

Abstract
Background and purpose:
In asthma, histamine contributes to bronchoconstriction, vasodilatation and oedema, and is associated with the late phase response. The current study investigates possible inflammatory effects of histamine acting on nuclear factor κB (NF-κB)-dependent transcription and cytokine release.
Experimental approach:
Using BEAS-2B bronchial epithelial cells, NF-κB-dependent transcription and both release and mRNA expression of IL-6 and IL-8 were examined by reporter assay, ELISA and quantitative RT-PCR. Histamine receptors were detected using qualitative RT-PCR and function examined using selective agonists and antagonists.
Key results:
Addition of histamine to TNFα-stimulated BEAS-2B cells maximally potentiated NF-κB-dependent transcription 1.8 fold, whereas IL-6 and IL-8 protein release were enhanced 7.3- and 2.7-fold respectively. These responses were, in part, NF-κB-dependent and were associated with 2.6- and 1.7-fold enhancements of IL-6 and IL-8 mRNA expression. The H1 receptor antagonist, mepyramine, caused a rightward shift in the concentration-response curves of TNFα-induced NF-κB-dependent transcription (pA2=9.91) and release of IL-6 (pA2=8.78) and IL-8 (pA2=8.99). Antagonists of histamine H2, H3 and H4 receptors were without effect. Similarly, H3 and H4 receptor agonists did not affect TNFα-induced NF-κB-dependent transcription, or IL-6 and IL-8 release at concentrations below 10 μM. The anti-inflammatory glucocorticoid, dexamethasone, inhibited the histamine enhanced NF-κB-dependent transcription and IL-6 and IL-8 release.
Conclusions and implications:
Potentiation of NF-κB-dependent transcription and inflammatory cytokine release by histamine predominantly involves receptors of the H1 receptor subtype. These data support an anti-inflammatory role for H1 receptor antagonists by preventing the transcription and release of pro-inflammatory cytokines.
Keywords: histamine, NF-κB, inflammation, IL-6, IL-8, mepyramine, glucocorticoid, BEAS-2B epithelial cells
Introduction
Chronic inflammation is a hallmark of airway diseases, including asthma, chronic obstructive pulmonary disease and allergic rhinitis (Chung, 2006; Jeffery and Haahtela, 2006). The production of histamine (2-[4-imidazole]-ethylamine) through activation of histidine decarboxylase in mast cells and basophils and, to a lesser extent, in neurons and lymphocytes, plays a major role in the establishment of the inflamed state (Jutel et al., 2005). In 1911, histamine was shown to have smooth muscle-stimulating and vasodepressor properties, which mimicked symptoms observed during anaphylaxis (Dale and Laidlaw, 1911). Since this time, histamine has been found to exert its effects via four different G-protein-coupled receptors, termed H1–H4 (Akdis and Simons, 2006; Alexander et al., 2006). These receptors can couple to different G-proteins and activate divergent signalling pathways. For example, the H1 histamine receptor couples via Gq/11 to phospholipase C and promotes the hydrolysis of phosphatidylinositol(4,5)bisphosphate into diacylglycerol and inositol(1,4,5)trisphosphate (Tilly et al., 1990; Li et al., 1995). This stimulates calcium mobilization and results in the activation of calcium- and diacylglycerol-dependent signalling molecules such as protein kinase C (Liu and Heckman, 1998). In contrast, the histamine H2 receptor is linked to Gs and couples to the adenylyl cyclase–cyclic AMP pathway to activate protein kinase A (Flamand et al., 2004). Both the H3 and H4 receptors have been shown to be linked to Gi/o and may inhibit adenylyl cyclase as well as activating mitogen-activated protein kinases (Lovenberg et al., 1999; Morse et al., 2001; Giovannini et al., 2003; Lim et al., 2005).
The transcription factor nuclear factor kappa B (NF-κB) promotes the transcription of over 150 genes, including cytokines, chemokines, adhesion molecules and inflammatory enzymes such as cyclooxygenase-2 (Barnes and Karin, 1997; Pahl, 1999). NF-κB typically exists as a heterodimer of two proteins, p65 (relA) and p50, which, in resting cells, is held in the cytoplasm through its association with one of the proteins from the inhibitor of κB (IκB) family (Karin and Ben-Neriah, 2000). Pro-inflammatory cytokines, such as tumour necrosis factor α (TNFα) or interleukin (IL)-1β, activate the IκB kinase complex, which then phosphorylates the IκB protein, usually IκBα, to target it for ubiquitination and degradation by the 26S proteosome (Karin and Ben-Neriah, 2000). This reveals a nuclear localization signal allowing NF-κB to translocate into the nucleus where it binds to κB sites in the promoters of NF-κB-dependent genes to upregulate transcription (Barnes and Karin, 1997).
The airways epithelium extends from the nose to the terminal bronchioles and acts as the interface between the inhaled air and the respiratory system (Davies and Holgate, 2002). These cells are a point of first contact for pro-inflammatory insults, including airborne allergens, pollutants and pathogens that may exacerbate inflammatory airway diseases (Davies and Holgate, 2002). Importantly, epithelial cells contribute to disease exacerbations by producing pro-inflammatory mediators, cytokines, chemokines and expressing adhesion molecules (Mills et al., 1999). The epithelial layer is also targeted by inhaled therapies such as glucocorticoids, making this cell type a critical site of therapeutic action.
In addition to inducing bronchoconstriction and oedema, histamine is also implicated in the induction of inflammatory cytokines, chemokines and other pro-inflammatory factors (Marone et al., 2003). In the present study, histamine, acting primarily via the H1 receptor on BEAS-2B bronchial epithelial cells, potentiated TNFα-induced NF-κB-dependent transcription and the release of IL-6 and IL-8.
Materials and methods
Cell culture, cytokines and drugs
BEAS-2B cells were originally isolated from normal human bronchial epithelial cells obtained from a non-cancerous individual and which had been infected with an adenovirus 12-SV40 virus hybrid (Ad12SV40) (Reddel et al., 1988). An individual colony (BEAS-2B) with unlimited proliferative potential was obtained, which did not show evidence of viral production and which retained the morphological characteristics of bronchial epithelial cells. BEAS-2B cells were grown to confluence in 6 or 24-well plates using Dulbecco's modified Eagle's medium/F12 medium (Invitrogen, Burlington, ON, Canada) in 10% fetal calf serum, as described previously (Catley et al., 2004). Cells were cultured overnight in serum-free media before changing to fresh serum-free media containing drugs and stimuli.
Adenovirus infection
BEAS-2B cells were either infected with empty (null) Ad5 expression vector or a vector expressing an IκBα protein with an N-terminal deletion (IκBαΔN) (Krappmann et al., 1996). This deletion causes the IκBα to be a constitutive inhibitor of NF-κB. Cells were infected with Ad5-IκBαΔN and null adenoviruses at a multiplicity of infection (MOI) of 30, which results in infection of >95% of cells, as described previously (Meja et al., 2004).
NF-κB reporter cells and luciferase assay
The NF-κB-dependent reporter pGL3.neo.TATA.3κBu was based on the parent vector pGL3.neo.TATA, which contains a neomycin-resistance gene and a modified minimal β-globin promoter driving luciferase (Catley et al., 2004). This was digested at the SmaI site, upstream of the minimal β-globin promoter, and a double-stranded oligonucleotide (sense strand: 5′-AGGGGATTCCCTAGGGGATTCCCTAGGGGATTCCCT-3′), containing three copies of the upstream NF-κB site (underlined) derived from the human cyclooxygenase-2 promoter (Newton et al., 1997), was inserted to produce pGL3.neo.TATA.3κBu (referred to as 3κBu-luc). A mutated version (3κBu(mut)-luc) of the reporter was generated as above, but using modified oligonucleotides (sense strand: 5′-AGGccATTCCCTAGGccATTCCCTAGGccATTCCCT-3′) (mutated bases in lower case), which do not bind NF-κB (Newton et al., 1997). In preliminary experiments, neither the mutated construct nor the parent construct, pGL3.neo.TATA, was responsive to TNFα (data not shown). This indicates that TNFα responsiveness of the 3κBu-luc construct is due to the NF-κB-binding sites. Stably transfected BEAS-2B reporter cells were generated by transient transfection of 8 μg of each plasmid into pre-confluent BEAS-2B cells in T-162 flasks. Cells were cultured in the presence of 0.1 mg ml−1 G-418 until foci of resistant cells appeared. These were harvested to create a heterogeneous population, randomized for integration site. The lines were expanded for generation of stocks and experimental procedures. Stably transfected BEAS-2B cells with a cyclic AMP response element (CRE)-dependent luciferase reporter that contains six tandem CRE motifs upstream of a β-globin promoter driving a luciferase gene were grown as described previously (Meja et al., 2004). Confluent BEAS-2B reporter cells in 24-well plates were changed to serum-free medium and treated for 6 h before harvesting in 1 × reporter lysis buffer (Promega, Madison, WI, USA). Luciferase activity was measured using the luciferase assay system (Promega).
Reverse transcription-PCR
RNA isolation, reverse transcription, primers, PCR conditions and cycling parameters for glyceraldehyde-3-phosphate dehydrogenase (GAPDH) were as described previously (Bergmann et al., 2000). Primer pairs (5′>3′) for the histamine H1 receptor were AACGGACTCAGATACCACCA (forward) and TCATTGCACAAGGGGTAGAT (reverse), and cycling parameters were 94 °C for 30 s, 58 °C for 30 s and 72 °C for 30 s for 30 cycles. Histamine H2 receptor primers were TGACCAATTGTTTCA TCGTG (forward) and TGAAGTAGGGAAACCAGCAG (reverse), and cycling parameters were 94 °C for 30 s, 58 °C for 30 s and 72 °C for 30 s for 44 cycles. Histamine H3 receptor primers were CCTCCTCTGCCTTCAACATC (forward) and GGAAGGGCGTAAAGAACTCC (reverse), and cycling parameters were 94 °C for 30 s, 58 °C for 30 s and 72 °C for 30 s for 44 cycles. Histamine H4 receptor primers were TCGAATGGGATTTTGGAAAG (forward) and TGGCCCATTCACTAAGAAGG (reverse), and cycling parameters were 94 °C for 30 s, 58 °C for 30 s and 72 °C for 30 s for 44 cycles. Following amplification, PCR products (10 μl) were size fractionated on 1.8% agarose gels stained with ethidium bromide.
Real-time TaqMan PCR analysis
After cDNA synthesis, TaqMan PCR was performed using 2.5 μl of cDNA in a reaction volume of 20 μl, essentially according to the manufacturer's specification (Applied Biosystems Inc., Foster City, CA, USA) using a pre-made master mix and an ABI 7900HT instrument (Applied Biosystems). Analysis of GAPDH was carried out using the validated off-the-shelf assay 432631E (Applied Biosystems). IL-8 was amplified using the primers (5′>3′) CTGGCCGTGGCTCTCTTG (forward) and TTAGCACTCCTTGGCAAAACTG (reverse) with the 5-carboxyfluorescein/5-carboxytetramethylrhodamine-linked probe 5′-CCTTCCTGATTTCTGCAGCTCTGTGTGAA-3′. IL-6 was amplified using the primers (5′>3′) TGGCTGAAAAAGATGGATGCT (forward) and AACTCCAAAAGACCAGTGATGATTT (reverse) with the 5-carboxyfluorescein/minor-groove-binding protein-linked probe CAATGAGGAGACTTG. All primers were designed using Primer Express version 2 software (Applied Biosystems). Samples were analysed in duplicate and relative cDNA concentrations were determined from a cDNA standard curve that was analysed simultaneously with the test samples.
Western blot analysis
Cells were harvested in 1 × reporter lysis buffer (Promega) containing 1 × Complete Protease Inhibitor Cocktail (Roche, Laval, QC, Canada). Samples were run on 4–12% gradient SDS polyacrylamide gels (Invitrogen) and transferred to Hybond-ECL nitrocellulose paper (GE Healthcare Bio-Sciences Inc., Baie d'Urfé, QC, Canada) using standard techniques. Membranes were probed with an anti-GAPDH (no. 4699-9555) (AbD Serotec, Raleigh, NC, USA) and anti-IκBα (sc-371) (Santa Cruz Biotechnology, Santa Cruz, CA, USA) antibodies. Proteins were visualized using ECL (GE Healthcare Bio-Sciences Inc.).
Determination of antagonist affinity
Antagonist affinity was determined by nonlinear regression (Lew and Angus, 1995; Motulsky and Christopoulos, 2005). Each pair of concentration–response curves (that is, the control concentration–response curve and the concentration–response curve constructed in the presence of antagonist) was fitted simultaneously to a form of the Hill and Gaddum–Schild equation (Equation (1)) derived by Waud et al. (1978). Thus, where [A] and [B] are the molar concentrations of agonist and antagonist, respectively, S is the Schild slope factor and pA2 is the affinity of the antagonist when S=1, which is equivalent to the pKB. To determine whether S deviated significantly from unity, the mean family of response (E) to concentration ([A]) (that is, E/[A]) curves that made up the entire experiment was fitted globally to the Hill and Gaddum–Schild equation (Equation (1)) under two conditions: one where S was constrained to a constant equal to 1 and the other where it was a shared value for all data sets (Motulsky and Christopoulos, 2005). The F-test was applied to determine statistically which equation gave the best fit and this was then used for the analysis (Prizm 4, GraphPad Software Inc., San Diego, CA, USA). In each case, S did not deviate from unity and, therefore, was constrained to a value of 1.
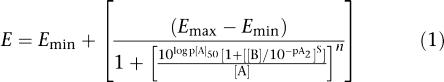 |
Statistics
Data are presented as means±s.e.m. of n independent observations. Comparison between groups of experimental data was performed using either a one-way analysis of variance with a Bonferroni post-test or Student's t-test, as appropriate. Significant differences between means are shown as *P<0.05, **P<0.01 and ***P<0.001.
Materials
TNFα (R&D Systems, Hornby, ON, Canada) and histamine (Sigma, Oakville, ON, Canada) were dissolved in sterile phosphate-buffered saline. Mepyramine (Sigma), tiotidine (Tocris Bioscience, Ellisville, MI, USA), thioperamide (Tocris Bioscience), Rα-methylhistamine (Tocris Bioscience) and 4-methylhistamine (Tocris Bioscience) were all dissolved in water; JNJ7777120 (Sigma) was dissolved in dimethylsulphoxide. Final concentrations of dimethylsulphoxide added to cells were <0.1% and this had no effect on any of the responses (data not shown).
Results
Role of histamine and H1 receptor in pro-inflammatory gene expression
Stimulation of BEAS-2B cells with histamine alone had no significant effect on either IL-6 or IL-8 expression at 1 and 2 h post-stimulation (Table 1). However, at 6 h after histamine stimulation, both IL-6 and IL-8 were significantly induced by 1.9- and 1.8-fold, respectively (Table 1). At 12 and 24 h, while IL-8 production remained significantly increased over basal levels, histamine-induced IL-6 was not significantly induced (Table 1). In addition, the enhancement of IL-6 and IL-8 release by TNFα in the presence of histamine was maximal at 6 h. In the presence of TNFα, histamine concentration-dependently increased IL-6 (EC50=4.69 × 10−7 M) and IL-8 (EC50=2.59 × 10−7 M) release from BEAS-2B cells to a maximum of 7.3- and 2.7-fold, respectively, at 100 μM (Figures 1a and c). This enhancement occurred at all concentrations of TNFα studied (Figures 1b and d). Prior addition of the H1 receptor antagonist, mepyramine, at 5 nM, had no obvious effect on either IL-6 or IL-8 release (data not shown), whereas at 50 nM, mepyramine resulted in 22- and 45-fold rightward shifts in the EC50 values to histamine for IL-6 and IL-8, respectively, and produced pA2 values of 8.78 and 8.99, respectively.
Table 1.
Time-course analysis of IL-6 and IL-8 release in BEAS-2B cells
| Time (h) |
IL-6 release (pg ml−1) |
IL-8 release (pg ml−1) |
||||||
|---|---|---|---|---|---|---|---|---|
| NS | His | TNFα | TNFα+His | NS | His | TNFα | TNFα+His | |
| 1 | 11.2 (±2.7) | 13.1 (±4.9) | 22.6 (±4.4) | 48.0 (±13.0) | 7.5 (±1.4) | 10.3 (±1.2) | 36.3 (±5.0) | 65.1 (±8.3) |
| 2 | 9.4 (±2.7) | 14.6 (±2.1) | 65.9 (±11.3) | 135 (±8.1) | 6.8 (±1.3) | 9.0 (±0.5) | 101 (±14.0) | 172 (±14.2) |
| 6 | 10.9 (±3.1) | 21.2** (±2.1) | 180* (±35.2) | 666*** (±68.5)††† | 6.9 (±0.7) | 12.4* (±2.5) | 503*** (±57.3) | 1980*** (±351)††† |
| 12 | 22.3 (±5.6) | 33.6 (±5.2) | 796*** (±87.0) | 1130*** (±92.4)††† | 6.5 (±1.0) | 13.7** (±1.6) | 3020*** (±328) | 6043*** (±913)††† |
| 24 | 30.7 (±5.5) | 35.7 (±3.8) | 1140*** (±129) | 1310*** (±89.0) | 7.6 (±0.2) | 14.0*** (±1.0) | 2950*** (±967) | 3620*** (±257)††† |
BEAS-2B cells were either not stimulated (NS) or were treated with either histamine (100 μM) (His), tumour necrosis factor (TNF) α (10 ng ml−1) or a combination of TNFα and histamine as indicated. Cells were harvested at the indicated times and IL-6 and IL-8 release was measured by enzyme-linked immunosorbent assay. Data (n=6) are expressed as means±s.e.m. Significance between NS and each of His, TNFα or TNFα+His is indicated as *P<0.05, **P<0.01 and ***P<0.001. Significance between TNFα and TNFα+His is indicated as †††P<0.001.
Figure 1.

Role of histamine and the histamine H1 receptor in tumour necrosis factor (TNF) α-stimulated release of interleukin (IL)-6 and IL-8. (a and c) BEAS-2B cells were stimulated with TNFα (10 ng ml−1) and various concentrations of histamine (0.03–1000 μM) in the presence or absence of mepyramine (50 nM). (b and d) BEAS-2B cells were stimulated with various concentrations of TNFα (0.003–30 and 0.03–30 ng ml−1 for IL-6 and IL-8, respectively) in the presence or absence of histamine (100 μM). Cells were harvested after 6 h and supernatants were analysed by enzyme-linked immunosorbent assay for IL-6 and IL-8 release. Data (n=6) are expressed either as a percentage of cells treated with TNFα alone or as picograms per millilitre. All data are plotted as means±s.e.m. and basal levels of IL-6 and IL-8 were 5.27±0.58 and 28.52±2.81 pg ml−1, respectively.
To investigate mechanisms involved in histamine-dependent potentiation of IL-6 and IL-8 release, cells were treated with TNFα (10 ng ml−1) in the presence or absence of histamine (100 μM). RNA was harvested at 6 h and subjected to real-time PCR. In each case, histamine potentiated the TNFα-induced IL-6 and IL-8 mRNA expression 2.6- and 1.7-fold, respectively (Figure 2).
Figure 2.

Effect of histamine on interleukin (IL)-6 and IL-8 mRNA expression. BEAS-2B cells were either left untreated or treated with histamine (100 μM) or tumour necrosis factor (TNF) α (10 ng ml−1), or a combination of histamine (100 μM) plus TNFα (10 ng ml−1). Cells were harvested after 6 h and TaqMan real-time PCR was carried out for IL-6, IL-8 and glyceraldehyde-3-phosphate dehydrogenase (GAPDH). Relative cDNA concentrations (n=3) were normalized to GAPDH and are plotted as means±s.e.m.
Since the expression of IL-6 and IL-8 is dependent upon the activation of NF-κB in epithelial cells (Catley et al., 2005; Newton et al., 2007), the effect of histamine on NF-κB-dependent transcription in BEAS-2B 3κBu-luc reporter cells was examined. Consistent with the cytokine results described above, histamine potentiated NF-κB-dependent transcription in a concentration-dependent manner (EC50=1.90 × 10−6 M), with a maximum induction of 1.8-fold (Figure 3a). Histamine alone also increased luciferase activity twofold over basal activity and this effect was maintained for all concentrations of TNFα (Figure 3b). Pre-incubation (1 h) with 50 nM mepyramine prevented the potentiation of TNFα-stimulated luciferase gene expression by histamine at concentrations of less than 100 μM (Figure 3a). Treatment with 5 nM of mepyramine caused a 19-fold rightward shift of the histamine response curve (Figure 3a). Analysis of the shift in the EC50 value for the histamine in the presence of both 50 nM (Figure 3a) and 5 nM (Figure 3a) mepyramine gave rise to a pA2 value of 9.91.
Figure 3.

Role of histamine and the histamine H1 receptor in tumour necrosis factor (TNF) α-stimulated nuclear factor (NF)-κB-dependent transcription. BEAS-2B 3κBu cells were stimulated with (a) TNFα (10 ng ml−1) and various concentrations of histamine (0.03–1000 μM) in the presence or absence of either 5 or 50 nM mepyramine and (b) various concentrations of TNFα (0.001–10 ng ml−1) in the presence or absence of histamine (100 μM). Cells were harvested after 6 h and lysates analysed for luciferase activity. Data (n=5) are expressed as either a percentage of cells treated with TNFα alone or as fold activation of untreated cells. All data are plotted as means±s.e.m.
Role of NF-κB in IL-6 and IL-8 release
The role of NF-κB was investigated using the dominant inhibitor IκBαΔN which was delivered using adenoviral-mediated gene transfer. As before, NF-κB-dependent transcription was strongly induced by TNFα (10 ng ml−1) and this was reduced to basal levels by increasing MOIs of the IκBαΔN-expressing virus (Figure 4). A null (empty) adenoviral vector had no significant effect at equivalent MOIs (Figure 4).
Figure 4.

Effect of dominant IκBαΔN on nuclear factor (NF)-κB-dependent transcription in BEAS-2B cells. BEAS-2B 3κBu cells were grown to approximately 70% confluence before infection either with a dominant IκBαΔN-expressing adenovirus or a null adenoviral expression vector at the indicated multiplicities of infection (MOI). After 24 h, cells were changed to serum-free media and incubated overnight before being either not stimulated or stimulated with TNFα (10 ng ml−1). After 6 h, cells were harvested for luciferase assay. Data (n=6) are plotted as a percentage of tumour necrosis factor (TNF) α-stimulated cells as means±s.e.m.
To examine the role of NF-κB in TNFα, and TNFα plus histamine-induced IL-6 and IL-8 expression, BEAS-2B 3κBu-luc cells were treated with IκBαΔN virus or null virus at an MOI of 30. Consistent with the above data, TNFα-induced NF-κB reporter activity was reduced to near basal levels by the IκBαΔN adenovirus (Figure 5a). Similarly, TNFα-induced release of IL-6 and IL-8 was also significantly reduced by IκBαΔN, which confirmed dependence of these responses on the activation of NF-κB (Figure 5a). Likewise, cells treated with histamine alone appeared to show small increases in IL-6 and IL-8 release, which decreased in the presence of the IκBαΔN adenovirus. However, this was again at the limit of detection and no significance was attained. The addition of Ad5-IκBαΔN also significantly repressed TNFα plus histamine-induced luciferase activity, IL-6 and IL-8 release (Figure 5a). However, while both luciferase activity and IL-8 were reduced to near basal levels, IL-6 release was reduced by 43% (Figure 5a). In all cases, the null adenovirus was without effect (Figure 5a). Infection of BEAS-2B cells by the IκBαΔN adenovirus was confirmed by western blot analysis for IκBα (Figure 5b). IκBαΔN expression was identified as a band of greater electrophoretic mobility than endogenous IκBα (Figure 5b). Additionally, whereas cells treated with TNFα revealed no endogenous IκBα, cells treated with histamine alone exhibited similar IκBα expression to untreated cells, suggesting that histamine had no marked effect on the IκB kinase–IκBα activation pathway (Figure 5b).
Figure 5.

Role of nuclear factor (NF)-κB in the histamine potentiation of luciferase activity and interleukin (IL)-6 and IL-8 release. BEAS-2B 3κBu cells grown to 70% confluence in 24-well plates were either left uninfected or were infected at a multiplicity of infection (MOI) of 30 with either a dominant IκBαΔN-expressing adenovirus or a null adenoviral expression vector. After 24 h, cells were changed to serum-free media and incubated overnight before being stimulated with either histamine (100 μM) (His), TNFα (10 ng ml−1) or a combination of TNFα (10 ng ml−1) plus histamine (100 μM) (T+H) as indicated. After 6 h, cells were harvested and supernatants were analysed for IL-6 and IL-8 expression. Cell lysates were analysed for (a) luciferase activity and (b) expression of IκBα and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) by western blot. Data (n=6) are plotted as a percentage of TNFα-stimulated naïve cells as means±s.e.m. and basal levels of IL-6 and IL-8 were 7.61±0.91 and 13.62±2.41 pg ml−1, respectively.
Expression of histamine receptors in BEAS-2B cells
The expression of histamine receptor mRNA in BEAS-2B cells was determined using conventional end-point reverse transcription-PCR. Analysis of either untreated or TNFα-treated cells revealed constitutive expression of H1 receptor mRNA (Figure 6). Histamine H2 receptor mRNA was not detected at 44 cycles of amplification (Figure 6). Despite a DNase digestion step (Qiagen, Mississauga, ON, Canada) in the preparation of RNA, further cycles of amplification resulted in detection of product in both cDNA as well as in RNA samples, which had not been subject to reverse transcription (data not shown). We are, therefore, unable to exclude the possibility that low levels of H2 receptor mRNA may exist, but are masked by the genomic signal arising from the intronless H2 receptor gene (see National Centre for Biotechnology Information (NCBI) accession no. NM_022304). Transcripts encoding the histamine H3 and H4 receptor subtypes were also detected in unstimulated cells, but were absent in cells treated with TNFα (10 ng ml−1) (Figure 6).
Figure 6.

Expression of histamine receptors in BEAS-2B cells. BEAS-2B cells were either not stimulated or were treated with tumour necrosis factor (TNF) α (10 ng ml−1). After 6 h, RNA was isolated and reverse transcription-PCR in the presence or absence of the AMV reverse transcriptase (AMV RT), as indicated, was carried out using primers for the histamine H1, H2, H3 and H4 receptors as well as glyceraldehyde-3-phosphate dehydrogenase (GAPDH). In addition, water and genomic DNA were analysed in parallel. Ethidium bromide-stained gels representative of three such experiments are shown.
No role for the H2 receptor in the potentiation of NF-κB-dependent transcription and cytokine release
To functionally test for the presence of the Gs-coupled histamine H2 receptor subtype, BEAS-2B cells harbouring a CRE-dependent luciferase reporter construct were treated with histamine (from 100 nM to 1 mM). As shown in Figure 7a, histamine activated CRE-dependent transcription in a concentration-dependent manner (EC50=1.41 × 10−5 M), with a maximum response of 2.4-fold (Figure 7a). In contrast, the β2-adrenoceptor agonist salbutamol stimulated luciferase activity by 13- or 14-fold, indicating that the responsiveness of BEAS-2B cells to histamine was in fact relatively modest (Figure 7b). Prior addition of the histamine H2 receptor-selective antagonist, tiotidine (5 μM) to either unstimulated cells or cells treated with salbutamol had no significant effect on CRE-dependent transcription (Figure 7b). Conversely, histamine-stimulated CRE-dependent transcription was significantly inhibited to a level that was not significantly different from basal level by the prior addition of tiotidine to cells (Figure 7b).
Figure 7.

Effect of histamine or a β2-adrenoceptor agonist on cyclic AMP response element (CRE)-dependent transcription. (a) BEAS-2B CRE reporter cells were either left unstimulated or were stimulated with various concentrations of histamine (0.1–1000 μM). (b) BEAS-2B CRE reporter cells were either left unstimulated or were stimulated with either salbutamol (1 μM) or histamine (100 μM) in the presence or absence of tiotidine (5 μM). In each case, cells were harvested after 6 h and luciferase activity was analysed. Data (n=3–5) are plotted as the fold activation of unstimulated cells as means±s.e.m.
To investigate a role for H2-dependent signalling in the potentiation of NF-κB-dependent transcription by histamine and on the release of IL-6 and IL-8, BEAS-2B cells were treated with TNFα and increasing concentrations of histamine in the presence or absence of tiotidine (5 μM). Tiotidine was without any significant effect on any of these histamine-induced responses (Figure 8).
Figure 8.

Effect of the histamine H2 receptor antagonist tiotidine on the histamine potentiation of nuclear factor (NF)-κB-dependent transcription and release of interleukin (IL)-6 and IL-8. BEAS-2B 3κBu cells were incubated in the presence or absence of 5 μM tiotidine. After 1 h, cells were stimulated with tumour necrosis factor (TNF) α (10 ng ml−1) in the presence of increasing concentrations of histamine. Cells were harvested after 6 h and were analysed for luciferase activity. In addition, supernatants were analysed for IL-6 and IL-8 release. Data (n=4–7) are plotted as a percentage of TNFα-stimulated cells as means±s.e.m. Basal levels of IL-6 and IL-8 were 6.33±0.54 and 34.41±1.91 pg ml−1, respectively.
Role of histamine H3 and H4 receptors in potentiating NF-κB-dependent transcription and cytokine release
The effect of H3 and H4 receptors in mediating the effects of histamine was investigated using the dual-selective H3/H4 receptor antagonist thioperamide and the selective H4 receptor antagonist JNJ7777120. BEAS-2B 3κBu-luc cells were stimulated with 10 ng ml−1 of TNFα in the presence of 30 μM histamine. Co-incubation with increasing concentrations of thioperamide or JNJ7777120 had no significant effect on luciferase activity or on IL-6 and IL-8 release (Figure 9a). However, stimulation with TNFα and high concentrations (30–100 μM) of either the selective H3 receptor agonist Rα-methylhistamine or the selective H4 receptor agonist 4-methylhistamine significantly increased TNFα-induced release of IL-6 and IL-8 (Figure 9b). Neither Rα-methylhistamine nor 4-methylhistamine agonist promoted NF-κB-dependent luciferase activity (Figure 9b).
Figure 9.

Role of the H3 and H4 histamine receptors in the histamine potentiation of nuclear factor (NF)-κB-dependent transcription and interleukin (IL)-6 and IL-8 release. (a) BEAS-2B 3κBu cells were incubated in the presence or absence of various concentrations of either thioperamide or JNJ7777120. After 1 h, cells were stimulated with a combination of tumour necrosis factor (TNF) α (10 ng ml−1) plus histamine (30 μM). (b) Cells were treated with TNFα (10 ng ml−1) and increasing concentrations of either Rα-methylhistamine or 4-methylhistamine. Cells were harvested after 6 h and lysates were analysed for luciferase activity. In addition, supernatants were analysed for IL-6 and IL-8 release. Data (n=4–7) are expressed (a) as a percentage of TNFα plus histamine-stimulated cells, where basal levels of IL-6 and IL-8 were 7.52±1.42 and 16.03±3.04 pg ml−1, respectively, or (b) as a percentage of TNFα-stimulated cells, where basal levels of IL-6 and IL-8 were 8.09±0.74 and 13.04±0.93 pg ml−1, respectively. All data are plotted as means±s.e.m.
Effect of the glucocorticoid dexamethasone on NF-κB, IL-6 and IL-8
To examine the effect of anti-inflammatory glucocorticoids on histamine-enhanced inflammatory responses, BEAS-2B cells were pretreated with a maximally effective concentration (1 μM) of dexamethasone prior to stimulation with TNFα and histamine. Under these conditions, NF-κB-dependent transcription induced by TNFα or TNFα plus histamine was decreased by 33 and 44%, respectively (Figure 10). In contrast, inhibition of both IL-6 and IL-8 release was considerably greater, with TNFα and TNFα plus histamine-induced IL-6 being repressed by 65 and 70%, respectively, whereas IL-8 release was reduced by ∼90% for both treatments (Figure 10).
Figure 10.

Effect of dexamethasone on the histamine potentiation of nuclear factor (NF)-κB-dependent transcription and on the release of interleukin (IL)-6 and IL-8. BEAS-2B 3κBu cells were incubated in the presence or absence of dexamethasone (1 μM) for 1 h before stimulation with either tumour necrosis factor (TNF) α (10 ng ml−1) or TNFα (10 ng ml−1) plus histamine (100 μM). Cells were harvested after 6 h and lysates were analysed for luciferase activity. In addition, supernatants were analysed for IL-6 and IL-8 release. Data (n=5–12) are expressed as a percentage of cells treated with TNFα alone as means±s.e.m. Basal levels of IL-6 and IL-8 were 7.80±0.69 and 21.57±1.26 pg ml−1, respectively.
Discussion and conclusions
Previous studies in several different cell types have demonstrated NF-κB activation and release of cytokines elicited by histamine acting via the H1 receptor subtype (Bruysters et al., 2004; Wu et al., 2004; Matsubara et al., 2005; Jin et al., 2006; Muller et al., 2006). However, whereas inflammation is complex and involves multiple mediators, including histamine and pro-inflammatory cytokines, most investigations have examined the effects of histamine acting alone. In the current study, we examined the ability of histamine to enhance pro-inflammatory responses in BEAS-2B cells elicited by TNFα as a representative, but key, inflammatory cytokine. Thus, as described by Muller et al. (2006), stimulation of BEAS-2B cells with histamine alone induced IL-6 and IL-8 production. However, this response was very modest when compared with cells treated with TNFα. Likewise, histamine alone produced little or no activation of an NF-κB reporter construct, yet significantly (synergistically) enhanced both TNFα-induced NF-κB-dependent transcription and TNFα-induced release of IL-6 and IL-8 at 6 h. For IL-6 and IL-8 release, this effect was maximal at 6 h, whereas by 24 h, the histamine-dependent enhancement had disappeared for IL-6 and was greatly reduced for IL-8. Reasons for this are not presently clear, but it is possible that histamine, in addition to stimulating positive regulatory processes, may also lead to enhanced feedback control processes and these may attenuate the responses with increasing time.
Similar enhancement phenomena have been described in coronary artery endothelial cells where TNFα- and LPS-induced IL-6 and IL-8 release was also enhanced by histamine (Li et al., 2001). As in our present study, maximal responses were achieved at ∼10 μM histamine and these were blocked by the H1 receptor antagonist, diphenhydramine but not the H2 receptor antagonist famotidine. Likewise, lipoteichoic acid- and peptidoglycan-induced IL-6 release was also increased by histamine in a diphenhydramine-sensitive, but famotidine-insensitive manner (Talreja et al., 2004). In both these studies, the enhancing effect of histamine was mediated through the H1 receptor subtype and involved NF-κB-dependent transcription. However, in our current study, the apparent affinity of the H1 receptor antagonist mepyramine was ∼10-fold higher for blocking histamine-induced NF-κB-dependent transcription (pA2=9.91) than for suppressing the release of IL-6 and IL-8 (pA2=8.78 and 8.99, respectively). The pA2 of mepyramine on NF-κB-dependent transcription is broadly consistent with previously published values (Tayo and Bevan, 1986; Impicciatore et al., 1987; Kamei et al., 1990; Djuric and Andjelkovic, 1995). However, the lower affinity of mepyramine, when IL-6 and IL-8 release is used as a measure, suggests that while the enhancement of NF-κB-dependent transcription is entirely H1 receptor-dependent, the enhancement by histamine of IL-8 and, particularly, IL-6 release may also involve additional histamine receptor subtypes. This supposition is supported by examining the role of NF-κB in the histamine-dependent enhancement of IL-6. Thus, the dominant inhibitor IκBαΔN completely suppressed NF-κB-dependent transcription and IL-8 release, but only inhibited IL-6 release by ∼50% in cells stimulated with TNFα plus histamine. Analysis of the IL-6 promoter has revealed not only the presence of an NF-κB site (Libermann and Baltimore, 1990) but also interferon regulatory factor-1 (IRF-1)- (Faggioli et al., 1997), activator protein-1 (AP-1)- (Dendorfer et al., 1994), CCAAT/enhancer-binding protein (C/EBP)- (Isshiki et al., 1990; Akira and Kishimoto, 1997) and Sp1-binding sites (Kang et al., 1996; Armenante et al., 1999), through which histamine could potentially act. In addition, histamine potentiation of IL-6 mRNA was no more than 3-fold compared to 5–7-fold for protein release. Thus, histamine may also modulate IL-6 release via post-transcriptional processes, including changes in mRNA stability, translation or secretion.
In the current study, mRNA for the histamine H1 receptor was constitutively expressed and was the most readily detectable, suggesting that the H1 receptor is the most abundant subtype expressed by BEAS-2B cells. In contrast, our inability to detect mRNA for the H2 receptor gene correlated with a lack of effect of tiotidine on the histamine-dependent enhancements of NF-κB, IL-6 and IL-8. However, this does not preclude the existence of H2 receptors on the cell, by virtue of low levels of H2 receptor mRNA. Indeed, histamine activation of the CRE reporter construct, and inhibition by tiotidine, suggests that low levels of H2 receptor may be present. Analysis of H3 and H4 receptor mRNA revealed basal expression that was profoundly downregulated by TNFα. These data suggest the existence of novel negative control mechanisms. In this respect, the downregulation of H4 receptor mRNA was particularly surprising, since promoter analysis reveals binding sites for inflammatory factors, including both AP-1 and NF-κB (Coge et al., 2001).
Despite the lack of effect of H3 and H4 receptor antagonists on the histamine enhancement of inflammatory gene expression, this does not necessarily exclude a role for these receptors. Indeed, we observed that both H3 and H4 agonists, at high concentrations, increased the release of IL-6 and IL-8 in TNFα-stimulated cells. Likewise, a number of reports suggest a pro-inflammatory role of the H4 receptor (O'Reilly et al., 2002; Ling et al., 2004; Zhang et al., 2006), including the release of IL-6 in the mouse (Dunford et al., 2006). In contrast, activation of the histamine H3 receptor is mainly linked to anti-inflammatory effects, including inhibition of substance P release (Ohkubo et al., 1995), of neurogenic vascular inflammation (McLeod et al., 1998) and of paw oedema (Rouleau et al., 2000). In the current study, the concentrations of histamine H3 and H4 receptor agonists that elicited upregulation of IL-6 and IL-8 release were at the point where their selectivity of action must be called into question. Indeed, 4-methylhistamine, the selective H4 receptor agonist, which had the greatest effect on IL-6 and IL-8 release, has been shown in agonist displacement assays to have effects on not only the H4 receptor but also via H2 and H3 receptors, and at concentrations of 30–100 μM via the H1 receptor (Lim et al., 2005).
Given the above data, how does one reconcile the suggested involvement of other receptors in addition to H1 receptor-mediated events in the enhancement of IL-6 and IL-8 release? A number of possibilities for this discrepancy do exist and have been reported previously. First, the histamine H3 receptor exists as at least six splice variants (Wellendorph et al., 2002). In addition, the relatively newly discovered histamine H4 receptor was found through its homology to the H3 receptor and may also be expected to exist as multiple isoforms (although to date there are little data addressing this possibility). Furthermore, much of the data relating to H3 and H4 receptor agonists and antagonists relies upon transfected reporter systems and the overexpression of specific receptor isoforms (for example, see Lim et al., 2005). Thus, uncharacterized effects of agonists and antagonists against different receptor isoform variants could account for our current observations (Wellendorph et al., 2002). Second, it has been suggested that rather than existing as monomers, G-protein-coupled receptors may exist as homo- or heterodimers or even higher order oligomers (see Milligan, 2004 for review). Thus it is possible to speculate that as with other G-protein-coupled receptors, histamine receptors may also exist as dimers and differential dimerization could lead to nonstandard effects of agonists and antagonists.
In previous studies, we hypothesized the existence of a protein kinase C-dependent pathway, which potentiated TNFα-stimulated NF-κB-dependent transcription in A549 and BEAS-2B cells via enhanced transactivation rather than via increased NF-κB DNA binding (Catley et al., 2004). Since activation of the H1 receptor results in the production of diacylglycerol and release of calcium, both of which activate protein kinase C, the H1-dependent enhancement of NF-κB-dependent transcription described in the current study may also act through this same pathway. In addition, the finding that histamine-enhanced IL-6 expression is not completely NF-κB-dependent suggests additional mechanisms of potentiation. In terms of inflammatory responses, all these effects may be expected to enhance inflammation as a result of the increases in both IL-6 and IL-8 expression. While these data indicate the possible therapeutic utility of blockade of the histamine H1 receptor, we also find that the anti-inflammatory glucocorticoid dexamethasone is effective in reducing these enhanced responses. Therefore, our data suggest that the enhanced production of inflammatory mediators, in this case IL-6 and IL-8, will be effectively targeted by existing anti-inflammatory strategies that involve glucocorticoids.
In summary, we have documented a histamine-dependent increase in both TNFα-stimulated NF-κB-dependent transcription and the release of the pro-inflammatory mediators IL-6 and IL-8. These effects were determined to be wholly dependent, in the case of NF-κB-dependent transcription, and predominantly dependent, in the case of IL-6 and IL-8, on the histamine H1 receptor. In addition, the histamine-dependent enhancement of NF-κB-dependent transcription and IL-8 release was completely ablated by the dominant IκBαΔN, whereas IL-6 release was only partially inhibited, suggesting the existence of additional regulatory steps aside from NF-κB. However, antagonists of the H2, H3 and H4 histamine receptors had no impact on the histamine enhancement, and selective stimulation of the H3 and H4 receptors was effective at enhancing IL-6 and IL-8 release only at concentrations that may yield nonspecific effects. Finally, despite showing that histamine can contribute to the inflammatory response by increasing NF-κB activation and inflammatory cytokine release, these effects are effectively combated by glucocorticoids, suggesting that existing anti-inflammatory therapies may already repress these enhanced responses due to histamine.
Acknowledgments
This study was supported by an establishment grant from the Alberta Heritage Foundation for Medical Research (AHFMR) and operating funds from the Canadian Institutes of Health Research (CIHR). RN is an AHFMR Scholar and CIHR New Investigator. MAG is an AHFMR Senior Scholar.
Abbreviations
- CRE
cyclic AMP response element
- GAPDH
glyceraldehyde-3-phosphate dehydrogenase
- IκB
inhibitor of kappa B
- MOI
multiplicity of infection
- NF-κB
nuclear factor kappa B
- PKA
protein kinase A
- TNFα
tumour necrosis factor α
Conflict of interest
RN and MAG have received research funding from GlaxoSmithKline, AstraZeneca and Altana Pharmaceuticals. The authors are unaware of any other conflicts of interest.
References
- Akdis CA, Simons FE. Histamine receptors are hot in immunopharmacology. Eur J Pharmacol. 2006;533:69–76. doi: 10.1016/j.ejphar.2005.12.044. [DOI] [PubMed] [Google Scholar]
- Akira S, Kishimoto T. NF-IL6 and NF-kappa B in cytokine gene regulation. Adv Immunol. 1997;65:1–46. [PubMed] [Google Scholar]
- Alexander SP, Mathie A, Peters JA. Guide to receptors and channels, 2nd edition. Br J Pharmacol. 2006;147 Suppl 3:S1–S168. doi: 10.1038/sj.bjp.0706651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armenante F, Merola M, Furia A, Tovey M, Palmieri M. Interleukin-6 repression is associated with a distinctive chromatin structure of the gene. Nucleic Acids Res. 1999;27:4483–4490. doi: 10.1093/nar/27.22.4483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes PJ, Karin M. Nuclear factor-kappaB: a pivotal transcription factor in chronic inflammatory diseases. N Engl J Med. 1997;336:1066–1071. doi: 10.1056/NEJM199704103361506. [DOI] [PubMed] [Google Scholar]
- Bergmann M, Barnes PJ, Newton R. Molecular regulation of granulocyte macrophage colony-stimulating factor in human lung epithelial cells by interleukin (IL)-1beta, IL-4, and IL-13 involves both transcriptional and post-transcriptional mechanisms. Am J Respir Cell Mol Biol. 2000;22:582–589. doi: 10.1165/ajrcmb.22.5.3889. [DOI] [PubMed] [Google Scholar]
- Bruysters M, Pertz HH, Teunissen A, Bakker RA, Gillard M, Chatelain P, et al. Mutational analysis of the histamine H1-receptor binding pocket of histaprodifens. Eur J Pharmacol. 2004;487:55–63. doi: 10.1016/j.ejphar.2004.01.028. [DOI] [PubMed] [Google Scholar]
- Catley MC, Cambridge LM, Nasuhara Y, Ito K, Chivers JE, Beaton A, et al. Inhibitors of protein kinase C (PKC) prevent activated transcription: role of events downstream of NF-kappaB DNA binding. J Biol Chem. 2004;279:18457–18466. doi: 10.1074/jbc.M400765200. [DOI] [PubMed] [Google Scholar]
- Catley MC, Chivers JE, Holden NS, Barnes PJ, Newton R. Validation of IKK beta as therapeutic target in airway inflammatory disease by adenoviral-mediated delivery of dominant-negative IKK beta to pulmonary epithelial cells. Br J Pharmacol. 2005;145:114–122. doi: 10.1038/sj.bjp.0706170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung KF. Cytokines as targets in chronic obstructive pulmonary disease. Curr Drug Targets. 2006;7:675–681. doi: 10.2174/138945006777435263. [DOI] [PubMed] [Google Scholar]
- Coge F, Guenin SP, Rique H, Boutin JA, Galizzi JP. Structure and expression of the human histamine H4-receptor gene. Biochem Biophys Res Commun. 2001;284:301–309. doi: 10.1006/bbrc.2001.4976. [DOI] [PubMed] [Google Scholar]
- Dale HH, Laidlaw PP. Further observations on the action of beta-iminazolylethylamine. J Physiol. 1911;43:182–195. doi: 10.1113/jphysiol.1911.sp001464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies DE, Holgate ST. Asthma: the importance of epithelial mesenchymal communication in pathogenesis. Inflammation and the airway epithelium in asthma. Int J Biochem Cell Biol. 2002;34:1520–1526. doi: 10.1016/s1357-2725(02)00048-1. [DOI] [PubMed] [Google Scholar]
- Dendorfer U, Oettgen P, Libermann TA. Multiple regulatory elements in the interleukin-6 gene mediate induction by prostaglandins, cyclic AMP, and lipopolysaccharide. Mol Cell Biol. 1994;14:4443–4454. doi: 10.1128/mcb.14.7.4443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Djuric DM, Andjelkovic IZ. The evidence for histamine H3 receptor-mediated endothelium-dependent relaxation in isolated rat aorta. Mediators Inflamm. 1995;4:217–221. doi: 10.1155/S0962935195000354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunford PJ, O'Donnell N, Riley JP, Williams KN, Karlsson L, Thurmond RL. The histamine H4 receptor mediates allergic airway inflammation by regulating the activation of CD4+ T cells. J Immunol. 2006;176:7062–7070. doi: 10.4049/jimmunol.176.11.7062. [DOI] [PubMed] [Google Scholar]
- Faggioli L, Merola M, Hiscott J, Furia A, Monese R, Tovey M, et al. Molecular mechanisms regulating induction of interleukin-6 gene transcription by interferon-gamma. Eur J Immunol. 1997;27:3022–3030. doi: 10.1002/eji.1830271140. [DOI] [PubMed] [Google Scholar]
- Flamand N, Plante H, Picard S, Laviolette M, Borgeat P. Histamine-induced inhibition of leukotriene biosynthesis in human neutrophils: involvement of the H2 receptor and cAMP. Br J Pharmacol. 2004;141:552–561. doi: 10.1038/sj.bjp.0705654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giovannini MG, Efoudebe M, Passani MB, Baldi E, Bucherelli C, Giachi F, et al. Improvement in fear memory by histamine-elicited ERK2 activation in hippocampal CA3 cells. J Neurosci. 2003;23:9016–9023. doi: 10.1523/JNEUROSCI.23-27-09016.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Impicciatore M, Morini G, Chiavarini M, Barocelli E, Bordi F, Plazzi PV, et al. Are histamine receptors involved in the stimulant activities of thiazolylethylamines supposed as cyclic models of dimaprit. Agents Actions. 1987;20:262–264. doi: 10.1007/BF02074686. [DOI] [PubMed] [Google Scholar]
- Isshiki H, Akira S, Tanabe O, Nakajima T, Shimamoto T, Hirano T, et al. Constitutive and interleukin-1 (IL-1)-inducible factors interact with the IL-1-responsive element in the IL-6 gene. Mol Cell Biol. 1990;10:2757–2764. doi: 10.1128/mcb.10.6.2757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeffery PK, Haahtela T. Allergic rhinitis and asthma: inflammation in a one-airway condition. BMC Pulm Med. 2006;6 Suppl 1:S5. doi: 10.1186/1471-2466-6-S1-S5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin S, Tian D, Chen JG, Zhu LP, Liu SY, Wang DX. Passive sensitization increases histamine-stimulated calcium signaling and NF-kappaB transcription activity in bronchial epithelial cells. Acta Pharmacol Sin. 2006;27:708–714. doi: 10.1111/j.1745-7254.2006.00334.x. [DOI] [PubMed] [Google Scholar]
- Jutel M, Blaser K, Akdis CA. Histamine in allergic inflammation and immune modulation. Int Arch Allergy Immunol. 2005;137:82–92. doi: 10.1159/000085108. [DOI] [PubMed] [Google Scholar]
- Kamei C, Chung YH, Tasaka K. Influence of certain H1-blockers on the step-through active avoidance response in rats. Psychopharmacology (Berl) 1990;102:312–318. doi: 10.1007/BF02244096. [DOI] [PubMed] [Google Scholar]
- Kang SH, Brown DA, Kitajima I, Xu X, Heidenreich O, Gryaznov S, et al. Binding and functional effects of transcriptional factor Sp1 on the murine interleukin-6 promotor. J Biol Chem. 1996;271:7330–7335. doi: 10.1074/jbc.271.13.7330. [DOI] [PubMed] [Google Scholar]
- Karin M, Ben-Neriah Y. Phosphorylation meets ubiquitination: the control of NF-[kappa]B activity. Annu Rev Immunol. 2000;18:621–663. doi: 10.1146/annurev.immunol.18.1.621. [DOI] [PubMed] [Google Scholar]
- Krappmann D, Wulczyn FG, Scheidereit C. Different mechanisms control signal-induced degradation and basal turnover of the NF-kappaB inhibitor IkappaB alpha in vivo. EMBO J. 1996;15:6716–6726. [PMC free article] [PubMed] [Google Scholar]
- Lew MJ, Angus JA. Analysis of competitive agonist-antagonist interactions by nonlinear regression. Trends Pharmacol Sci. 1995;16:328–337. doi: 10.1016/s0165-6147(00)89066-5. [DOI] [PubMed] [Google Scholar]
- Li H, Choe NH, Wright DT, Adler KB. Histamine provokes turnover of inositol phospholipids in guinea pig and human airway epithelial cells via an H1-receptor/G protein-dependent mechanism. Am J Respir Cell Mol Biol. 1995;12:416–424. doi: 10.1165/ajrcmb.12.4.7695921. [DOI] [PubMed] [Google Scholar]
- Li Y, Chi L, Stechschulte DJ, Dileepan KN. Histamine-induced production of interleukin-6 and interleukin-8 by human coronary artery endothelial cells is enhanced by endotoxin and tumor necrosis factor-alpha. Microvasc Res. 2001;61:253–262. doi: 10.1006/mvre.2001.2304. [DOI] [PubMed] [Google Scholar]
- Libermann TA, Baltimore D. Activation of interleukin-6 gene expression through the NF-kappa B transcription factor. Mol Cell Biol. 1990;10:2327–2334. doi: 10.1128/mcb.10.5.2327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim HD, van Rijn RM, Ling P, Bakker RA, Thurmond RL, Leurs R. Evaluation of histamine H1-, H2-, and H3-receptor ligands at the human histamine H4 receptor: identification of 4-methylhistamine as the first potent and selective H4 receptor agonist. J Pharmacol Exp Ther. 2005;314:1310–1321. doi: 10.1124/jpet.105.087965. [DOI] [PubMed] [Google Scholar]
- Ling P, Ngo K, Nguyen S, Thurmond RL, Edwards JP, Karlsson L, et al. Histamine H4 receptor mediates eosinophil chemotaxis with cell shape change and adhesion molecule upregulation. Br J Pharmacol. 2004;142:161–171. doi: 10.1038/sj.bjp.0705729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu WS, Heckman CA. The sevenfold way of PKC regulation. Cell Signal. 1998;10:529–542. doi: 10.1016/s0898-6568(98)00012-6. [DOI] [PubMed] [Google Scholar]
- Lovenberg TW, Roland BL, Wilson SJ, Jiang X, Pyati J, Huvar A, et al. Cloning and functional expression of the human histamine H3 receptor. Mol Pharmacol. 1999;55:1101–1107. [PubMed] [Google Scholar]
- Marone G, Granata F, Spadaro G, Genovese A, Triggiani M. The histamine-cytokine network in allergic inflammation. J Allergy Clin Immunol. 2003;112:S83–S88. doi: 10.1016/s0091-6749(03)01881-5. [DOI] [PubMed] [Google Scholar]
- Matsubara M, Tamura T, Ohmori K, Hasegawa K. Histamine H1 receptor antagonist blocks histamine-induced proinflammatory cytokine production through inhibition of Ca2+-dependent protein kinase C, Raf/MEK/ERK and IKK/I kappa B/NF-kappa B signal cascades. Biochem Pharmacol. 2005;69:433–449. doi: 10.1016/j.bcp.2004.10.006. [DOI] [PubMed] [Google Scholar]
- McLeod RL, Aslanian R, del Prado M, Duffy R, Egan RW, Kreutner W, et al. Sch 50971, an orally active histamine H3 receptor agonist, inhibits central neurogenic vascular inflammation and produces sedation in the guinea pig. J Pharmacol Exp Ther. 1998;287:43–50. [PubMed] [Google Scholar]
- Meja KK, Catley MC, Cambridge LM, Barnes PJ, Lum H, Newton R, et al. Adenovirus-mediated delivery and expression of a cAMP-dependent protein kinase inhibitor gene to BEAS-2B epithelial cells abolishes the anti-inflammatory effects of rolipram, salbutamol, and prostaglandin E2: a comparison with H-89. J Pharmacol Exp Ther. 2004;309:833–844. doi: 10.1124/jpet.103.060020. [DOI] [PubMed] [Google Scholar]
- Milligan G. G protein-coupled receptor dimerization: function and ligand pharmacology. Mol Pharmacol. 2004;66:1–7. doi: 10.1124/mol.104.000497.. [DOI] [PubMed] [Google Scholar]
- Mills PR, Davies RJ, Devalia JL. Airway epithelial cells, cytokines, and pollutants. Am J Respir Crit Care Med. 1999;160:S38–S43. doi: 10.1164/ajrccm.160.supplement_1.11. [DOI] [PubMed] [Google Scholar]
- Morse KL, Behan J, Laz TM, West RE, Jr, Greenfeder SA, Anthes JC, et al. Cloning and characterization of a novel human histamine receptor. J Pharmacol Exp Ther. 2001;296:1058–1066. [PubMed] [Google Scholar]
- Motulsky H, Christopoulos A. Fitting Models to Biological Data using Linear and Nonlinear Regression: a Practical Guide to Curve Fitting. GraphPad Software: San Diego; 2005. [Google Scholar]
- Muller T, Myrtek D, Bayer H, Sorichter S, Schneider K, Zissel G, et al. Functional characterization of histamine receptor subtypes in a human bronchial epithelial cell line. Int J Mol Med. 2006;18:925–931. [PubMed] [Google Scholar]
- Newton R, Holden NS, Catley MC, Oyelusi W, Leigh R, Proud D, et al. Repression of inflammatory gene expression in human pulmonary epithelial cells by small-molecule IkappaB kinase inhibitors. J Pharmacol Exp Ther. 2007;321:734–742. doi: 10.1124/jpet.106.118125. [DOI] [PubMed] [Google Scholar]
- Newton R, Kuitert LM, Bergmann M, Adcock IM, Barnes PJ. Evidence for involvement of NF-kappaB in the transcriptional control of COX-2 gene expression by IL-1beta. Biochem Biophys Res Commun. 1997;237:28–32. doi: 10.1006/bbrc.1997.7064. [DOI] [PubMed] [Google Scholar]
- O'Reilly M, Alpert R, Jenkinson S, Gladue RP, Foo S, Trim S, et al. Identification of a histamine H4 receptor on human eosinophils—role in eosinophil chemotaxis. J Recept Signal Transduct Res. 2002;22:431–448. doi: 10.1081/rrs-120014612. [DOI] [PubMed] [Google Scholar]
- Ohkubo T, Shibata M, Inoue M, Kaya H, Takahashi H. Regulation of substance P release mediated via prejunctional histamine H3 receptors. Eur J Pharmacol. 1995;273:83–88. doi: 10.1016/0014-2999(94)00668-w. [DOI] [PubMed] [Google Scholar]
- Pahl HL. Activators and target genes of Rel/NF-kappaB transcription factors. Oncogene. 1999;18:6853–6866. doi: 10.1038/sj.onc.1203239. [DOI] [PubMed] [Google Scholar]
- Reddel RR, Ke Y, Gerwin BI, McMenamin MG, Lechner JF, Su RT, et al. Transformation of human bronchial epithelial cells by infection with SV40 or adenovirus-12 SV40 hybrid virus, or transfection via strontium phosphate coprecipitation with a plasmid containing SV40 early region genes. Cancer Res. 1988;48:1904–1909. [PubMed] [Google Scholar]
- Rouleau A, Stark H, Schunack W, Schwartz JC. Anti-inflammatory and antinociceptive properties of BP 2-94, a histamine H(3)-receptor agonist prodrug. J Pharmacol Exp Ther. 2000;295:219–225. [PubMed] [Google Scholar]
- Talreja J, Kabir MH, B Filla M, Stechschulte DJ, Dileepan KN. Histamine induces Toll-like receptor 2 and 4 expression in endothelial cells and enhances sensitivity to Gram-positive and Gram-negative bacterial cell wall components. Immunology. 2004;113:224–233. doi: 10.1111/j.1365-2567.2004.01946.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tayo FM, Bevan JA. Pharmacological characterization of histamine receptors in the rabbit renal artery. Eur J Pharmacol. 1986;121:129–133. doi: 10.1016/0014-2999(86)90402-4. [DOI] [PubMed] [Google Scholar]
- Tilly BC, Tertoolen LG, Lambrechts AC, Remorie R, de Laat SW, Moolenaar WH. Histamine-H1-receptor-mediated phosphoinositide hydrolysis, Ca2+ signalling and membrane-potential oscillations in human HeLa carcinoma cells. Biochem J. 1990;266:235–243. doi: 10.1042/bj2660235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waud DR, Son SL, Waud BE. Kinetic and empirical analysis of dose–response curves illustrated with a cardiac example. Life Sci. 1978;22:1275–1285. doi: 10.1016/0024-3205(78)90096-6. [DOI] [PubMed] [Google Scholar]
- Wellendorph P, Goodman MW, Burstein ES, Nash NR, Brann MR, Weiner DM. Molecular cloning and pharmacology of functionally distinct isoforms of the human histamine H(3) receptor. Neuropharmacology. 2002;42:929–940. doi: 10.1016/s0028-3908(02)00041-2. [DOI] [PubMed] [Google Scholar]
- Wu RL, Anthes JC, Kreutner W, Harris AG, West RE., Jr Desloratadine inhibits constitutive and histamine-stimulated nuclear factor-kappaB activity consistent with inverse agonism at the histamine H1 Receptor. Int Arch Allergy Immunol. 2004;135:313–318. doi: 10.1159/000082325. [DOI] [PubMed] [Google Scholar]
- Zhang M, Thurmond RL, Dunford PJ. The histamine H(4) receptor: a novel modulator of inflammatory and immune disorders. Pharmacol Ther. 2006;113:594–606. doi: 10.1016/j.pharmthera.2006.11.008. [DOI] [PubMed] [Google Scholar]