BF00687584. [DOI] [PubMed] [Google Scholar]
- 22.Lopresti BJ, Klunk WE, Mathis CA, Hoge JA, Ziolko SK, Lu X, et al. Simplified quantification of Pittsburgh Compound B amyloid imaging PET studies: a comparative analysis. J Nucl Med. 2005;46(12):1959–72. [PubMed] [Google Scholar]
- 23.Rowe CC, Ng S, Ackermann U, Gong SJ, Pike K, Savage G, et al. Imaging beta-amyloid burden in aging and dementia. Neurology. 2007;68(20):1718–25. doi: 10.1212/01.wnl.0000261919.22630.ea. [DOI] [PubMed] [Google Scholar]
- 24.Hardy J, Allsop D. Amyloid deposition as the central event in the aetiology of Alzheimer’s disease. Trends Pharmacol Sci. 1991;12(10):383–8. doi: 10.1016/0165-6147(91)90609-v. [DOI] [PubMed] [Google Scholar]
- 25.Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science. 2002;297(5580):353–6. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- 26.Hardy JA, Higgins GA. Alzheimer’s disease: the amyloid cascade hypothesis. Science. 1992;256:184. doi: 10.1126/science.1566067. [DOI] [PubMed] [Google Scholar]
- 27.Hardy J. Has the amyloid cascade hypothesis for Alzheimer’s disease been proved? Curr Alzheimer Res. 2006;3(1):71–3. doi: 10.2174/156720506775697098. [DOI] [PubMed] [Google Scholar]
- 28.Hardy J, Duff K, Hardy KG, Perez-Tur J, Hutton M. Genetic dissection of Alzheimer’s disease and related dementias: amyloid and its relationship to tau. Nat Neurosci. 1998;1(5):355–8. doi: 10.1038/1565. [DOI] [PubMed] [Google Scholar]
- 29.Price DL, Sisodia SS. Mutant genes in familial Alzheimer’s disease and transgenic models. Annu Rev Neurosci. 1998;21:479–505. doi: 10.1146/annurev.neuro.21.1.479. [DOI] [PubMed] [Google Scholar]
- 30.Tanzi RE, Kovacs DM, Kim TW, Moir RD, Guenette SY, Wasco W. The gene defects responsible for familial Alzheimer’s disease. Neurobiol Dis. 1996;3(3):159–68. doi: 10.1006/nbdi.1996.0016. [DOI] [PubMed] [Google Scholar]
- 31.Xia W, Ostaszewski BL, Kimberly WT, Rahmati T, Moore CL, Wolfe MS, et al. FAD mutations in presenilin-1 or amyloid precursor protein decrease the efficacy of a gamma-secretase inhibitor: evidence for direct involvement of PS1 in the gamma-secretase cleavage complex. Neurobiol Dis. 2000;7(6 Pt B):673–81. doi: 10.1006/nbdi.2000.0322. [DOI] [PubMed] [Google Scholar]
- 32.Oddo S, Caccamo A, Kitazawa M, Tseng BP, LaFerla FM. Amyloid deposition precedes tangle formation in a triple transgenic model of Alzheimer’s disease. Neurobiol Aging. 2003;24(8):1063–70. doi: 10.1016/j.neurobiolaging.2003.08.012. [DOI] [PubMed] [Google Scholar]
- 33.Oddo S, Caccamo A, Shepherd JD, Murphy MP, Golde TE, Kayed R, et al. Triple-transgenic model of Alzheimer’s disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron. 2003;39(3):409–21. doi: 10.1016/s0896-6273(03)00434-3. [DOI] [PubMed] [Google Scholar]
- 34.Billings LM, Oddo S, Green KN, McGaugh JL, LaFerla FM. Intraneuronal Abeta causes the onset of early Alzheimer’s disease-related cognitive deficits in transgenic mice. Neuron. 2005;45(5):675–88. doi: 10.1016/j.neuron.2005.01.040. [DOI] [PubMed] [Google Scholar]
- 35.Oddo S, Billings L, Kesslak JP, Cribbs DH, LaFerla FM. Abeta immunotherapy leads to clearance of early, but not late, hyperphosphorylated tau aggregates via the proteasome. Neuron. 2004;43(3):321–32. doi: 10.1016/j.neuron.2004.07.003. [DOI] [PubMed] [Google Scholar]
- 36.Gotz J, Chen F, van Dorpe J, Nitsch RM. Formation of neurofibrillary tangles in P3011 tau transgenic mice induced by Abeta 42 fibrils. Science. 2001;293(5534):1491–5. doi: 10.1126/science.1062097. [DOI] [PubMed] [Google Scholar]
- 37.Lewis J, Dickson DW, Lin WL, Chisholm L, Corral A, Jones G, et al. Enhanced neurofibrillary degeneration in transgenic mice expressing mutant tau and APP. Science. 2001;293(5534):1487–91. doi: 10.1126/science.1058189. [DOI] [PubMed] [Google Scholar]
- 38.Klein WL, Stine WB, Jr, Teplow DB. Small assemblies of unmodified amyloid beta-protein are the proximate neurotoxin in Alzheimer’s disease. Neurobiol Aging. 2004;25(5):569–80. doi: 10.1016/j.neurobiolaging.2004.02.010. [DOI] [PubMed] [Google Scholar]
- 39.Stine WB, Jr, Dahlgren KN, Krafft GA, LaDu MJ. In vitro characterization of conditions for amyloid-beta peptide oligomerization and fibrillogenesis. J Biol Chem. 2003;278(13):11612–22. doi: 10.1074/jbc.M210207200. [DOI] [PubMed] [Google Scholar]
- 40.Schenk D, Barbour R, Dunn W, Gordon G, Grajeda H, Guido T, et al. Immunization with amyloid-beta attenuates Alzheimer-disease-like pathology in the PDAPP mouse. Nature. 1999;400(6740):173. doi: 10.1038/22124. [DOI] [PubMed] [Google Scholar]
- 41.Walker LC, Ibegbu CC, Todd CW, Robinson HL, Jucker M, LeVine H, 3rd, et al. Emerging prospects for the disease-modifying treatment of Alzheimer’s disease. Biochem Pharmacol. 2005;69(7):1001–8. doi: 10.1016/j.bcp.2004.12.015. [DOI] [PubMed] [Google Scholar]
- 42.Ferrer I, Boada R, Sanchez G, Rey MJ, Costa-Jussa F. Neuropathology and pathogenesis of encephalitis following amyloid-beta immunization in Alzheimer’s disease. Brain Pathology. 2004;14(1):11. doi: 10.1111/j.1750-3639.2004.tb00493.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Masliah E, Hansen L, Adame A, Crews L, Bard F, Lee C, et al. Abeta vaccination effects on plaque pathology in the absence of encephalitis in Alzheimer disease. Neurology. 2005;64(1):129–31. doi: 10.1212/01.WNL.0000148590.39911.DF. [DOI] [PubMed] [Google Scholar]
- 44.Nicoll JA, Wilkinson D, Holmes C, Steart P, Markham H, Weller RO. Neuropathology of human Alzheimer disease after immunization with amyloid-á peptide: a case report. Nature Medicine. 2003;9:448. doi: 10.1038/nm840. [DOI] [PubMed] [Google Scholar]
- 45.Glabe CG. Conformation-dependent antibodies target diseases of protein misfolding. Trends Biochem Sci. 2004;29(10):542–7. doi: 10.1016/j.tibs.2004.08.009. [DOI] [PubMed] [Google Scholar]
- 46.Zhou J, Fonseca MI, Kayed R, Hernandez I, Webster SD, Yazan O, et al. Novel Abeta peptide immunogens modulate plaque pathology and inflammation in a murine model of Alzheimer’s disease. J Neuroinflammation. 2005;2:28. doi: 10.1186/1742-2094-2-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kuo YM, Emmerling MR, Vigo-Pelfrey C, Kasunic TC, Kirkpatrick JB, Murdoch GH, et al. Water-soluble Abeta (N-40, N-42) oligomers in normal and Alzheimer disease brains. J Biol Chem. 1996;271(8):4077–81. doi: 10.1074/jbc.271.8.4077. [DOI] [PubMed] [Google Scholar]
- 48.Lambert MP, Barlow AK, Chromy BA, Edwards C, Freed R, Liosatos M, et al. Diffusible, nonfibrillar ligands derived from Abeta1–42 are potent central nervous system neurotoxins. Proc Natl Acad Sci U S A. 1998;95(11):6448–53. doi: 10.1073/pnas.95.11.6448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Walsh DM, Klyubin I, Fadeeva JV, Rowan MJ, Selkoe DJ. Amyloid-beta oligomers: their production, toxicity and therapeutic inhibition. Biochem Soc Trans. 2002;30(4):552. doi: 10.1042/bst0300552. [DOI] [PubMed] [Google Scholar]
- 50.Dodart JC, Bales KR, Gannon KS, Greene SJ, DeMattos RB, Mathis C, et al. Immunization reverses memory deficits without reducing brain Abeta burden in Alzheimer’s disease model. Nat Neurosci. 2002;5(5):452–7. doi: 10.1038/nn842. [DOI] [PubMed] [Google Scholar]
- 51.Klyubin I, Walsh DM, Lemere CA, Cullen WK, Shankar GM, Betts V, et al. Amyloid beta protein immunotherapy neutralizes Abeta oligomers that disrupt synaptic plasticity in vivo. Nat Med. 2005;11(5):556–61. doi: 10.1038/nm1234. [DOI] [PubMed] [Google Scholar]
- 52.Kotilinek LA, Bacskai B, Westerman M, Kawarabayashi T, Younkin L, Hyman BT, et al. Reversible memory loss in a mouse transgenic model of Alzheimer’s disease. J Neurosci. 2002;22(15):6331–5. doi: 10.1523/JNEUROSCI.22-15-06331.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Westerman MA, Cooper-Blacketer D, Mariash A, Kotilinek L, Kawarabayashi T, Younkin LH, et al. The relationship between Abeta and memory in the Tg2576 mouse model of Alzheimer’s disease. J Neurosci. 2002;22(5):1858–67. doi: 10.1523/JNEUROSCI.22-05-01858.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gong Y, Chang L, Viola KL, Lacor PN, Lambert MP, Finch CE, et al. Alzheimer’s disease-affected brain: presence of oligomeric A beta ligands (ADDLs) suggests a molecular basis for reversible memory loss. Proc Natl Acad Sci U S A. 2003;100(18):10417–22. doi: 10.1073/pnas.1834302100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.DeMattos RB, Bales KR, Cummins DJ, Dodart JC, Paul SM, Holtzman DM. Peripheral anti-A beta antibody alters CNS and plasma A beta clearance and decreases brain A beta burden in a mouse model of Alzheimer’s disease. Proc Natl Acad Sci U S A. 2001;98(15):8850. doi: 10.1073/pnas.151261398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Klafki HW, Staufenbiel M, Kornhuber J, Wiltfang J. Therapeutic approaches to Alzheimer’s disease. Brain. 2006;129(Pt 11):2840–55. doi: 10.1093/brain/awl280. [DOI] [PubMed] [Google Scholar]
- 57.Bard F, Cannon C, Barbour R, Burke RL, Games D, Grajeda H, et al. Peripherally administered antibodies against amyloid á-peptide enter the central nervous system and reduce pathology in a mouse model of Alzheimer disease. Nature Medicine. 2000;6:916. doi: 10.1038/78682. [DOI] [PubMed] [Google Scholar]
- 58.Gilman S, Koller M, Black RS, Jenkins L, Griffith SG, Fox NC, et al. Clinical effects of Abeta immunization (AN1792) in patients with AD in an interrupted trial. Neurology. 2005;64(9):1553–62. doi: 10.1212/01.WNL.0000159740.16984.3C. [DOI] [PubMed] [Google Scholar]
- 59.Olson RE, Copeland RA, Seiffert D. Progress towards testing the amyloid hypothesis: inhibitors of APP processing. Curr Opin Drug Discov Devel. 2001;4(4):390–401. [PubMed] [Google Scholar]
- 60.Nunan J, Small DH. Regulation of APP cleavage by alpha-, beta- and gamma-secretases. FEBS Lett. 2000;483(1):6–10. doi: 10.1016/s0014-5793(00)02076-7. [DOI] [PubMed] [Google Scholar]
- 61.Dovey HF, John V, Anderson JP, Chen LZ, de Saint AP, Fang LY, et al. Functional gamma-secretase inhibitors reduce beta-amyloid peptide levels in brain. J Neurochem. 2001;76(1):173–81. doi: 10.1046/j.1471-4159.2001.00012.x. [DOI] [PubMed] [Google Scholar]
- 62.Fraering PC, Ye W, LaVoie MJ, Ostaszewski BL, Selkoe DJ, Wolfe MS. gamma-Secretase substrate selectivity can be modulated directly via interaction with a nucleotide-binding site. J Biol Chem. 2005;280(51):41987–96. doi: 10.1074/jbc.M501368200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wolfe MS. The gamma-secretase complex: membrane-embedded proteolytic ensemble. Biochemistry. 2006;45(26):7931–9. doi: 10.1021/bi060799c. [DOI] [PubMed] [Google Scholar]
- 64.Roberds SL, Anderson J, Basi G, Bienkowski MJ, Branstetter DG, Chen KS, et al. BACE knockout mice are healthy despite lacking the primary beta-secretase activity in brain: implications for Alzheimer’s disease therapeutics. Hum Mol Genet. 2001;10(12):1317–24. doi: 10.1093/hmg/10.12.1317. [DOI] [PubMed] [Google Scholar]
- 65.Dewachter I, Van Leuven F. Secretases as targets for the treatment of Alzheimer’s disease: the prospects. Lancet Neurol. 2002;1(7):409–16. doi: 10.1016/s1474-4422(02)00188-6. [DOI] [PubMed] [Google Scholar]
- 66.Wang W, Reichert P, Beyer BM, Liu JJ, Lee J, Zhang L, et al. Crystallization of glycosylated human BACE protease domain expressed in Trichoplusia ni. Biochim Biophys Acta. 2004;1698(2):255–9. doi: 10.1016/j.bbapap.2003.11.035. [DOI] [PubMed] [Google Scholar]
- 67.Melnikova I. Therapies for Alzheimer’s disease. Nat Rev Drug Discov. 2007;6(5):341–2. doi: 10.1038/nrd2314. [DOI] [PubMed] [Google Scholar]
- 68.Siemers E, Skinner M, Dean RA, Gonzales C, Satterwhite J, Farlow M, et al. Safety, tolerability, and changes in amyloid beta concentrations after administration of a gamma-secretase inhibitor in volunteers. Clin Neuropharmacol. 2005;28(3):126–32. doi: 10.1097/01.wnf.0000167360.27670.29. [DOI] [PubMed] [Google Scholar]
- 69.Weggen S, Eriksen JL, Das P, Sagi SA, Wang R, Pietrzik CU, et al. A subset of NSAIDs lower amyloidogenic Abeta42 independently of cyclooxygenase activity. Nature. 2001;414(6860):212–6. doi: 10.1038/35102591. [DOI] [PubMed] [Google Scholar]
- 70.Weggen S, Eriksen JL, Sagi SA, Pietrzik CU, Ozols V, Fauq A, et al. Evidence that nonsteroidal anti-inflammatory drugs decrease amyloid beta 42 production by direct modulation of gamma-secretase activity. J Biol Chem. 2003;278(34):31831–7. doi: 10.1074/jbc.M303592200. [DOI] [PubMed] [Google Scholar]
- 71.Aisen PS, Schafer KA, Grundman M, Pfeiffer E, Sano M, Davis KL, et al. Effects of rofecoxib or naproxen vs placebo on Alzheimer disease progression: a randomized controlled trial. Jama. 2003;289(21):2819–26. doi: 10.1001/jama.289.21.2819. [DOI] [PubMed] [Google Scholar]
- 72.Refolo LM, Pappolla MA, LaFrancois J, Malester B, Schmidt SD, Thomas-Bryant T, et al. A cholesterol-lowering drug reduces beta-amyloid pathology in a transgenic mouse model of Alzheimer’s disease. Neurobiol Dis. 2001;8(5):890–9. doi: 10.1006/nbdi.2001.0422. [DOI] [PubMed] [Google Scholar]
- 73.Bard F, Barbour R, Cannon C, Carretto R, Fox M, Games D, et al. Epitope and isotype specificities of antibodies to beta -amyloid peptide for protection against Alzheimer’s disease-like neuropathology. Proc Natl Acad Sci U S A. 2003;100(4):2023–8. doi: 10.1073/pnas.0436286100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Bacskai BJ, Kajdasz ST, McLellan ME, Games D, Seubert P, Schenk D, et al. Non-Fc-mediated mechanisms are involved in clearance of amyloid-beta in vivo by immunotherapy. J Neurosci. 2002;22(18):7873–8. doi: 10.1523/JNEUROSCI.22-18-07873.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Solomon B, Koppel R, Frankel D, Hanan-Aharon E. Disaggregation of Alzheimer beta-amyloid by site-directed mAb. Proc Natl Acad Sci U S A. 1997;94(8):4109–12. doi: 10.1073/pnas.94.8.4109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Das P, Howard V, Loosbrock N, Dickson D, Murphy MP, Golde TE. Amyloid-beta immunization effectively reduces amyloid deposition in FcRgamma-/- knock-out mice. J Neurosci. 2003;23(24):8532–8. doi: 10.1523/JNEUROSCI.23-24-08532.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lemere CA, Spooner ET, LaFrancois J, Malester B, Mori C, Leverone JF, et al. Evidence for peripheral clearance of cerebral Abeta protein following chronic, active Abeta immunization in PSAPP mice. Neurobiol Dis. 2003;14(1):10–8. doi: 10.1016/s0969-9961(03)00044-5. [DOI] [PubMed] [Google Scholar]
- 78.Yang F, Lim GP, Begum AN, Ubeda OJ, Simmons MR, Ambegaokar SS, et al. Curcumin inhibits formation of amyloid beta oligomers and fibrils, binds plaques, and reduces amyloid in vivo. J Biol Chem. 2005;280(7):5892–901. doi: 10.1074/jbc.M404751200. [DOI] [PubMed] [Google Scholar]
- 79.Mason JM, Kokkoni N, Stott K, Doig AJ. Design strategies for anti-amyloid agents. Curr Opin Struct Biol. 2003;13(4):526–32. doi: 10.1016/s0959-440x(03)00100-3. [DOI] [PubMed] [Google Scholar]
- 80.Kisilevsky R, Lemieux LJ, Fraser PE, Kong X, Hultin PG, Szarek WA. Arresting amyloidosis in vivo using small-molecule anionic sulphonates or sulphates: implications for Alzheimer’s disease. Nat Med. 1995;1(2):143–8. doi: 10.1038/nm0295-143. [DOI] [PubMed] [Google Scholar]
- 81.Bush AI. Metal complexing agents as therapies for Alzheimer’s disease. Neurobiol Aging. 2002;23(6):1031–8. doi: 10.1016/s0197-4580(02)00120-3. [DOI] [PubMed] [Google Scholar]
- 82.Bush AI. The metallobiology of Alzheimer’s disease. Trends Neurosci. 2003;26(4):207–14. doi: 10.1016/S0166-2236(03)00067-5. [DOI] [PubMed] [Google Scholar]
- 83.Cherny RA, Atwood CS, Xilinas ME, Gray DN, Jones WD, McLean CA, et al. Treatment with a copper-zinc chelator markedly and rapidly inhibits beta-amyloid accumulation in Alzheimer’s disease transgenic mice. Neuron. 2001;30(3):665–76. doi: 10.1016/s0896-6273(01)00317-8. [DOI] [PubMed] [Google Scholar]
- 84.Orgogozo JM, Gilman S, Dartigues JF, Laurent B, Puel M, Kirby LC, et al. Subacute meningoencephalitis in a subset of patients with AD after Abeta42 immunization. Neurology. 2003;61(1):46–54. doi: 10.1212/01.wnl.0000073623.84147.a8. [DOI] [PubMed] [Google Scholar]
- 85.Gandy S, Walker L. Toward modeling hemorrhagic and encephalitic complications of Alzheimer amyloid-beta vaccination in nonhuman primates. Curr Opin Immunol. 2004;16(5):607–15. doi: 10.1016/j.coi.2004.08.002. [DOI] [PubMed] [Google Scholar]
- 86.Hock C, Konietzko U, Streffer JR, Tracy J, Signorell A, ller-Tillmanns BM, et al. Antibodies against á-Amyloid Slow Cognitive Decline in Alzheimer’s Disease. Neuron. 2003;38:547. doi: 10.1016/s0896-6273(03)00294-0. [DOI] [PubMed] [Google Scholar]
- 87.Fox NC, Black RS, Gilman S, Rossor MN, Griffith SG, Jenkins L, et al. Effects of Abeta immunization (AN1792) on MRI measures of cerebral volume in Alzheimer disease. Neurology. 2005;64(9):1563–72. doi: 10.1212/01.WNL.0000159743.08996.99. [DOI] [PubMed] [Google Scholar]
- 88.Lee EB, Leng LZ, Lee VM, Trojanowski JQ. Meningoencephalitis associated with passive immunization of a transgenic murine model of Alzheimer’s amyloidosis. FEBS Lett. 2005;579(12):2564–8. doi: 10.1016/j.febslet.2005.03.070. [DOI] [PubMed] [Google Scholar]
- 89.Styren SD, Hamilton RL, Styren GC, Klunk WE. X-34, a fluorescent derivative of Congo red: A novel histochemical stain for Alzheimer’s disease pathology. Journal of Histochemistry & Cytochemistry. 2000;48(9):1223. doi: 10.1177/002215540004800906. [DOI] [PubMed] [Google Scholar]
- 90.Mathis CA, Klunk WE, Price JC, DeKosky ST. Imaging technology for neurodegenerative diseases: progress toward detection of specific pathologies. Arch Neurol. 2005;62(2):196–200. doi: 10.1001/archneur.62.2.196. [DOI] [PubMed] [Google Scholar]
- 91.Small GW, Kepe V, Ercoli LM, Siddarth P, Bookheimer SY, Miller KJ, et al. PET of brain amyloid and tau in mild cognitive impairment. N Engl J Med. 2006;355(25):2652–63. doi: 10.1056/NEJMoa054625. [DOI] [PubMed] [Google Scholar]
- 92.Verhoeff NP, Wilson AA, Takeshita S, Trop L, Hussey D, Singh K, et al. In-Vivo Imaging of Alzheimer Disease {beta}-Amyloid With [11C]SB-13 PET. Am J Geriatr Psychiatry. 2004;12(6):584–595. doi: 10.1176/appi.ajgp.12.6.584. [DOI] [PubMed] [Google Scholar]
- 93.Gauthier S, Reisberg B, Zaudig M, Petersen RC, Ritchie K, Broich K, et al. Mild cognitive impairment. Lancet. 2006;367(9518):1262–70. doi: 10.1016/S0140-6736(06)68542-5. [DOI] [PubMed] [Google Scholar]
- 94.Klunk WE, Mathis CA, Price JC, Lopresti BJ, DeKosky ST. Two-year follow-up of amyloid deposition in patients with Alzheimer’s disease. Brain. 2006;129(Pt 11):2805–7. doi: 10.1093/brain/awl281. [DOI] [PubMed] [Google Scholar]