

Abstract
Myeloperoxidase (MPO), a member of the haem peroxidase-cyclooxygenase superfamily, is abundantly expressed in neutrophils and to a lesser extent in monocytes and certain type of macrophages. MPO participates in innate immune defence mechanism through formation of microbicidal reactive oxidants and diffusible radical species. A unique activity of MPO is its ability to use chloride as a cosubstrate with hydrogen peroxide to generate chlorinating oxidants such as hypochlorous acid, a potent antimicrobial agent. However, evidence has emerged that MPO-derived oxidants contribute to tissue damage and the initiation and propagation of acute and chronic vascular inflammatory disease. The fact that circulating levels of MPO have been shown to predict risks for major adverse cardiac events and that levels of MPO-derived chlorinated compounds are specific biomarkers for disease progression, has attracted considerable interest in the development of therapeutically useful MPO inhibitors. Today, detailed information on the structure of ferric MPO and its complexes with low- and high-spin ligands is available. This, together with a thorough understanding of reaction mechanisms including redox properties of intermediates, enables a rationale attempt in developing specific MPO inhibitors that still maintain MPO activity during host defence and bacterial killing but interfere with pathophysiologically persistent activation of MPO. The various approaches to inhibit enzyme activity of MPO and to ameliorate adverse effects of MPO-derived oxidants will be discussed. Emphasis will be put on mechanism-based inhibitors and high-throughput screening of compounds as well as the discussion of physiologically useful HOCl scavengers.
Keywords: MPO-H2O2-halide system; hypochlorite; inflammation; neutrophils, atherosclerosis; lipid oxidation; protein oxidation
Introduction
An intensely green protein containing iron that had peroxidase activity was originally isolated from canine pus and purulent fluid of patients with tuberculous empyema (Agner, 1941). Observations that expression of this enzyme is restricted to the myeloid series of haematopoietic cells where it is entirely located in azurophil (primary) granules, resulted in renaming of this protein, verdoperoxidase, to myeloperoxidase (MPO) (Yamada and Kurahashi, 1984; Koeffler et al., 1985). Promyelocytes and promyelomonocytes actively synthesize MPO during granulocyte differentiation in the bone marrow. MPO levels in neutrophilic polymorphonuclear leucocytes (neutrophils) make up in between 2 and 5% of the total cellular protein or 2 to 4 μg per 106 cells (Schultz and Kaminker, 1962; Bos et al., 1978). Human monocytes also contain MPO-positive granules although they are fewer in number than in neutrophils and lost during differentiation into tissue macrophages (for a review see: Klebanoff, 2005).
Observations that destruction of microorganisms occurs in the phagosome of neutrophils and that MPO is among the enzymes discharged into these phagocytic vacuoles from cytoplasmic granules suggest an important role of MPO in bacterial killing (Klebanoff, 2005). Among the antimicrobial systems present in the phagosome, a major proportion consists of MPO, hydrogen peroxide (H2O2, formed during the respiratory burst), and a halide (X−), particularly chloride (Cl−) (Hampton et al., 1998). The initial product of the MPO–H2O2–Cl− system is the potent antimicrobial oxidant hypochlorous acid/hypochlorite (HOCl/OCl−, pKa 7.53) (Figure 1). However, under pathological conditions, persistent activation of the MPO–H2O2 system of activated phagocytes may adversely affect tissues. HOCl is able to initiate modification reactions targeting lipids, DNA and (lipo)proteins, including halogenation, nitration and oxidative crosslinking (Figure 1).
Figure 1.

Primary and secondary MPO-reaction products and (patho)physiology. Adapted and modified from Malle et al. (2006a). CNS, central nervous system.
Chlorohydrins are formed by addition of HOCl to double bonds present either on cholesterol or various unsaturated ester- and ether-phospholipid species (Malle et al., 2006a; Leßig et al., 2007); however, the usefulness of chlorohydrins to be used as biomarkers is limited. Most importantly, the remaining lyso-phospholipid remnants formed by HOCl-mediated attack of ester-/ether-phospholipids (plasmalogens) may adversely affect membrane dynamics and cause cell lysis. In contrast to ester-phospholipids, HOCl-mediated attack of ether-phospholipids (present in biological membranes and lipoprotein particles) results in formation of 2-chlorohexadecanal (2-ClHDA), a chlorinated fatty aldehyde that is enriched in human atherosclerotic lesions (Thukkani et al., 2003) and infarcted myocardium (Thukkani et al., 2005). 2-ClHDA is a potent chemoattractant in vitro initiating recruitment of circulating neutrophils to areas of inflammation and promotes endothelial dysfunction (Marsche et al., 2004). After release from activated phagocytes, MPO colocalizes with HOCl-modified epitopes on the endothelial layer lining the blood vessel (Malle et al., 2006b) and contributes to endothelial dysfunction (Eiserich et al., 2002).
HOCl has also been reported to react with nucleobases resulting in the formation of 5-chlorouracil, a marker for DNA damage during inflammation, which is enriched in human atherosclerotic tissue (Takeshita et al., 2006).
Another indicator for MPO-catalysed oxidation and specific protein-associated biomarker, formed either directly by HOCl attack or via intermediate formation of chlorine gas from HOCl (Hazen et al., 1996), is 3-chlorotyrosine. 3-Chlorotyrosine was identified in human atherosclerotic lesion and lipoproteins extracted from lesions (Hazen and Heinecke, 1997; Zheng et al., 2004), in respiratory tract disease (Buss et al., 2003; Kettle et al., 2004), and neutrophil-induced liver injury (Gujral et al., 2004).
Immunocytochemistry and immunohistochemistry with specific monoclonal antibodies generated against HOCl-modified proteins/epitopes (Malle et al., 1995) provides an immunological tool to identify chlorinated biomarkers in kidney disease (Malle et al., 1997, 2003; Gröne et al., 2002), ischaemia reperfusion (Hasegawa et al., 2005), Parkinson's disease (Choi et al., 2005), and atherosclerosis (Hazell et al., 1996; Malle et al., 2000). Fractionation of human plaque homogenate by density gradient centrifugation and subsequent immunoblot analysis of the low-density lipoprotein-bouyant fraction revealed that apoB-100, the major apolipoprotein of low-density lipoprotein is modified by the MPO–H2O2–Cl− system of activated phagocytes in lesion material (Hazell et al., 1996). In vitro studies have shown that lipoproteins, modified by HOCl (added as reagent or generated by the MPO–H2O2–Cl− system), display a number of pathophysiological effects on phagocytes and vascular cells, contributing to initiation and maintenance of the inflammatory process during atherosclerotic lesion development (Daugherty et al., 1994; Malle et al., 2006a, 2006b). In particular, the uptake and degradation of HOCl-modified lipoproteins by monocyte-derived macrophages via scavenger receptor-mediated pathways (Marsche et al., 2001, 2003) is a leading event in the formation of cholesterol-enriched foam cells (Malle et al., 2006a), representing the hallmark of fatty streaks and the earliest recognisable lesion of atherosclerosis. HOCl-modified (lipo)proteins are also high-affinity signalling transducing ligands for RAGE (Marsche et al., 2007a, 2007b), a multiligand receptor that, originally reported to bind advanced glycation end products, promotes an array of inflammatory complications.
In general, oxidation of amino-acid side chain appears to play a specific role during HOCl-mediated protein backbone fragmentation. The first step involves chloramine formation (probably from lysine side chains), giving rise to nitrogen-centred radicals in a time- and HOCl-concentration-dependent manner. These radicals may further form carbon-centred radicals at peptide bonds thereby promoting protein fragmentation and/or promote lipid peroxidation (Hawkins and Davies, 1998).
Although several groups have detected accumulation of lipid peroxidation products in HOCl-modified liposomes and lipoproteins, the mechanisms of lipid-hydroperoxide (ROOH) formation is not entirely clear (Malle et al., 2006a). Panasenko et al. (1997) have suggested that this requires the presence of preformed ROOH able to induce further lipid peroxidation. During reaction with hydroperoxide, HOCl induces the formation of singlet molecular oxygen and comparable observations were reported for fatty acid and phospholipid hydroperoxides (Miyamoto et al., 2006). The reaction of HOCl with preformed ROOH may be of particular importance at sites of inflammation where both, MPO/HOCl and ROOH concentrations are significantly elevated leading to the formation of secondary lipid/protein oxidation products.
In the absence of physiological Cl− concentrations, formation of tyrosyl radicals promotes protein crosslinks via dityrosine formation, whereas nitrogen dioxide radical generates nitrated lipids. In addition, both radical species are able to induce lipid peroxidation (Figure 1).
As MPO levels of leucocyte- and blood-MPO are associated with the presence of coronary artery disease (Baldus et al., 2003; Brennan et al., 2003) and MPO considerably contributes to plaque vulnerability (for a review see: Nicholls and Hazen, 2005), quantitation of MPO mass and activity, as well as analysis of MPO-derived biomarkers (secondary MPO products, Figure 1) is of prognostic value to follow disease progression.
Development of therapeutic intervention strategies aiming at efficient MPO inhibition can be envisaged at different levels: (i) active site blockade of MPO, (ii) diversion of MPO from the chlorination cycle, (iii) irreversible suicide inhibition by use of oxidized inhibitors, and (iv) application of HOCl scavengers to prevent initiation and propagation of diseases with an inflammatory component. All these strategies have already been followed, but in a limited and non-systematic way. The provided information is distributed in journals of varying disciplines and often fragmentary or even contradictory. It is the aim of this review to provide a systematic overview about potential inhibitors and their mode of action that is based on our present knowledge about structure–function relationship and (patho)physiological relevance of MPO.
MPO transcription, translation, processing and release
MPO is encoded by a single gene (approximately 14 kb in size), composed of 11 introns and 12 exons, and located on the long arm of chromosome 17 in segment q12–24. The primary 80 kDa translation product preproMPO is processed in the endoplasmic reticulum undergoing cotranslational N-glycosylation (resulting in an 90 kDa apoproMPO), transient interaction with molecular chaperones, and haem incorporation to generate enzymatically active proMPO that is exported into the Golgi compartment (for a review see: Hansson et al., 2006). After exiting the Golgi, the propeptide is removed before final proteolytic processing in azurophilic granules. The processed MPO protein is a glycosylated, predominantly α-helical cationic 146 kDa dimer with a single disulphide bridge between symmetry-related halves (73 kDa), each containing two polypeptides of 108 (14.5 kDa, light chain) and 466 amino acids (58.5 kDa, heavy chain). Dimeric MPO is found in neutrophils and monocytes. All available structural data refer to this mature dimeric MPO (Zeng and Fenna, 1992; Fiedler et al., 2000; Blair-Johnson et al., 2001) and the majority of studies on MPO inhibitors have been performed with this protein.
However, some proMPO escapes granule targeting and becomes constitutively secreted to the extracellular environment as a monomer (Hansson et al., 2006). There is also evidence of de novo MPO synthesis in certain subtypes of macrophages and it has been suggested that foam cells newly engaged in MPO gene transcription lack the cellular machinery to proteolytically process proMPO into subunits of mature MPO. As a result, only proMPO enters the secretory pathway and is released in the extracellular environment. It is important to note that recombinant MPO overexpressed in Chinese hamster ovary cell lines (Moguilevsky et al., 1991) resembles monomeric and partially unprocessed proMPO. Nevertheless, several spectral and kinetic investigations clearly demonstrated that mature MPO and recombinant proMPO exhibit identical enzymatic features (Jacquet et al., 1991; Moguilevsky et al., 1991; Furtmüller et al., 2001), indicating identical molecular architecture of the haem cavity and substrate channel. Thus, recombinant proMPO represents a valid model for inhibitor studies and it is reasonable to assume that the action of inhibitors on mature dimeric MPO and monomeric (recombinant) proMPO is identical.
Structural information about MPO
The overall protein fold of MPO was first revealed by a 3 Å resolution crystal structure of the canine enzyme (Protein Data Bank (PDB) code: 1MYP) (Zeng and Fenna, 1992). By now, the structure of human MPO at 2.3 Å resolution is solved and refined to 1.8 Å using X-ray data recorded at −180°C (1CXP) (Fiedler et al., 2000). The structure of MPO and its interaction with bromide (Br−) (1D2V) and thiocyanate (SCN−) (1DNU) (Fiedler et al., 2000) as well as of the MPO-cyanide (MPO–CN−) complex (1D5L) and its interaction with Br− (1D7W) and SCN− (1DNW) was published (Blair-Johnson et al., 2001). In addition, there is one report on the crystal structure of salicylhydroxamic acid (SHA) bound to human MPO (Davey and Fenna, 1996), but unfortunately no entry in the PDB exists. Additionally, computational analysis of indole derivative binding to MPO by computational docking is reported (Hallingbäck et al., 2006).
Enzymatic properties and activity assays
Chlorination activity
The principal reaction catalysed by MPO under physiological conditions is the oxidation of X−, for example Cl− and Br− as well as SCN− to the corresponding hypohalous acids (HOX) and hypothiocyanus acid (HOSCN) according to equation 1 (halogenation activity). HOX/HOSCN can participate in subsequent nonenzymatic reactions such as oxidation and halogenation of compounds present in the immediate environment (Figure 1). There is no well-defined pH optimum for MPO-catalysed chlorination because it depends on the relative concentrations of Cl− and H2O2. However, at constant concentrations of these reaction partners, the rate of HOCl release increases with decreasing pH (Andrews and Krinsky, 1982). It is important to realize that at high concentrations, H2O2 inactivates MPO. To minimize inactivation, the concentration should be kept at ⩽100 μM. Because amine-containing buffer systems are potent scavengers of HOCl activity, measurements must be performed in phosphate buffers.
Two assays are routinely used to measure the chlorination activity of MPO. The first assay involves chlorination of monochlorodimedon (MCD) by HOCl, a process that is accompanied by a decrease in absorbance (ɛ290=1.99 × 104 M−1 cm−1) (Kettle and Winterbourn, 1988). However, MCD cannot be regarded as an inert detector of HOCl production by MPO because it has been shown to function as competitive electron donor (Kettle and Winterbourn, 1988). Thus, the MCD assay underestimates the chlorinating activity (Kettle and Winterbourn, 1994). Nevertheless, this method is useful for detecting HOCl, as shown by its complete inhibition by methionine, and thus is applicable in inhibitor studies.
The second assay involves chlorination of nitrogen compounds, particularly primary amines. The reaction of HOCl with these compounds yields chloramines containing covalent nitrogen-chlorine bonds. The taurine chloramine assay is based on the reaction of HOCl with taurine (TauNH2) to produce taurine chloramine (TauNHCl). Usually, chloramines are unstable and decompose to the corresponding aldehydes and nitriles. By contrast, TauNHCl is relatively stable and its formation can be either followed spectrophotometrically [ɛ252=429 M−1 cm−1] (Thomas et al., 1986) or by measuring the oxidation of yellow 5-thio-2-nitrobenzoic acid (ɛ412=14 000 M−1 cm−1) to colourless 5,5′-dithiobis(2-nitrobenzoic acid) (TNB) by TauNHCl (Weiss et al., 1982). This is an extremely sensitive assay and can accurately measure concentrations of HOCl as low as 5 μM.
Alternatively, I− can be used to detect TauNHCl produced by MPO (Dypbukt et al., 2005). TauNHCl oxidizes I− to HOI, which finally oxidizes 3,3′,5,5′-tetramethylbenzidine (TMB) to a strongly absorbing blue product or dihydrorhodamine to highly fluorescent rhodamine, respectively (Dypbukt et al., 2005). The advantage over the TNB assay is that in this assay a signal is produced rather than quenched, which enhances its sensitivity and accuracy and offers its application to microtitre plate format (Dypbukt et al., 2005).
Besides MCD and TauNH2, ascorbate (Chesney et al., 1991) and TMB (Suzuki et al., 1983) have been used as the basis for assaying the chlorination activity of MPO. The loss of ascorbate (ɛ267=15 000 M−1 cm−1) can be related to its oxidation by HOCl because the reaction is inhibited by methionine. However, results must be interpreted with caution because ascorbate can act directly as peroxidase substrate (equation 2). Similarly, oxidation of TMB (ɛ285=2.1 × 104 M−1 cm−1) by HOCl or via the peroxidation cycle of MPO gives a blue product with an absorbance maximum (ɛ652=3.9 × 104 M−1 cm−1) (Josephy et al., 1982). Generally, the TMB assay is extremely sensitive and has been used to quantitate the total utilization of H2O2 by MPO. However, under usual assay conditions, the lack of inhibition by methionine indicates that the peroxidation activity completely overrides the chlorination activity.
Peroxidase activity
MPO is also able to catalyse typical peroxidation reactions (equation 2) with AH2 representing the peroxidase substrate oxidized to the corresponding radical (•AH). Principally, each peroxidase assay described in the literature can also be performed with MPO.
Oxidation of tyrosine to dityrosine can be followed spectrofluorimetrically using an excitation wavelength of 325 nm and an emission wavelength at 405 nm (Marquez and Dunford, 1995). As already mentioned, TMB functions both as scavenger of HOCl and as peroxidase substrate. In the absence of Cl−, the TMB assay can be used to monitor the peroxidatic activity of MPO (Marquez and Dunford, 1997). Another assay, following the oxidation of guaiacol to tetraguaiacol (ɛ470=2.6 × 104 M−1 cm−1) (Maehly and Chance, 1954) is also often used to follow MPO activity. Furthermore, a luciferin derivative (2-methyl-6-(p-methoxylphenyl)-3,7-dihydroimidazol[1,2-α]pyrazin-3-one) has also been used for the determination of singlet oxygen produced from the reaction of HOBr with H2O2 (Nakano and Koga, 1994).
Hydrogen peroxide electrode assay
The loss of H2O2 catalysed by MPO during halogenation and peroxidatic cycles (equations 1 and 2) can be continuously monitored using a H2O2 electrode (Kettle and Winterbourn, 1994). In the presence of Cl− (up to 100 mM) as the only electron donor, this method allows direct assessment of the chlorination activity without the need for a detector compound that might potentially interfere with MPO. The formation of HOCl accounts completely for the loss of H2O2. It is recommended to include TauNH2 (10 mM) or methionine (500 μM) in the reaction buffer to scavenge HOCl and prevent it from inactivating MPO (Kettle and Winterbourn, 1994).
Assays for inhibitor studies
In our opinion screening for putative MPO inhibitors should include both, the use of the H2O2 electrode and the TauNHCl assay, respectively. Polarographic monitoring of the loss of H2O2 should be followed either in the presence of Cl− or a peroxidase substrate (for example, tyrosine). This approach allows a first conclusion of (i) whether the inhibitor specifically interferes with the chlorination and/or peroxidatic cycle, (ii) whether the inhibitory mode is reversible or irreversible, and (iii) whether the putative inhibitor simply acts as a scavenger for HOCl or TauNH2. In any case, measuring inhibition of only peroxidative activity is an inappropriate strategy in searching for inhibitors of HOCl production because many reversible inhibitors act by diverting MPO from the chlorinating cycle to the peroxidase cycle where they act as competitive electron donors for compounds I and II (see below).
In high-throughput screenings, the polarographic method is inapplicable. Here, remaining H2O2, which has not been consumed either in the chlorination (equation 1) or peroxidase activity (equation 2) due to inhibitory effects, can be monitored photometrically or fluorimetrically (Tarpey et al., 2004). Caution is advised when horseradish peroxidase and a peroxidase substrate is used for H2O2 visualisation, because the inhibitor of MPO could also impair the activity of horseradish peroxidase. Alternatively, I−-mediated oxidation of TMB or dihydrorhodamine by TauNHCl can be used to elucidate the effect of putative inhibitors.
Redox intermediates of MPO
To understand the underlying mechanism of MPO inhibition it is necessary to investigate the interaction of potential inhibitors with the individual reaction steps both in the halogenation (equation 1) and peroxidation (equation 2) reactions. Firstly, the inhibitor could block the access of H2O2 or its binding and reduction. As a consequence it would inhibit both cycles because both start by reaction of the Fe(III) form of MPO with H2O2 to form compound I, which contains two oxidizing equivalents more than the resting enzyme (Figure 2, reaction 1). The reaction occurs at the distal haem cavity and amino acids His95 and Arg239 (Figure 3b) plays critical roles in the binding/orientation/activation and finally heterolytic cleavage of H2O2. Compound I is an oxoiron(IV) intermediate [Fe(IV)=O] containing a porphyrin π-cation radical (Por•+) (Dolphin et al., 1971).
Figure 2.

General reaction scheme of MPO. In the first step H2O2 is used for compound I formation (reaction 1). Compound I is two oxidizing equivalents above the native enzyme with a porphyrin π-cation radical in combination with an oxoiron(IV) centre. Compound I can react with halides (X−) reducing the enzyme back to the ferric state (reaction 2, halogenation activity). In the peroxidase reaction, compound I is transformed in the first one-electron reduction to compound II, which contains an oxoiron(IV) centre (reaction 3). Compound II is finally reduced back to ferric peroxidase in a second one-electron reduction (reaction 4). Compound III (oxyperoxidase) is formed either from ferric peroxidase with superoxide anion (reaction 9), from ferrous MPO with O2 (reaction 8) or from compound II with H2O2 (reaction 6). Compound III is a complex of ferrous-dioxygen in resonance with ferric superoxide. Ferrous [Fe(II)]MPO can be formed from ferric [Fe(III)]MPO by reducing radicals produced in the peroxidase cycle (reaction 11).
Figure 3.

(a) The hydrogen bonding network and locations of five water molecules (W1–W5) at the distal haem cavity of ferric high-spin MPO. (b) The non-planar porphyrin ring in MPO and its covalent attachments to the protein via two ester bonds (Glu242 and Asp94) and one sulfonium ion linkage (Met243). In addition, the proximal His336 and the distal catalytic residues His95, Arg239, and Gln91 are shown, the latter being important in halide binding. The figure was constructed using the coordinates deposited in the Protein Data Bank (accession code 1CXP).
Once produced, compound I of MPO is a strong oxidant that mediates both one- and two-electron oxidation reactions. Depending on the apparent bimolecular rate constant and donor concentration, the enzyme follows either the halogenation (Figure 2, reactions 1 and 2) or the peroxidase cycle (Figure 2, reactions 1, 3 and 4). In the halogenation cycle X− and SCN− directly reduce compound I to the Fe(III) form of MPO (Figure 2, reaction 2) releasing the corresponding HOX/HOSCN. The standard reduction potential of the couple compound I/native MPO is quite high (+1.16 V) (Arnhold et al., 2001, 2006) enabling the enzyme to oxidize Cl−. The putative binding site(s) of these small anionic inorganic two-electron donors and the impact of inhibiting substances on compound I reduction by X−/SCN−are described below.
In the peroxidase cycle, compound I is reduced by two successive one-electron steps via compound II, a protonated oxoiron(IV) species (Figure 2, reactions 3 and 4). Many inorganic and organic substrates were found to act as electron donors for both compounds I and II (for a review see: Dunford, 1999; Furtmüller et al., 2006). MPO compound I is an extremely strong one-electron oxidant, because the standard reduction potential of the couple compound I/compound II is 1.35 V; the standard reduction potential for the couple compound II/native enzyme is only 0.97 V (Furtmüller et al., 2003). This marked difference in the oxidation capacity of MPO compounds I and II is strongly reflected by the actual reaction rate constants of these intermediates with substrates of one-electron reduction potentials >0.95 V, for example indole and tryptamine derivatives (Jantschko et al., 2005). The latter molecules are good electron donors for compound I but do not react with compound II thus diverting MPO from the chlorination cycle.
Owing to its strong oxidation capacity, MPO compound I is responsible for drug bioactivation (Yang et al., 2006) including oxidation of phenytoin (Uetrecht and Zahid, 1988; Kubow and Wells, 1989; Mays et al., 1995), carbamazepine (Furst and Uetrecht, 1993), L-dopa (Nappi and Vass, 2001), clozapine (Uetrecht et al., 1997; Williams et al., 1997), trimethoprim (Lai et al., 1999), fluperlapine (Lai et al., 2000) and procainamide (Uetrecht and Zahid, 1991) to reactive metabolites/intermediates (for example, nitroso, nitrogen free radical and iminium species). Diclofenac (Miyamoto et al., 1997) undergoes p-hydroxylation by MPO, resulting in p-aminophenols, which can be further oxidized to reactive quinoneimines. However, MPO itself also oxidizes many potential inhibitors. The present knowledge concerning their binding and electron transfer site(s) is summarized below.
With respect to inhibitory mechanisms further redox intermediates have to be discussed, which neither participate in the halogenation nor in the peroxidase cycle, namely the Fe(II) form of MPO and compound III, the latter being the ferrous-dioxy/ferric-superoxide anion complex of MPO (Figure 2). Similar to higher oxidation states the reduction potential of Fe(III)/Fe(II) of MPO is significantly higher (5 mV) (Battistuzzi et al., 2006) than that of other haem peroxidases. Thus, MPO is very susceptible to reduction to its Fe(II) form, that is by radical intermediates formed during the peroxidase cycle (Figure 2, reaction 11). In the presence of oxygen Fe(II), MPO is readily converted into compound III (Figure 2, reaction 8). Alternatively, compound III can be formed by reaction of ferric MPO with superoxide anion (Figure 2, reaction 9) or of compound II with H2O2 (Figure 2, reaction 6). It is important to note that compound III should be regarded as a complex that can decay to ferric MPO and superoxide anion (Figure 2, reaction 10) or ferrous MPO and oxygen (Figure 2, reaction 7).
Substrate and inhibitor binding sites
To be able to rationally design therapeutically useful MPO inhibitors, a detailed knowledge of the structural features of the enzyme is essential. Each monomer or catalytic domain of dimeric MPO or monomeric proMPO contains one iron atom (present as covalently bound ferri-protoporphyrin IX derivative) and one Ca2+ion. The Ca2+-binding site has typical pentagonal bipyramidal coordination (Zeng and Fenna, 1992; Fiedler et al., 2000). Because one of its ligand, Asp96, is in close vicinity to the distal His95 (Figure 3b), Ca2+ is important in maintaining the distal architecture but does not participate immediately in substrate and/or inhibitor binding. A unique structural feature of MPO concerns the modification of the 1- and 5-methyl groups on pyrrole ring A and C of the haem group (Figures 3a and b) enabling formation of ester linkages with the carboxyl groups of an Asp94 and Glu242. In addition the β-carbon of the vinyl group on pyrrole ring A forms a covalent bond with the sulphur atom of methionine 243, giving rise to a sulphonium ion linkage (Figures 3a and b) (Zeng and Fenna, 1992; Fiedler et al., 2000), which is responsible for the peculiar redox properties of MPO (Arnhold et al., 2006; Zederbauer et al., 2007b). As a consequence, the haem porphyrine ring is considerably distorted from planarity (Figure 3b). Although pyrrole rings B and D are nearly coplanar, ring A and, to a lesser extent ring C are tilted toward the distal side, resulting in a bow-shaped haem structure. The proximal haem ligand is a histidine, which is hydrogen bound with an asparagine.
The haem of MPO is located in a crevice, about 15 Å in depth, with access to the solvent via an open channel, approximately 10 Å in diameter (Figure 5). It discharges into the distal cavity that accommodates the amino-acid triade histidine, arginine and glutamine (Figure 3b). The distal His95 is hydrogen bonded to a water molecule (W1), which is positioned approximately mid-way between the Nɛ atom of His95 and the haem iron (Figures 3a and b). Its distance from both the histidine nitrogen and the iron atom suggests that it is hydrogen bonded to the histidine and not strongly coordinated to the haem iron. Four additional water molecules (W2–W5) form hydrogen bonds with His95, Arg239, Gln91, the haem pyrrole ring C propionate and between themselves (Figure 3a). Water W2 is hydrogen bonded to the Nɛ atom of Gln91, W3 to NH2 of Arg239, W4 to haem propionate of pyrrole ring C and W5 having no additional hydrogen bonding (Figure 3a) (Fiedler et al., 2000). Generally, the covalent haem attachment in MPO causes small solvent reorganisation effects in the reduction reaction of Fe(III) MPO (Battistuzzi et al., 2006), suggesting a high rigidity of the hydrogen bond network involving water molecules in the substrate channel leading to the distal haem site.
Figure 5.
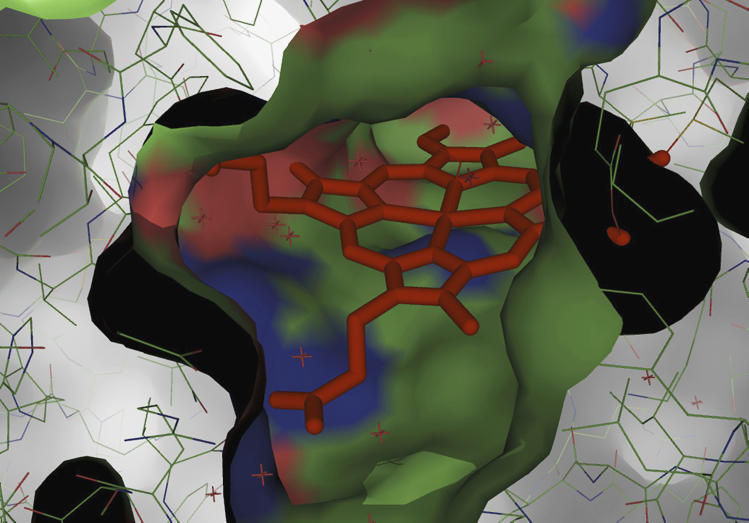
View through the access channel to the active site (complexed with cyanide) showing exposure of the haem pyrrole ring D with its 8-methyl and of the δ-methine bridge (that is, electron transfer site). The figure was constructed using PyMol Viewer (version 0.99).
Halide binding site
In the MPO–Br– complex (data not shown), Br– replaces water molecule W2, which is hydrogen bonded to the amide of Gln91 in the proximity of the Nɛ atom of His95. Unfortunately, there is no structure of a Cl− bound to the distal haem cavity. Figures 4a and b allow a comparison of the distal cavities of the MPO–CN− complex with the MPO–CN––Br– double complex. In the MPO–CN––Br– double complex, which could be regarded as a model for compound I with bound Br−, W2 is present and Br− binds in place of W5 thereby preventing direct hydrogen bonding to protonated His95. When CN− is bound to the iron, the W2 site appears to be inaccessible for larger anions like Br− or I−. However, it is possible that the W2 position could accommodate Cl− equally well in both native MPO and its CN− complex, because Cl− (1.81 Å) is significantly smaller in radius than Br− (1.96 Å) or I− (2.2 Å). The sterical hindrance of Br– and I– to bind at the W2 position in compound I might be reflected by the fact that with increasing radius of the anionic substrates, the increase in the rate of compound I reduction by these donors at acidic pH is less pronounced compared to Cl− oxidation (Furtmüller et al., 1998). Binding of Cl− at position W2 together with the MPO typical distal water network of low mobility could be crucial in fixing the position of Cl− and in favouring the transfer and incorporation of the oxyferryl oxygen into HOCl. In any case at neutral pH, Cl− is bound weakly (KD>100 mM) and needs a rigid hydrogen-bonding network, which is easily destabilized by peroxidase substrates or inhibitor binding at the haem edge (see below).
Figure 4.

(a) Structure of the myeloperoxidase-cyanide (MPO–CN−) complex (accession code 1D5L), which can be regarded as a model of compound I. In the CN−-complex water molecule W1 is displaced by CN−. (b) Structure of the MPO–CN−–bromide (Br−) double complex (accession code 1D7W). CN− and Br− are located at positions of W1 and W5. Note that in the MPO–Br− complex (data not shown), the halide is binding at W2. (c) Structure of the MPO–CN−–thiocyanate (SCN−) double complex (accession code 1DNW). In this complex CN− displaces W1, whereas SCN− displaces both W2 and W5.
There is evidence from kinetic studies that the Cl− level (2 mM) used to obtain the X-ray structure of the MPO–Cl− complex could be insufficient to occupy the distal binding site (Proteasa et al., 2007). Moreover, based on the biphasic behaviour of nitric oxide binding to MPO in the presence of increasing Cl− concentrations, two distal Cl− binding sites of different affinities have been proposed (Proteasa et al., 2007).
In the X-ray structure, the closest haem atom of X− bound at the position of W2 is the δ-methine bridge between pyrrole rings A and D (Figures 4a and b) at a distance of 3.8 Å, and the haem iron is at a distance of 5.0 Å. A X− bound at the position of W2 is also in vicinity to the side chain of Glu242, which is only 3.5 Å away from W2 in the native enzyme. Although Met243 is relatively distant and not involved in positioning of W2 in native MPO or Br– in the MPO–Br– complex (data not shown), its exchange has a tremendous effect on the Cl– affinity. The affinity is decreased by 100-fold, compared to recombinant proMPO (Kooter et al., 1997). In contrast, the exchange of Glu242 and Asp94 by 242Gln and 94Val in MPO mutants is relatively small (Zederbauer et al., 2005, 2007a). A specific role of Met243 in X− binding cannot be inferred by inspection of the structures but might be again related to its role in maintaining a rigid structural environment at the distal site that enables the binding of small anionic ligands (Battistuzzi et al., 2006).
In the MPO–CN−–Br− double complex (Figure 4b) (Blair-Johnson et al., 2001) the Br− binding site is close to the δ-methine bridge carbon and does not appear to form any readily identifiable electrostatic interactions with the enzyme, the haem, or the bound CN−. The closest amino acid is Glu242 and the nearest neighbouring haem atom is the pyrrole ring D methyl carbon (Blair-Johnson et al., 2001). The close proximity of Glu242 to positions of both W2 and W5 is underlined by the fact that compound I reduction by X− is generally decreased in Glu242Gln mutants irrespective of the nature of the X− (Zederbauer et al., 2005). By contrast, disruption of the haem to Asp94 ester linkage affected the oxidation of Cl− and Br− but not of I− and SCN− (Zederbauer et al., 2007b).
In the MPO–SCN− complex (data not shown), SCN− is arranged almost parallel with the plane of the haem replacing water molecules W2 and W5 in the native enzyme. The nitrogen of SCN−, at the W2 position, forms hydrogen bonds with the amide NH2-group of Gln91 and water molecule W2 as well as a weak interaction with the Nɛ atom of distal His95, whereas the sulphur makes van der Waals contacts with Cγ of Glu242, Cδ of Arg239, and the haem pyrrole ring D methyl carbon. SCN− in the distal cavity of the MPO–CN− complex (Figure 4c) (Blair-Johnson et al., 2001) occupies the same position as in the native enzyme, except that it is inclined ≈15° from its former orientation parallel with the haem. In the presence of CN−, the nitrogen atom in SCN− can form only a single hydrogen bond with Gln91.
Aromatic substrate/inhibitor binding site
Binding studies have indicated that aromatic molecules bind near to the distal haem pocket in MPO (Hori et al., 1994). On the basis of electron paramagnetic spectroscopy and molecular modelling it was proposed that the hydroxamic acid side chains of benzylhydroxamic acid (BHA) and SHA interact with the haem iron in MPO (Ikeda-Saito et al., 1991). Finally, the 2.3 Å resolution X-ray structure of the MPO-SHA complex was published (Davey and Fenna, 1996) showing the aromatic ring of SHA binding to a hydrophobic region at the entrance to the distal haem pocket between pyrrole ring D and the side chain of Arg239 (Figure 6a). The hydroxamic acid moiety is hydrogen bonded to both the distal His95 and Gln91 amide group but is not coordinated to the haem iron. Although SHA binding displaces three water molecules from the distal cavity (W1, W2 and W3), no significant conformational differences between the active site regions of the complex and the native enzyme became apparent.
Figure 6.

Docking models for SHA (a) and melatonin (b) bound to MPO. The positions of the three oxygen atoms of SHA are close to the positions occupied by the three water molecules W1–W3 in the native enzyme (see Figure 3a). Both models were calculated using LigandScout 1.03 (Wolber and Langer, 2005) from Inte:Ligand GmbH (www.inteligand.com). SHA, salicylhydroxamic acid.
The hydrophobic region at the entrance to the distal cavity is conserved among mammalian peroxidases (Figure 5) (Furtmüller et al., 2006). The aromatic ring of SHA (Figure 6a) is tilted about 20° with respect to that of the haem pyrrole ring D which forms the lower surface of the hydrophobic cavity, whereas the β, γ and δ carbons of Arg239 form the upper surface (Davey and Fenna, 1996). In the MPO–SHA complex, the aromatic ring of SHA is almost centred above the pyrrole ring D 8-methyl group. Additional van der Waals interactions occur with adjacent phenylalanines, that is Phe99, Phe366 and Phe407, each of which has an aromatic ring within 4.2–4.6 Å of the aromatic ring of the SHA molecule.
It is reasonable to assume that substrate binding to compounds I and II (Figure 2, reactions 3 and 4) is identical. The proposed electron transfer most probably occurs near the δ-methine bridge carbon (Davey and Fenna, 1996). Mutation of neighboured Glu242Gln decreases the overall rate of guaiacol and tyrosine oxidation compared to non-mutated recombinant proMPO (Zederbauer et al., 2005). Similar to the negative effect on X− oxidation, disruption of the neighboured Glu242 ester bond seems to decrease the electron transfer between the donor molecule and the haem moiety. Interestingly, oxidation of the hydrophilic donor ascorbate by both compounds I and II was enhanced in Glu242Gln mutated compared to non-mutated recombinant proMPO (Zederbauer et al., 2005), suggesting that the described binding scenario fits to aromatic but not to hydrophilic one-electron donors.
Recently, the binding of melatonin (N-acetyl-5-methoxytryptamine) and serotonin (5-hydroxytryptamine) to MPO in its Fe(III) compounds I and II state was studied by computational docking (Hallingbäck et al., 2006). Both indole derivatives could be docked into all three forms with their indole rings oriented parallel to the haem plane and close enough to the D pyrrole ring to achieve stacking (Figure 6b). The distances of the closest indole atom to the centre of the D ring were about 3.4 Å for both substrates. The ligand side chain was never directed towards the distal cavity, whereas the indole substituent at position 5 was close to the haem centre (Hallingbäck et al., 2006). In detail, the distance between the oxygen of the 5-methoxy or 5-hydroxyl substituent on the indole ring and the haem iron ion was calculated to be around 3.0 Å.
Computational docking of compounds I and II states of MPO (data not shown) demonstrated, that both, melatonin and serotonin were pushed about 1 Å away from the ferryl oxygen, which abolished the 5-substituent to point to the haem centre thus favouring an alternative binding mode with a rotated indole still parallel to pyrrole ring D (Hallingbäck et al., 2006).
MPO inhibitors
Hydroxamic acids
Hydroxamic acids [RCNOHOH or RC(O)NHOH] are known to act as reducing substrates for compounds I and II of haem peroxidases (Figure 2, reactions 3 and 4) and can also substitute for H2O2 in compound I formation (reaction 3) (Schonbaum and Lo, 1972; Schonbaum, 1973). SHA has been reported to inhibit the luminol-dependent chemiluminescence of human neutrophils stimulated by phorbol 12-myristate 13-acetate or the chemotactic peptide N-formyl-methionyl-leucyl-phenyl-alanine (Davies and Edwards, 1989). Furthermore, SHA had no inhibitory effect on the kinetics of superoxide anion generation or O2 uptake during the respiratory burst of phagocytes, but inhibited the peroxidase activity of purified MPO (IC50 value of 3–5 μM). SHA also prevented the formation of compounds I and II but only at low H2O2 concentrations, suggesting that it may compete for the H2O2 binding site on the enzyme (Davies and Edwards, 1989).
Binding of SHA and BHA to dimeric leucocytic MPO, as studied by optical absorption (Ikeda-Saito et al., 1991) and electron paramagnetic spectroscopy (Hori et al., 1994), showed dissociation constants of 2 μM (SHA) and 5 mM (BHA) as well as the involvement of an ionizable group on the protein with a pKa of approximately 4. In the SHA–MPO inhibitory complex determined at 2.3 Å resolution (Davey and Fenna, 1996), it is clearly shown that this residue is the distal histidine, because the hydroxamic acid moiety is hydrogen bonded to His95 (Figure 6b). The ability of the three SHA oxygen atoms to closely duplicate the hydrogen-bonding pattern of W1–W3 in the native state (Figure 3a) could account for the strong binding of SHA to MPO. The salicyl-ring hydroxyl oxygen participates in hydrogen bonding to the guanidinium group of Arg239 and to water molecule W2 that remains hydrogen bonded to the pyrrole ring C propionate carboxyl group. As described above, a separate hydrophobic interaction occurs between the aromatic ring and a hydrophobic region at the entrance to the distal haem cavity suggesting that oxidation occurs at the haem edge, in the vicinity of the δ-meso carbon and the 8-methyl group on pyrrole ring D (Davey and Fenna, 1996).
In any case, SHA plays dual inhibitory roles in peroxidase catalysis by inhibiting hydroperoxide binding to the iron and by competing with other donors in reactions with higher oxidation states. However, this inhibition is not irreversible and strongly depends both on H2O2 and competing electron donor concentration.
Benzoic acid hydrazides
Hydrazines (RNHNH2) and hydrazides (RCONHNH2) have been reported to be suicide substrates of other haem peroxidases than MPO (Ator et al., 1987). The antituberculous agent isonicotinic acid hydrazide (isoniazid) inhibits both the chlorination and peroxidation activity of MPO irreversibly. Oxidation of isoniazid or hydrazine sulphate between pH values 6.5 and 7.8 by the MPO–H2O2 system caused irreversible loss of haem absorbance associated with compound III formation (van Zyl et al., 1989a). Finally, during a study using a series of benzoic acid hydrazides, it was demonstrated that these are general inhibitors of the peroxidation and chlorination activity of MPO (Kettle et al., 1995). Unsubstituted benzoic acid hydrazide and its 4-chloroderivative were poor inhibitors, whereas derivatives with substitutents containing oxygen or nitrogen were much better inhibitors. Benzoic acid hydrazides are readily oxidized by compound I (Figure 2, reaction 3) and the rate constants for these reactions are relatively insensitive to the substituents on the aromatic ring (Burner et al., 1999). By contrast, reduction of compound II (Figure 2, reaction 4) by benzoic acid hydrazides strongly correlated with the substitution patterns and to the Hammett rule, and there were also significant correlations with Brown–Okamoto substituent constants (Hansch and Leo, 1979; Burner et al., 1999). This indicates that the rates of these reactions were simply determined by the redox potential of the substrates and that the incipient free radical carried a positive charge, which is delocalized via resonance between the substituent on the aromatic ring and the hydrazide group.
4-Aminobenzoic acid hydrazide (ABAH) was the best electron donor of compound II and also the most potent inhibitor of peroxidation. ABAH irreversibly inhibited HOCl production of MPO isolated from leucocytes with an IC50 value of 0.3 μM. The kinetic analysis of the inactivation conformed to that for a mechanism-based inhibitor (Kettle et al., 1997). ABAH is oxidized in the peroxidase cycle by compounds I and II and the produced radicals readily convert the enzyme into ferrous MPO (Figure 2, reaction 11) and compound III (Figure 2, reaction 8). Compound III is neither part of the peroxidase nor of the chlorination cycle thus impeding oxidation of reducing substrates reversibly. Its turnover is slow and ABAH or an ABAH radical may reduce it in analogy to the reduction of oxyhaemoglobin by phenylhydrazine (Misra and Fridovich, 1976). In the absence of oxygen, ferrous MPO accumulates and irreversible inactivation is exacerbated by destruction of the haem prosthetic group (Kettle et al., 1997). Protection by dioxygen suggests that ferrous MPO, permanently formed by reactions 11 and 7 (Figure 2) (that is, dissociation of the dioxygen–Fe(II) complex), is the actual target of irreversible inactivation but the detailed mechanism is still unknown (Burner et al., 1999).
With neutrophils stimulated with opsonized zymosan or phorbol myristate acetate, ABAH inhibited HOCl production by up to 90% and corresponding IC50 values were 16 and 2.2 μM, respectively. However, in the presence of superoxide dismutase, these values decreased to 6.4 and 0.6 μM, indicating that superoxide anion similar to molecular oxygen protects MPO to some extent from irreversible inactivation. ABAH was shown to have no effect on superoxide anion production and degranulation of neutrophils, nor did it inhibit catalase or glutathione peroxidase (Kettle et al., 1995).
Indoles and tryptamines
Indole derivatives act as one-electron donors of compounds I and II of haem peroxidases (Figure 2, reactions 3 and 4) (Jantschko et al., 2002). These molecules have been shown to act as reversible inhibitors of MPO when they exhibit a one-electron reduction potential E°′(A•, H+/AH)>E°′ (compound II/ferric MPO)=0.97 V (Jantschko et al., 2005). Generally, tryptamines were more effective in inhibition of chlorination than indoles and this strongly correlated with the determined ratios of rates of compound I to compound II reduction. 5-Fluorotryptamine and 5-chlorotryptamine were found to act as the most effective chlorination inhibitors during this study.
The putative binding mode of indole derivatives has been described above (Hallingbäck et al., 2006). In ferric MPO, the indole ring binds parallel to the pyrrole ring D and the 5-OH group (serotonin, an excellent substrate) and 5-OCH3 group (melatonin, a poor substrate) most probably points to the haem centre. In both cases, the indole ring seems to be in the same place. The orientation of the side chain constitutes the largest difference between melatonin and serotonin. In case of serotonin it points out into the access channel, whereas in case of melatonin it points into a pocket along the periphery of the distal cavity (Hallingbäck et al., 2006). The ring substituent determines the actual rates of compounds I and II reduction with electron-donating groups by increasing the aromatic ring reactivity (Jantschko et al., 2005). Electron-withdrawing substituents, for example −Cl and −F atoms, will increase the redox potential of the corresponding indole derivatives and, as a consequence, disqualify them to act as a peroxidase substrate. Owing to the extremely positive reduction potential of the redox couple compound I/compound II (Arnhold et al., 2006), these substituents can still serve as electron donor for compound I but are unreactive towards compound II (Jantschko et al., 2005). As a consequence, compound II accumulates and MPO is diverted from its chlorination cycle (Figure 2).
In effect these indole and tryptamine derivatives are poor peroxidase substrates and reversible inhibitors. Under in vivo conditions compound II can easily be reduced by alternative electron donors thus diminishing the efficacy of these reversible-inhibiting compounds. This has been demonstrated by the effect of tryptophan on formation of HOCl by human neutrophils (Kettle and Candaeis, 2000). Using neutrophil-derived MPO, IC50 values increased from 5 to 80 μM, because physiological substrates such as ascorbate, tyrosine, urate or superoxide anion are able to reduce compound II enabling the enzyme to cycle.
Drugs that promote compound II formation
As outlined above, hydroxamic acid, indole and tryptamine derivatives promote compound II formation of MPO thus diverting the enzyme from the chlorination cycle. Kettle and Winterbourn (1991) have demonstrated that many anti-inflammatory drugs that are already in clinical application, follow the same reaction pattern. By using the H2O2 electrode to assess the ability of these compounds to inhibit the conversion of H2O2 into HOCl, dapsone, mefenamic acid, sulphapyridine, quinacrine, primaquine and aminopyrine were found to induce 50% inhibition of the initial rate of H2O2 loss at concentrations of approximately 1 μM or less. Phenylbutazone, piroxicam, salicylate, olsalazine, benzocaine, sulfasalazine (Kettle and Winterbourn, 1991), diclofenac (Zuurbier et al., 1990), chlorpromazine (van Zyl et al., 1990), acetaminophen (van Zyl et al., 1989b), deferoxamine (Klebanoff and Waltersdorph, 1988) were also effective inhibitors (IC50 <30 μM). However, inhibition of MPO by all these drugs was reversed by ascorbate, which is known to reduce compound II to ferric MPO (Figure 2, reaction 4) (Hsuanyu and Dunford, 1999). Additionally, the inhibitory effects of these molecules were significantly diminished in the presence of a superoxide anion generating system, because superoxide anion acts as electron donor for compound II (Kettle et al., 1993).
The mechanism of inhibition of HOCl production, which relies on MPO being trapped as inactive compound II, was also supported by the findings that dapsone and indomethacin are competitive inhibitors with respect to I− and Cl− (Stendahl et al., 1978; Shacter et al., 1991). All these drugs are efficiently oxidized by compound I but have a limited capacity to reduce compound II. This indicates that they are unable to compete with high-affinity peroxidase substrates indicating that measurement of inhibited peroxidative activity is an inappropriate strategy to screen for potential inhibitors of HOCl production by MPO.
The IC50 values are influenced by the electrochemical properties of these compounds. Their reduction potential determines their reactivity towards MPO compounds I and II and thus their effectiveness as inhibitors of HOCl production. Compounds with reduction potentials >1.1 V could be relatively specific for MPO because they would react poorly with other haem enzymes. For all these reversible inhibitors it is unlikely that size or hydrophobicity have a major impact on inhibition properties. This is best demonstrated by salicylate analogues, sulphasalazine and olsalazine, which have similar IC50 values as salicylate, even though they are much larger and more hydrophobic (Kettle and Winterbourn, 1991).
In a recent study, recombinant proMPO has been shown to be convenient for pharmacological purposes (Neve et al., 2001). Fourteen drugs representative of a family of various nonsteroidal anti-inflammatory drugs were tested for their ability to inhibit the MPO-mediated chlorination of TauNH2. Most of them induced a dose-dependent inhibition of TauNH2 and IC50 values could be calculated. The most efficient inhibitors (IC50<40 μM) were found to be flufenamic acid, diclofenac, niflumic acid, tenoxicam and piroxicam. In all cases, these drugs interacted with recombinant proMPO in the same way as already described for dimeric MPO from neutrophils (Kettle and Winterbourn, 1991).
In any case, within the phagosome inhibition of intracellular HOCl formation by reversible inhibitors will be limited by superoxide anion and other electron donors of compound II. To achieve more efficient inhibition, drugs that irreversibly inhibit MPO (like benzoic acid hydrazides, see above) are needed.
Drugs that promote compound III formation
Compound III is positioned outside both of the chlorination and peroxidation cycle of MPO (Figure 2). Thus, drugs that promote compound III formation divert MPO also from HOCl production. Many drugs and xenobiotics are oxidized by MPO via the peroxidase cycle (Figure 2, reactions 1, 3 and 4) but in some cases the kinetics of oxidation cannot adequately be explained by this pathway alone. Hydroquinones are known to be excellent substrates for MPO compounds I and II (Kettle and Winterbourn, 1992; Burner et al., 2000). Semiquinone radicals are formed in a high flux, which efficiently reduce ferric MPO to ferrous MPO that rapidly binds oxygen to form compound III (Figure 2, reactions 11 and 8). The same reaction pathway seems to be followed in the oxidation of the anticancer drug amsacrine by the MPO–H2O2 system (Kettle et al., 1992); in both cases production of HOCl by MPO is inhibited. However, in contrast to hydrazides that follow the same mechanism but finally irreversibly modify the active site (see above), the hydroquinone derivatives and amsacrine are reversible inhibitors because compound III is in dynamic equilibrium with the native enzyme or ferrous MPO (Figure 2, reactions 10 and 7) or can even be reduced to the native enzyme by ascorbate (Marquez et al., 1990).
Further studies on putative MPO inhibitors
In a comparative study of the effects of nonsteroidal anti-inflammatory cyclooxygenase 1 and 2 inhibitors on MPO activity, it has been shown that indomethacin and other representative tricyclic aromatics display only poor inhibition of the peroxidase activity of MPO (EC50 >100 μM) (Rider et al., 1996).
More promising molecules with inhibitory effects on the TauNH2 chlorination activity of MPO are 2(3H)-benzoxazolone derivatives. In a recent study 20 ω-[2-oxo-3H-benzoxazol-3-yl]-N-phenylacetamide and propionamide derivatives (carrying substituents of different lipophilic and electrophilic nature on the N-phenyl ring) have been synthesized and shown to exhibit inhibitory effects on the chlorinating capacity of MPO (IC50 2–95 μM) (Soyer et al., 2005). In general, propionamide derivatives with chlorine and nitrosubstituents and non-substituted ones were more active than the acetamide counterparts. The authors reported that these compounds were unable to react with HOCl suggesting that they interact directly with MPO. However, there are no detailed mechanistic studies available.
The antithyroid drug methimazole (2-mercapto-1-methylimidazole) (Bandyopadhyay et al., 1993, 1995) and the selenium analogue (Roy and Mugesh, 2005) have been shown to be irreversible mechanism-based inhibitors of human peroxidases (Bandyopadhyay et al., 1993, 1995). Methimazole is oxidized by thyroid peroxidase, lactoperoxidase and MPO, because no inactivation occurred in the absence of H2O2. With lactoperoxidase and a gastric peroxidase the kinetics of the inactivation process were described to be consistent with a mechanism-based mode. With I− (good electron donor for lactoperoxidase) and addition of the spin-trap 5,5′-dimethyl-1-pyrroline N-oxide, but not with Cl− (poor electron donor for lactoperoxidase), lactoperoxidase could be protected against inactivation. This clearly demonstrates that methimazole is oxidized to thiyl radicals, which finally could be responsible for irreversible modification of the haem cavity and inhibition of lactoperoxidase (Bandyopadhyay et al., 1993, 1995). Inhibition of MPO-mediated chlorination by methimazole is also based on the oxidation of methimazole but in contrast to lactoperoxidase inhibition of MPO seems to be only reversible (McGirr et al., 1990; Sayo and Saito, 1991).
Propylthiouracil (also used as antithyroid drug) was also reported to inhibit the peroxidase and chlorination activity of MPO (Lee et al., 1990). The inactivation was dependent on H2O2 and was still observed after propylthiouracil and H2O2 were removed by gelfiltration, which suggests an irreversible inactivation mode. With a gastric peroxidase it has been shown that propylthiouracil is a mechanism-based inhibitor but with MPO detailed mechanistic studies are lacking.
The last group of sulphur-centred compounds with inhibitory effects on MPO are thiourea derivatives. Dimethylthiourea (1–10 mM) has been reported to completely block formation of HOCl by phorbol myristate acetate- and zymosan-stimulated neutrophils (Sagone et al., 1989). The compound has been shown to act both as a substrate for MPO and a scavenger for HOCl. From these data there is no evidence that dimethylthiourea mediates an irreversible inactivation of MPO (Sagone et al., 1989).
Alternative therapeutic options to influence the MPO–H2O2–chloride system
Subjects with profound deficiencies in MPO (approximately one out of 4000 subjects) appear to have a significant increase in risk for infection (Nauseef et al., 1998; Klebanoff, 2005); observations that parallel severe impairment in early host defence in MPO−/− mice against Candida albicans (Aratani et al., 1999) and enhanced inflammation after allogeneic marrow transplantation (Milla et al., 2004). In contrast, cardiovascular problems appear to be very rare among MPO-deficient individuals in comparison to their high frequency among the reference population (Kutter et al., 2000). Hoy et al. (2001) reported that subjects carrying the A allele of the G-463A MPO polymorphism display higher levels of triglycerides, total and low-density lipoprotein-cholesterol, and apoB-100 which are known risk factors for atherosclerosis. Recent reports demonstrated that lipid-lowering drugs downregulate MPO on gene (Kumar and Reynolds, 2005) and protein level (Stenvinkel et al., 2006; Zhou et al., 2006); statins block 3-hydroxy-3-methylglutaryl-CoA-reductase, the key enzyme of endogenous cholesterol biosynthesis pathway, thereby apparently blocking production of isoprenoid intermediates of the mevalonate pathway, which is required for human and mouse MPO expression. Observations that an Alu receptor response element in the human MPO promoter is regulated by T0901317 and GW9578, respectively, suggest that MPO regulation is under tight control of LXR and PPARα, which normally regulate genes involved in cholesterol metabolism and inflammation (Reynolds et al., 2006).
Neutrophils protect themselves from HOCl by maintaining an exceptionally high cytoplasmic concentration of TauNH2 (approximately 50 mM). TauNH2 reacts with HOCl to yield the relatively innocuous compound TauNHCl that may act as immunomodulator by compensating adverse effects of primary and secondary MPO-derived products (Schuller-Levis and Park, 2003). Therapeutic approaches using TauNH2 have revealed that this sulphur-containing amino acid may blunt the contribution of phagocyte activation to acute coronary events (McCarty, 1999). Consequently, TauNH2 was used as HOCl scavenger in a number of animal studies. Diminution of atherosclerotic lesions has been reported in control and WHHL rabbits (lacking functional low-density lipoprotein receptor) (Petty et al., 1990; Murakami et al., 2002) when dietary-induced hypercholesterolaemia was accompanied by TauNH2 supplementation. In the absence of TauNH2, abundant staining for HOCl-modified (lipo)proteins is present in these animals (Malle et al., 2001; Bräsen et al., 2003). Although endogenous MPO levels in rodents are much lower than in humans, TauNH2 supplementation leads to diminution and regression of lipid accumulation in murine lesions after cholesterol feeding (Murakami et al., 1999a, 1999b). Most importantly, overexpression of human MPO in murine models promoted formation of atherosclerosis (McMillen et al., 2005; Castellani et al., 2006). Hyperlipidemia-induced renal damage in mesangioproliferative glomerulonephritis is paralleled by occurrence of HOCl-modified (lipo)proteins in rats (Scheuer et al., 2000). The protective effect of TauNH2 in rat models of human kidney disease is paralleled by a decrease in total proteinuria and albuminuria, prevention of glomerular hypertrophy, and diminuition of glomerulosclerosis and tubulointerstitial fibrosis (Malle et al., 2003).
Another possibility is the application of antioxidants, for example, ambroxol (Cho et al., 1999), dithiocarbamate (Zhu et al., 2002) and vitamin C (Carr et al., 2000), which protect against or even may reverse HOCl- and chloramine-dependent modification of proteins by pathways probably involving reduced glutathione and ATP. Even phenolic compounds, for example chlorogenic acid (Kono et al., 1995) might be considered as HOCl scavengers. Chlorogenic acid is approved as food additive (Svetol) and used in coffee, chewing gum, and mints to promote weight reduction.
Very efficient HOCl scavengers are antiarthritic drugs containing thiol groups, such as D-penicillamine, tiopronin, sodium aurothiomalate and aurothioglucose; it has been suggested that the therapeutic effect of these drugs may be due to the protection of tissues against reactive HOCl released by activated granulocytes at inflamed sites (Cuperus et al., 1985). D-penicillamine, tiopronin, aurothiomalate or aurothioglucose (10–50 μM) are relatively potential inhibitors of MCD chlorination by HOCl. It has to be mentioned that besides effectively scavenging HOCl, D-penicillamine and tiopronin inhibit the enzyme itself (Cuperus et al., 1983, 1985). Extrapolation to in vivo conditions is possible because the concentration of D-penicillamine and tiopronin in serum from patients treated with these drugs is about 100 μM (van der Korst et al., 1981). The concentration of gold compounds in patients treated with aurothiomalate and aurothioglucose is about 50 μM (Lewis et al., 1983). Interestingly, all four antiarthritic drugs develop their beneficial effects over a period of weeks of therapy. Although the therapeutic effect of these antirheumatics may be multifactorial, scavenging of HOCl and inhibition of MPO offers an attractive explanation for their mechanism of action.
Another drug already applied in practice is thiacetazone. It is used to treat tuberculosis and has been reported to be also an effective HOCl scavenger as well as to promote compound III formation of MPO (van Zyl and van der Walt, 1996).
Future perspectives
MPO inhibitory properties of a variety of compounds were assessed with different methodologies. The information on promising MPO inhibiting compounds in the literature is sparse making a comparison and critical evaluation of their actual inhibitory potential difficult. Additionally, only a few compounds were investigated in detail to clarify the underlying molecular mechanism of inactivation. Nevertheless, the knowledge of (i) the three-dimensional structure of human MPO and (ii) binding sites of its endogenous substrates (that is, two-electron donating anionic halides) and of artificial aromatic one-electron donors/inhibitors (for example, SHA or indoles) as well as our knowledge of (iii) its reaction mechanisms and redox properties will accelerate the process for a rationale design of specific MPO inhibitors in the future.
Almost all known MPO inhibitors are oxidized at least by compound I, which is one of the strongest oxidizing enzyme intermediates found in vivo. Thus, these inhibitors divert the enzyme from the chlorination cycle and suppress HOCl production. In case the reaction products do not interact with the protein, compound II accumulates, which can be easily reduced by many electron donors found in vivo, for example superoxide anion, tyrosine or ascorbate. This inhibition mode is reversible and the in vivo inhibitory effect of these drugs on HOCl is questionable.
The interaction of MPO with some drugs results in the formation of radicals that either attack the haem prosthetic group or functionally important active site residues (mechanism-based irreversible inactivation). Alternatively, they can mediate the reduction of ferric MPO to ferrous MPO, which is readily converted into its dioxygen complex (compound III) under aerobic conditions. In case these radicals are unreactive toward the enzyme this inhibition mode is also reversible. With benzoic acid hydrazides the products generated in the peroxidase cycle mediate the irreversible destruction of the haem group of MPO in its ferrous form, with ABAH being the most efficient suicide substrate (irreversible inhibitor). However, we still do not fully understand the molecular basis of this suicide inactivation nor do we know the nature of the modification at the haem cavity. Understanding these features will facilitate the design of more specific and more potent irreversible MPO inhibitors.
Acknowledgments
This work was supported by grants from the Austrian Science Fund (FWF) to EM/WS (P17013-B05, P19074-B05, and F3007-B05) and CO/PGF (P14187-B11, P15660-B10). We thank Inte:Ligand for help with three-dimensional MPO structure models.
Abbreviations
- 2-ClHDA
2-chlorohexadecanal
- ABAH
4-aminobenzoic acid hydrazide
- BHA
benzylhydroxamic acid
- E°′
standard reduction potential
- HCN/CN−
hydrocyanic acid (hydrogen cyanide)/cyanide
- HOCl/OCl−
hypochlorous acid/hypochlorite
- HOSCN/OSCN−
hypothiocyanous acid/hypothiocyanate
- HSCN/SCN−
thiocyanous acid/thiocyanate
- HOX/X−
hypohalous acid/halide
- MCD
monochlorodimedon
- MPO
myeloperoxidase
- PDB
Protein Data Bank
- ROOH
lipid-hydroperoxide
- SHA
salicylhydroxamic acid (2-hydroxybenzohydraxamic acid)
- TauNH2
taurine
- TauNHCl
taurine chloramine
- TMB
3,3′,5,5′-tetramethylbenzidine
- TNB
5,5′-dithiobis(2-nitrobenzoic acid)
Conflict of interest
E Malle has no conflict of interest. PG Furtmüller is coinvestigator of a project (protec-TRANS sponsered by the Austrian ERP Fonds) executed by Planta Naturstoffe Vertriebs GmbH that investigates natural anti-inflammatory substances (January 1, 2005–July 31, 2007). W Sattler has no conflict of interest. C Obinger is consultant to the Myeloperoxidase Programme Team of GlaxoSmithKline Research & Development Ltd (September 1, 2005–August 31, 2007).
References
- Agner K. Verdoperoxidase. A ferment isolated from leukocytes. Acta Chem Scand A. 1941;2 Suppl. 8:1–62. [Google Scholar]
- Andrews PC, Krinsky NI. A kinetic analysis of the interaction of human myeloperoxidase with hydrogen peroxide, chloride ions, and protons. J Biol Chem. 1982;257:13240–13245. [PubMed] [Google Scholar]
- Aratani Y, Koyama H, Nyui S, Suzuki K, Kura F, Maeda N. Severe impairment in early host defense against Candida albicans in mice deficient in myeloperoxidase. Infect Immun. 1999;67:1828–1836. doi: 10.1128/iai.67.4.1828-1836.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnhold J, Furtmüller PG, Regelsberger G, Obinger C. Redox properties of the couple compound I/native enzyme of human myeloperoxidase and eosinophil peroxidase. Eur J Biochem. 2001;268:5142–5148. doi: 10.1046/j.0014-2956.2001.02449.x. [DOI] [PubMed] [Google Scholar]
- Arnhold J, Monzani E, Furtmüller PG, Zederbauer M, Casella L, Obinger C. Kinetics and thermodynamics of halide and nitrite oxidation by mammalian peroxidases. Eur J Inorg Chem. 2006;19:3801–3811. [Google Scholar]
- Ator MA, David SK, Ortiz de Montellano PR. Structure and catalytic mechanism of horseradish peroxidase. Regiospecific meso alkylation of the prosthetic heme group by alkylhydrazines. J Biol Chem. 1987;262:14954–14960. [PubMed] [Google Scholar]
- Baldus S, Heeschen C, Meinertz T, Zeiher AM, Eiserich JP, Munzel T, on behalf of the CAPTURE investigators et al. Myeloperoxidase serum levels predict risk in patients with acute coronary syndromes. Circulation. 2003;108:1440–1445. doi: 10.1161/01.CIR.0000090690.67322.51. [DOI] [PubMed] [Google Scholar]
- Bandyopadhyay U, Bhattacharyya DK, Banerjee RK. Mechanism-based inactivation of gastric peroxidase by mercaptomethylimidazole. Biochem J. 1993;296:79–84. doi: 10.1042/bj2960079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bandyopadhyay U, Bhattacharyya DK, Chatterjee R, Banerjee RK. Irreversible inactivation of lactoperoxidase by mercaptomethylimidazole through generation of a thiyl radical: its use to probe the active site. Biochem J. 1995;306:751–757. doi: 10.1042/bj3060751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Battistuzzi G, Bellei M, Zederbauer M, Furtmüller PG, Sola M, Obinger C. Redox thermodynamics of the Fe(III)/Fe(II) couple of human myeloperoxidase in its high-spin and low-spin forms. Biochemistry. 2006;45:12750–12755. doi: 10.1021/bi061647k. [DOI] [PubMed] [Google Scholar]
- Blair-Johnson M, Fiedler T, Fenna R. Human myeloperoxidase: structure of a cyanide complex and its interaction with bromide and thiocyanate substrates at 1.9 Å resolution. Biochemistry. 2001;40:13990–13997. doi: 10.1021/bi0111808. [DOI] [PubMed] [Google Scholar]
- Bos A, Wever R, Roos D. Characterization and quantification of the peroxidase in human monocytes. Biochim Biophys Acta. 1978;525:37–44. doi: 10.1016/0005-2744(78)90197-3. [DOI] [PubMed] [Google Scholar]
- Bräsen JH, Hakkinen T, Malle E, Beisiegel U, Ylä-Herttuala S. Patterns of oxidized epitopes, but not NF-kB expression, change during atherogenesis in WHHL rabbits. Atherosclerosis. 2003;166:13–21. doi: 10.1016/s0021-9150(02)00130-2. [DOI] [PubMed] [Google Scholar]
- Brennan ML, Penn MS, Van Lente F, Nambi V, Shishehbor MH, Aviles RJ, et al. Prognostic value of myeloperoxidase in patients with chest pain. N Engl J Med. 2003;349:1595–1604. doi: 10.1056/NEJMoa035003. [DOI] [PubMed] [Google Scholar]
- Burner U, Krapfenbauer G, Furtmüller PG, Regelsberger G, Obinger C. Oxidation of hydroquinone, 2,3-dimethylhydroquinone and 2,3,5-trimethylhydroquinone by human myeloperoxidase. Redox Rep. 2000;5:185–190. doi: 10.1179/135100000101535735. [DOI] [PubMed] [Google Scholar]
- Burner U, Obinger C, Paumann M, Furtmüller PG, Kettle AJ. Transient and steady-state kinetics of the oxidation of substituted benzoic acid hydrazides by myeloperoxidase. J Biol Chem. 1999;274:9494–9502. doi: 10.1074/jbc.274.14.9494. [DOI] [PubMed] [Google Scholar]
- Buss IH, Senthilmohan R, Darlow BA, Mogridge N, Kettle AJ, Winterbourn CC. 3-Chlorotyrosine as a marker of protein damage by myeloperoxidase in tracheal aspirates from preterm infants: association with adverse respiratory outcome. Pediatr Res. 2003;53:455–462. doi: 10.1203/01.PDR.0000050655.25689.CE. [DOI] [PubMed] [Google Scholar]
- Carr AC, Tijerina T, Frei B. Vitamin C protects against and reverses specific hypochlorous acid- and chloramine-dependent modifications of low-density lipoprotein. Biochem J. 2000;346:491–499. [PMC free article] [PubMed] [Google Scholar]
- Castellani LW, Chang JJ, Wang X, Lusis AJ, Reynolds WF. Transgenic mice express human MPO −463G/A alleles at atherosclerotic lesions, developing hyperlipidemia and obesity in −463G males. J Lipid Res. 2006;47:1366–1377. doi: 10.1194/jlr.M600005-JLR200. [DOI] [PubMed] [Google Scholar]
- Chesney JA, Mahoney JR, Eaton JW. A spectrophotometric assay for chlorine-containing compounds. Anal Biochem. 1991;196:262–266. doi: 10.1016/0003-2697(91)90463-4. [DOI] [PubMed] [Google Scholar]
- Cho Y, Jang YY, Han ES, Lee CS. The inhibitory effect of ambroxol on hypochlorous acid-induced tissue damage and respiratory burst of phagocytic cells. Eur J Pharmacol. 1999;383:83–91. doi: 10.1016/s0014-2999(99)00585-3. [DOI] [PubMed] [Google Scholar]
- Choi DK, Pennathur S, Perier C, Tieu K, Teismann P, Wu DC, et al. Ablation of the inflammatory enzyme myeloperoxidase mitigates features of Parkinson's disease in mice. J Neurosci. 2005;25:6594–6600. doi: 10.1523/JNEUROSCI.0970-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuperus RA, Muijsers AO, Wever R. The effect of D-penicillamine on human myeloperoxidase: a mechanism for the efficacy of the drug in rheumatoid arthritis. Biochim Biophys Acta. 1983;749:18–23. doi: 10.1016/0167-4838(83)90145-0. [DOI] [PubMed] [Google Scholar]
- Cuperus RA, Muijsers AO, Wever R. Antiarthritic drugs containg thiol groups scavenge hypochlorite and inhibit its formation by myeloperoxidase from human leukocytes. Arthritis Rheum. 1985;28:1228–1233. doi: 10.1002/art.1780281106. [DOI] [PubMed] [Google Scholar]
- Daugherty A, Rateri DL, Dunn JL, Heinecke JW. Myeloperoxidase, a catalyst for lipoprotein oxidation, is expressed in human atherosclerotic lesions. J Clin Invest. 1994;94:437–444. doi: 10.1172/JCI117342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davey CA, Fenna RE. 2.3 Å resolution X-ray crystal structure of the bisubstrate analogue inhibitor salicylhydroxamic acid bound to human myeloperoxidase: a model for a prereaction complex with hydrogen peroxide. Biochemistry. 1996;35:10967–10973. doi: 10.1021/bi960577m. [DOI] [PubMed] [Google Scholar]
- Davies B, Edwards SW. Inhibition of myeloperoxidase by salicylhydroxamic acid. Biochem J. 1989;258:801–806. doi: 10.1042/bj2580801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolphin D, Forman A, Borg DC, Fajer J, Felton RH. Compounds I of catalase and horseradish peroxidase: p-cation radicals. Proc Natl Acad Sci USA. 1971;68:614–618. doi: 10.1073/pnas.68.3.614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunford HB.Myeloperoxidase and eosinophil peroxidase: phagocytosis and microbial killing Heme Peroxidases 1999Wiley-VCH: New York; 349–385.In: Dunford HB (ed). [Google Scholar]
- Dypbukt JM, Bishop C, Brooks WM, Thong B, Eriksson H, Kettle AJ. A sensitive and selective assay for chloramine production by myeloperoxidase. Free Radic Biol Med. 2005;39:1468–1477. doi: 10.1016/j.freeradbiomed.2005.07.008. [DOI] [PubMed] [Google Scholar]
- Eiserich JP, Baldus S, Brennan ML, Ma W, Zhang C, Tousson A, et al. Myeloperoxidase, a leukocyte-derived vascular NO oxidase. Science. 2002;296:2391–2394. doi: 10.1126/science.1106830. [DOI] [PubMed] [Google Scholar]
- Fiedler TJ, Davey CA, Fenna RE. X-ray crystal structure and characterization of halide-binding sites of human myeloperoxidase at 1.8 Å resolution. J Biol Chem. 2000;275:11964–11971. doi: 10.1074/jbc.275.16.11964. [DOI] [PubMed] [Google Scholar]
- Furst SM, Uetrecht JP. Carbamazepin metabolism to a reactive intermediate by the myeloperoxidase system of activated neutrophils. Biochem Pharmacol. 1993;45:1267–1275. doi: 10.1016/0006-2952(93)90279-6. [DOI] [PubMed] [Google Scholar]
- Furtmüller PG, Arnhold J, Jantschko W, Pichler H, Obinger C. Redox properties of the couples compound I/compound II and compound II/native enzyme of human myeloperoxidase. Biochem Biophys Res Commun. 2003;301:551–557. doi: 10.1016/s0006-291x(02)03075-9. [DOI] [PubMed] [Google Scholar]
- Furtmüller PG, Burner U, Obinger C. Reaction of human myeloperoxidase compound I with chloride, bromide, iodide, and thiocyanate. Biochemistry. 1998;37:17923–17930. doi: 10.1021/bi9818772. [DOI] [PubMed] [Google Scholar]
- Furtmüller PG, Jantschko W, Regelsberger G, Jakopitsch C, Moguilevsky N, Obinger C. A transient kinetic study on the reactivity of recombinant unprocessed monomeric myeloperoxidase. FEBS Lett. 2001;503:147–150. doi: 10.1016/s0014-5793(01)02725-9. [DOI] [PubMed] [Google Scholar]
- Furtmüller PG, Zederbauer M, Jantschko W, Helm J, Bogner M, Jakopitsch C, et al. Active site structure and catalytic mechanisms of human peroxidases. Arch Biochem Biophys. 2006;445:199–213. doi: 10.1016/j.abb.2005.09.017. [DOI] [PubMed] [Google Scholar]
- Gröne HJ, Gröne EF, Malle E. Immunohistochemical detection of hypochlorite-modified proteins in glomeruli of human membranous glomerulonephritis. Lab Invest. 2002;82:5–14. doi: 10.1038/labinvest.3780390. [DOI] [PubMed] [Google Scholar]
- Gujral JS, Liu J, Farhood A, Hinson JA, Jaeschke H. Functional importance of ICAM-1 in the mechanism of neutrophil-induced liver injury in bile duct-ligated mice. Am J Physiol Gastrointest Liver Physiol. 2004;286:G499–G507. doi: 10.1152/ajpgi.00318.2003. [DOI] [PubMed] [Google Scholar]
- Hallingbäck HR, Gabdoulline RR, Wade RC. Comparison of the binding and reactivity of plant and mammalian peroxidases to indole derivatives by computational docking. Biochemistry. 2006;45:2940–2950. doi: 10.1021/bi051510e. [DOI] [PubMed] [Google Scholar]
- Hampton MB, Kettle AJ, Winterbourn CC. Inside the neutrophil phagosome: oxidants, myeloperoxidase, and bacterial killing. Blood. 1998;92:3007–3017. [PubMed] [Google Scholar]
- Hansch C, Leo A. Substituent Constants for Correlation Analysis in Chemistry and Biology. John Wiley & Sons, Inc.: New York; 1979. [DOI] [PubMed] [Google Scholar]
- Hansson M, Olsson I, Nauseef WM. Biosynthesis, processing, and sorting of human myeloperoxidase. Arch Biochem Biophys. 2006;445:214–224. doi: 10.1016/j.abb.2005.08.009. [DOI] [PubMed] [Google Scholar]
- Hasegawa T, Malle E, Farhood A, Jaeschke H. Generation of hypochlorite-modified proteins by neutrophils during ischemia-reperfusion injury in rat liver: attenuation by ischemic preconditioning. Am J Physiol Gastrointest Liver Physiol. 2005;289:G760–G767. doi: 10.1152/ajpgi.00141.2005. [DOI] [PubMed] [Google Scholar]
- Hawkins CL, Davies MJ. Hypochlorite-induced damage to proteins: formation of nitrogen-centred radicals from lysine residues and their role in protein fragmentation. Biochem J. 1998;332:617–625. doi: 10.1042/bj3320617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hazell LJ, Arnold L, Flowers D, Waeg G, Malle E, Stocker R. Presence of hypochlorite-modified proteins in human atherosclerotic lesions. J Clin Invest. 1996;97:1535–1544. doi: 10.1172/JCI118576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hazen SL, Heinecke JW. 3-Chlorotyrosine, a specific marker of myeloperoxidase-catalyzed oxidation, is markedly elevated in low density lipoprotein isolated from human atherosclerotic intima. J Clin Invest. 1997;99:2075–2081. doi: 10.1172/JCI119379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hazen SL, Hsu FF, Mueller DM, Crowley JR, Heinecke JW. Human neutrophils employ chlorine gas as an oxidant during phagocytosis. J Clin Invest. 1996;98:1283–1289. doi: 10.1172/JCI118914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hori H, Fenna RE, Kimura S, Ikeda-Saito M. Aromatic substrate molecules bind at the distal heme pocket of myeloperoxidase. J Biol Chem. 1994;269:8388–8392. [PubMed] [Google Scholar]
- Hoy A, Trégouet D, Leininger-Muller B, Poirier O, Maurice M, Sass C, et al. Serum myeloperoxidase concentration in a healthy population: biological variations, familial resemblance and new genetic polymorphisms. Eur J Hum Genet. 2001;9:780–786. doi: 10.1038/sj.ejhg.5200702. [DOI] [PubMed] [Google Scholar]
- Hsuanyu Y, Dunford HB. Oxidation of clozapine and ascorbate by myeloperoxidase. Arch Biochem Biophys. 1999;368:413–420. doi: 10.1006/abbi.1999.1328. [DOI] [PubMed] [Google Scholar]
- Ikeda-Saito M, Shelley DA, Lu L, Booth KS, Caughey WS, Kimura S. Salicylhydroxamic acid inhibits myeloperoxidase activity. J Biol Chem. 1991;266:3611–3616. [PubMed] [Google Scholar]
- Jacquet A, Deby C, Mathy M, Moguilevsky N, Deby-Dupont G, Thirion A, et al. Spectral and enzymatic properties of human recombinant myeloperoxidase: comparison with the mature enzyme. Arch Biochem Biophys. 1991;291:132–138. doi: 10.1016/0003-9861(91)90115-y. [DOI] [PubMed] [Google Scholar]
- Jantschko W, Furtmüller PG, Allegra M, Livrea MA, Jakopitsch C, Regelsberger G, et al. Redox intermediates of plant and mammalian peroxidases: a comparative transient-state kinetic study of their reactivity toward indole derivatives. Arch Biochem Biophys. 2002;398:12–22. doi: 10.1006/abbi.2001.2674. [DOI] [PubMed] [Google Scholar]
- Jantschko W, Furtmüller PG, Zederbauer M, Neugschwandtner K, Lehner I, Jakopitsch C, et al. Exploitation of the unusual properties of human myeloperoxidase in inhibitor design. Biochem Pharmacol. 2005;69:1149–1157. doi: 10.1016/j.bcp.2005.02.006. [DOI] [PubMed] [Google Scholar]
- Josephy PD, Eling T, Mason RP. The horseradish peroxidase-catalyzed oxidation of 3,5,3′,5′-tetramethylbenzidine. Free radical and charge-transfer complex intermediates. J Biol Chem. 1982;257:3669–3675. [PubMed] [Google Scholar]
- Kettle AJ, Candaeis LP. Oxidation of tryptophan by redox intermediates of myeloperoxidase and inhibition of hypochlorous acid production. Redox Rep. 2000;5:179–184. doi: 10.1179/135100000101535726. [DOI] [PubMed] [Google Scholar]
- Kettle AJ, Chan T, Osberg I, Senthilmohan R, Chapman AL, Mocatta TJ, et al. Myeloperoxidase and protein oxidation in the airways of young children with cystic fibrosis. Am J Respir Crit Care Med. 2004;170:1317–1323. doi: 10.1164/rccm.200311-1516OC. [DOI] [PubMed] [Google Scholar]
- Kettle AJ, Gedye CA, Hampton MB, Winterbourn CC. Inhibition of myeloperoxidase by benzoic acid hydrazides. Biochem J. 1995;308:559–563. doi: 10.1042/bj3080559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kettle AJ, Gedye CA, Winterbourn CC. Superoxide is an antagonist of anti-inflammatory drugs that inhibit hypochlorous acid production by myeloperoxidase. Biochem Pharmacol. 1993;45:2003–2010. doi: 10.1016/0006-2952(93)90010-t. [DOI] [PubMed] [Google Scholar]
- Kettle AJ, Gedye CA, Winterbourn CC. Mechanism of inactivation of myeloperoxidase by 4-aminobenzoic acid hydrazide. Biochem J. 1997;321:503–508. doi: 10.1042/bj3210503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kettle AJ, Robertson IG, Palmer BD, Anderson RF, Patel KB, Winterbourn CC. Oxidative metabolism of amsacrine by the neutrophil enzyme myeloperoxidase. Biochem Pharmacol. 1992;44:1731–1738. doi: 10.1016/0006-2952(92)90066-r. [DOI] [PubMed] [Google Scholar]
- Kettle AJ, Winterbourn CC. The mechanism of myeloperoxidase-dependent chlorination of monochlorodimedon. Biochim Biophys Acta. 1988;957:185–191. doi: 10.1016/0167-4838(88)90271-3. [DOI] [PubMed] [Google Scholar]
- Kettle AJ, Winterbourn CC. Mechanism of inhibition of myeloperoxidase by anti-inflammatory drugs. Biochem Pharmacol. 1991;41:1485–1492. doi: 10.1016/0006-2952(91)90565-m. [DOI] [PubMed] [Google Scholar]
- Kettle AJ, Winterbourn CC. Oxidation of hydroquinone by myeloperoxidase. Mechanism of stimulation by benzoquinone. J Biol Chem. 1992;267:8319–8324. [PubMed] [Google Scholar]
- Kettle AJ, Winterbourn CC. Assays for the chlorination activity of myeloperoxidase. Methods Enzymol. 1994;233:502–512. doi: 10.1016/s0076-6879(94)33056-5. [DOI] [PubMed] [Google Scholar]
- Klebanoff SJ. Myeloperoxidase: friend and foe. J Leukoc Biol. 2005;77:598–625. doi: 10.1189/jlb.1204697. [DOI] [PubMed] [Google Scholar]
- Klebanoff SJ, Waltersdorph AM. Inhibition of peroxidase-catalyzed reactions by desferoxamine. Arch Biochem Biophys. 1988;264:600–606. doi: 10.1016/0003-9861(88)90326-8. [DOI] [PubMed] [Google Scholar]
- Koeffler HP, Ranyard J, Pertcheck M. Myeloperoxidase: its structure and expression during myeloid differentiation. Blood. 1985;65:484–491. [PubMed] [Google Scholar]
- Kono Y, Shibata H, Kodama Y, Ueda A, Sawa Y. Chlorogenic acid as a natural scavenger for hypochlorous acid. Biochem Biophys Res Commun. 1995;217:972–978. doi: 10.1006/bbrc.1995.2865. [DOI] [PubMed] [Google Scholar]
- Kooter IM, Moguilevsky N, Bollen A, Sijtsema NM, Otto C, Wever R. Site-directed mutagenesis of Met243, a residue of myeloperoxidase involved in binding of the prosthetic group. J Biol Inorg Chem. 1997;2:191–197. [Google Scholar]
- Kubow S, Wells PG. In vitro bioactivation of phenytoin to a reactive free radical intermediate by prostaglandin synthetase, horseradisch peroxidase, and thyroid peroxidase. Mol Pharmacol. 1989;35:504–511. [PubMed] [Google Scholar]
- Kumar AP, Reynolds WF. Statins downregulate myeloperoxidase gene expression in macrophages. Biochem Biophys Res Commun. 2005;331:442–451. doi: 10.1016/j.bbrc.2005.03.204. [DOI] [PubMed] [Google Scholar]
- Kutter D, Devaquet P, Vanderstocken G, Paulus JM, Marchal V, Gothot A. Consequences of total and subtotal myeloperoxidase deficiency: risk or benefit. Acta Haematol. 2000;104:10–15. doi: 10.1159/000041062. [DOI] [PubMed] [Google Scholar]
- Lai WG, Gardner I, Zahid N, Uetrecht JP. Bioactivation and covalent binding of hydroxyfluperlapine in human neutrophils: implications for fluperlapine-induced agranulocytosis. Drug Metab Dispos. 2000;28:255–263. [PubMed] [Google Scholar]
- Lai WG, Zahid N, Uetrecht JP. Metabolism of trimethoprim to a reactive iminoquinone methide by activated human neutrophils and hepatic microsomes. J Pharmacol Exp Ther. 1999;291:292–299. [PubMed] [Google Scholar]
- Leßig J, Schiller J, Arnhold J, Fuchs B. Hypochlorous acid-mediated generation of glycerophosphocholine from unsaturated plasmalogen glycerophosphocholine lipids. J Lipid Res. 2007;48:1316–1324. doi: 10.1194/jlr.M600478-JLR200. [DOI] [PubMed] [Google Scholar]
- Lee E, Miki Y, Katsura H, Kariya K. Mechanism of inactivation of myeloperoxidase by propylthiouracil. Biochem Pharmacol. 1990;39:1467–1471. doi: 10.1016/0006-2952(90)90428-n. [DOI] [PubMed] [Google Scholar]
- Lewis D, Capell HA, McNeil CJ, Iqbal MS, Brown DH, Smith WE. Gold levels produced by treatment with auranofin and sodium-aurothiomalate. Ann Rheum Dis. 1983;42:566–570. doi: 10.1136/ard.42.5.566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maehly AC, Chance B. The assay of catalases and peroxidases. Methods Biochem Anal. 1954;1:357–424. doi: 10.1002/9780470110171.ch14. [DOI] [PubMed] [Google Scholar]
- Malle E, Buch T, Grone HJ. Myeloperoxidase in kidney disease. Kidney Int. 2003;64:1956–1967. doi: 10.1046/j.1523-1755.2003.00336.x. [DOI] [PubMed] [Google Scholar]
- Malle E, Hazell L, Stocker R, Sattler W, Esterbauer H, Waeg G. Immunologic detection and measurement of hypochlorite-modified LDL with specific monoclonal antibodies. Arterioscler Thromb Vasc Biol. 1995;15:982–989. doi: 10.1161/01.atv.15.7.982. [DOI] [PubMed] [Google Scholar]
- Malle E, Marsche G, Arnhold J, Davies MJ. Modification of low-density lipoprotein by myeloperoxidase-derived oxidants and reagent hypochlorous acid. Biochim Biophys Acta. 2006a;1761:392–415. doi: 10.1016/j.bbalip.2006.03.024. [DOI] [PubMed] [Google Scholar]
- Malle E, Marsche G, Panzenboeck U, Sattler W. Myeloperoxidase-mediated oxidation of high-density lipoproteins: fingerprints of newly recognized potential proatherogenic lipoproteins. Arch Biochem Biophys. 2006b;445:245–255. doi: 10.1016/j.abb.2005.08.008. [DOI] [PubMed] [Google Scholar]
- Malle E, Waeg G, Schreiber R, Gröne EF, Sattler W, Gröne HJ. Immunohistochemical evidence for the myeloperoxidase/H2O2/halide system in human atherosclerotic lesions. Eur J Biochem. 2000;267:4495–4503. doi: 10.1046/j.1432-1327.2000.01498.x. [DOI] [PubMed] [Google Scholar]
- Malle E, Wäg G, Thiery J, Sattler W, Gröne HJ. Hypochlorite-modified (lipo)proteins are present in rabbit lesions in response to dietary cholesterol. Biochem Biophys Res Commun. 2001;289:894–900. doi: 10.1006/bbrc.2001.6074. [DOI] [PubMed] [Google Scholar]
- Malle E, Woenckhaus C, Waeg G, Esterbauer H, Grone EF, Grone HJ. Immunological evidence for hypochlorite-modified proteins in human kidney. Am J Pathol. 1997;150:603–615. [PMC free article] [PubMed] [Google Scholar]
- Marquez LA, Dunford HB. Kinetics of oxidation of tyrosine and dityrosine by myelo-peroxidase compounds I and II. Implications for lipoprotein peroxidation studies. J Biol Chem. 1995;270:30434–30440. doi: 10.1074/jbc.270.51.30434. [DOI] [PubMed] [Google Scholar]
- Marquez LA, Dunford HB. Mechanism of the oxidation of 3,5,3′,5′-tetramethylbenzidine by myeloperoxidase determined by transient- and steady-state kinetics. Biochemistry. 1997;36:9349–9355. doi: 10.1021/bi970595j. [DOI] [PubMed] [Google Scholar]
- Marquez LA, Dunford HB, Van Wart H. Kinetic studies on the reaction of compound II of myeloperoxidase with ascorbic acid. Role of ascorbic acid in myeloperoxidase function. J Biol Chem. 1990;265:5666–56670. [PubMed] [Google Scholar]
- Marsche G, Heller R, Fauler G, Kovacevic A, Nuszkowski A, Graier W, et al. 2-Chlorohexadecanal derived from hypochlorite-modified high-density lipoprotein-associated plasmalogen is a natural inhibitor of endothelial nitric oxide biosynthesis. Arterioscler Thromb Vasc Biol. 2004;24:2302–2306. doi: 10.1161/01.ATV.0000148703.43429.25. [DOI] [PubMed] [Google Scholar]
- Marsche G, Levak-Frank S, Quehenberger O, Heller R, Sattler W, Malle E. Identification of the human analog of SR-BI and LOX-1 as receptors for hypochlorite-modified high density lipoprotein on human umbilical venous endothelial cells. FASEB J. 2001;15:1095–1097. doi: 10.1096/fj.00-0532fje. [DOI] [PubMed] [Google Scholar]
- Marsche G, Semlitsch M, Hammer A, Frank S, Weigle B, Demling N, et al. Hypochlorite-modified albumin colocalizes with RAGE in the artery wall and promotes MCP-1 expression via the RAGE-Erk1/2 MAP-kinase pathway. FASEB J. 2007a;21:1145–1152. doi: 10.1096/fj.06-7439com. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsche G, Weigle B, Sattler W, Malle E.Soluble RAGE blocks scavenger receptor CD-36-mediated uptake of hypochlorite-modified low-density lipoprotein FASEB J 2007b21 10.1096/fj.07-8316com(in press)Doi [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsche G, Zimmermann R, Horiuchi S, Tandon NN, Sattler W, Malle E. Class B scavenger receptors CD36 and SR-BI are receptors for hypochlorite-modified low density lipoprotein. J Biol Chem. 2003;278:47562–47570. doi: 10.1074/jbc.M308428200. [DOI] [PubMed] [Google Scholar]
- Mays DC, Pawluk LJ, Apseloff G, Davis WB, She ZW, Sagone AL, et al. Metabolism of phenytoin and covalent binding of reactive intermediates in activated human neutrophils. Biochem Pharmacol. 1995;50:367–380. doi: 10.1016/0006-2952(95)00151-o. [DOI] [PubMed] [Google Scholar]
- McCarty MF. The reported clinical utility of taurine in ischemic disorders may reflect a down-regulation of neutrophil activation and adhesion. Med Hypotheses. 1999;53:290–299. doi: 10.1054/mehy.1998.0760. [DOI] [PubMed] [Google Scholar]
- McGirr LG, Jatoe SD, O'Brien PJ. Myeloperoxidase catalysed cooxidative metabolism of methimazole: oxidation of glutathione and NADH by free radical intermediates. Chem Biol Interact. 1990;73:279–295. doi: 10.1016/0009-2797(90)90009-c. [DOI] [PubMed] [Google Scholar]
- McMillen TS, Heinecke JW, LeBoeuf RC. Expression of human myeloperoxidase by macrophages promotes atherosclerosis in mice. Circulation. 2005;111:2798–2804. doi: 10.1161/CIRCULATIONAHA.104.516278. [DOI] [PubMed] [Google Scholar]
- Milla C, Yang S, Cornfield DN, Brennan ML, Hazen SL, Panoskaltsis-Mortari A, et al. Myeloperoxidase deficiency enhances inflammation after allogeneic marrow transplantation. Am J Physiol Lung Cell Mol Physiol. 2004;287:L706–L714. doi: 10.1152/ajplung.00015.2004. [DOI] [PubMed] [Google Scholar]
- Misra HP, Fridovich I. The oxidation of phenylhydrazine: superoxide and mechanism. Biochemistry. 1976;10:681–687. doi: 10.1021/bi00648a036. [DOI] [PubMed] [Google Scholar]
- Miyamoto G, Zahid N, Uetrecht JP. Oxidation of diclofenac to reactive intermediates by neutrophils, myeloperoxidase, and hypochlorous acid. Chem Res Toxicol. 1997;10:414–419. doi: 10.1021/tx960190k. [DOI] [PubMed] [Google Scholar]
- Miyamoto S, Martinez GR, Rettori D, Augusto O, Medeiros MH, Di Mascio P. Linoleic acid hydroperoxide reacts with hypochlorous acid, generating peroxyl radical intermediates and singlet molecular oxygen. Proc Natl Acad Sci USA. 2006;103:293–298. doi: 10.1073/pnas.0508170103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moguilevsky N, Garcia-Quintana L, Jacquet A, Tournay C, Fabry L, Pierard L, et al. Structural and biological properties of human recombinant myeloperoxidase produced by Chinese hamster ovary cell lines. Eur J Biochem. 1991;197:605–614. doi: 10.1111/j.1432-1033.1991.tb15950.x. [DOI] [PubMed] [Google Scholar]
- Murakami S, Kondo Y, Sakurai T, Kitajima H, Nagate T. Taurine suppresses development of atherosclerosis in Watanabe heritable hyperlipidemic (WHHL) rabbits. Atherosclerosis. 2002;163:79–87. doi: 10.1016/s0021-9150(01)00764-x. [DOI] [PubMed] [Google Scholar]
- Murakami S, Kondo Y, Tomisawa K, Nagate T. Prevention of atherosclerotic lesion development in mice by taurine. Drugs Exp Clin Res. 1999a;25:227–234. [PubMed] [Google Scholar]
- Murakami S, Kondo-Ohta Y, Tomisawa K. Improvement in cholesterol metabolism in mice given chronic treatment of taurine and fed a high-fat diet. Life Sci. 1999b;64:83–91. doi: 10.1016/s0024-3205(98)00536-0. [DOI] [PubMed] [Google Scholar]
- Nakano M, Koga S. Luciferin derivative for assay of myeloperoxidase and dopamine metabolism. Methods Enzymol. 1994;233:495–501. doi: 10.1016/s0076-6879(94)33055-7. [DOI] [PubMed] [Google Scholar]
- Nappi AJ, Vass E. The effects of nitric oxide on the oxidations of L-dopa and dopamine mediated by tyrosinase and peroxidase. J Biol Chem. 2001;276:11214–11222. doi: 10.1074/jbc.M009872200. [DOI] [PubMed] [Google Scholar]
- Nauseef WM, Cogley M, Bock S, Petrides PE. Pattern of inheritance in hereditary myeloperoxidase deficiency associated with the R569W missense mutation. J Leukoc Biol. 1998;63:264–269. doi: 10.1002/jlb.63.2.264. [DOI] [PubMed] [Google Scholar]
- Neve J, Parij N, Moguilevsky N. Inhibition of the myeloperoxidase chlorinating activity by non-steroidal anti-inflammatory drugs investigated with a human recombinant enzyme. Eur J Pharmacol. 2001;417:37–43. doi: 10.1016/s0014-2999(01)00895-0. [DOI] [PubMed] [Google Scholar]
- Nicholls SJ, Hazen SL. Myeloperoxidase and cardiovascular disease. Arterioscler Thromb Vasc Biol. 2005;25:1102–1111. doi: 10.1161/01.ATV.0000163262.83456.6d. [DOI] [PubMed] [Google Scholar]
- Panasenko OM, Arnhold J, Vladimirov YuA, Arnhold K, Sergienko VI. Hypochlorite-induced peroxidation of egg yolk phosphatidylcholine is mediated by hydroperoxides. Free Radic Res. 1997;27:1–12. doi: 10.3109/10715769709097832. [DOI] [PubMed] [Google Scholar]
- Petty MA, Kintz J, DiFrancesco GF. The effects of taurine on atherosclerosis development in cholesterol-fed rabbits. Eur J Pharmacol. 1990;180:119–127. doi: 10.1016/0014-2999(90)90599-2. [DOI] [PubMed] [Google Scholar]
- Proteasa G, Tahboub YR, Galijasevic S, Raushel FM, Abu-Soud HM. Kinetic evidence supports the existence of two halide binding sites that have a distinct impact on the heme iron microenvironment in myeloperoxidase. Biochemistry. 2007;46:398–405. doi: 10.1021/bi0609725. [DOI] [PubMed] [Google Scholar]
- Reynolds WF, Kumar AP, Piedrafita FJ. The human myeloperoxidase gene is regulated by LXR and PPARα ligands. Biochem Biophys Res Commun. 2006;349:846–854. doi: 10.1016/j.bbrc.2006.08.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rider NL, Pinto D, Covington M, Orwat MJ, Giannaras J, Nurnberg S, et al. Comparative effects of selective cyclooxygenase 1 and cyclooxygenase 2 inhibitors on myeloperoxidase and 3α-hydroxysteroid dehydrogenase. J Enzyme Inhib. 1996;10:73–79. doi: 10.3109/14756369609020160. [DOI] [PubMed] [Google Scholar]
- Roy G, Mugesh G. Anti-thyroid drugs and thyroid hormone synthesis: effect of methimazole derivatives on peroxidase-catalyzed reactions. J Am Chem Soc. 2005;127:15207–15217. doi: 10.1021/ja054497u. [DOI] [PubMed] [Google Scholar]
- Sagone AL, Husney RM, Wevers MD, Herzyk DJ, Davis WB. Effect of dimethylthiourea on the neutrophil myeloperoxidase pathway. J Appl Physiol. 1989;67:1056–1062. doi: 10.1152/jappl.1989.67.3.1056. [DOI] [PubMed] [Google Scholar]
- Sayo H, Saito M. Hydrogen peroxide-dependent oxidation metabolism of 1-methyl-2-mercaptoimidazole (methimazole) catalysed by myeloperoxidase. Xenobiotica. 1991;21:1217–1224. doi: 10.3109/00498259109039562. [DOI] [PubMed] [Google Scholar]
- Scheuer H, Gwinner W, Hohbach J, Gröne EF, Brandes RP, Malle E, et al. Oxidant stress in hyperlipidemia-induced renal damage. Am J Physiol Renal Physiol. 2000;278:F63–F74. doi: 10.1152/ajprenal.2000.278.1.F63. [DOI] [PubMed] [Google Scholar]
- Schonbaum GR. New complexes of peroxidases with hydroxamic acids, hydrazides, and amides. J Biol Chem. 1973;248:502–511. [PubMed] [Google Scholar]
- Schonbaum GR, Lo S. Interaction of peroxidases with aromatic peracids and alkyl peroxides. J Biol Chem. 1972;247:3353–3360. [PubMed] [Google Scholar]
- Schuller-Levis GB, Park E. Taurine: new implications for an old amino acid. FEMS Microbiol Lett. 2003;226:195–202. doi: 10.1016/S0378-1097(03)00611-6. [DOI] [PubMed] [Google Scholar]
- Schultz J, Kaminker K. Myeloperoxidase of the leucocyte of normal human blood. I. Content and localization. Arch Biochem Biophys. 1962;96:465–467. doi: 10.1016/0003-9861(62)90321-1. [DOI] [PubMed] [Google Scholar]
- Shacter E, Lopez RL, Pati S. Inhibition of the myeloperoxidase-H2O2-Cl−system of neutrophils by indomethacin and other non-steroidal anti-inflammatory drugs. Biochem Pharmacol. 1991;41:975–984. doi: 10.1016/0006-2952(91)90204-i. [DOI] [PubMed] [Google Scholar]
- Soyer Z, Bas M, Pabuccuoglu A, Pabuccuoglu V. Synthesis of some 2(3H)-benzoxazolone derivatives and their in-vitro effects on human leukocyte myeloperoxidase activity. Arch Pharm Chem Life Sci. 2005;338:405–410. doi: 10.1002/ardp.200500106. [DOI] [PubMed] [Google Scholar]
- Stendahl O, Molin L, Dahlgren C. The inhibition of polymorphonuclear leukocyte cytotoxicity by dapsone: a possible mechanism in the treatment of dermatitis herpetiformis. J Clin Invest. 1978;62:214–220. doi: 10.1172/JCI109109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenvinkel P, Rodríguez-Ayala E, Massy ZA, Qureshi AR, Barany P, Fellström B, et al. Strain treatment and diabetes affect myeloperoxidase activity in maintenance hemodialysis patients. Clin J Am Soc Nephrol. 2006;1:281–287. doi: 10.2215/CJN.01281005. [DOI] [PubMed] [Google Scholar]
- Suzuki K, Ota H, Sasagawa S, Sakatani T, Fujikura T. Assay method for myeloperoxidase in human polymorphonuclear leukocytes. Anal Biochem. 1983;132:345–352. doi: 10.1016/0003-2697(83)90019-2. [DOI] [PubMed] [Google Scholar]
- Takeshita J, Byun J, Nhan TQ, Pritchard DK, Pennathur S, Schwartz SM, et al. Myeloperoxidase generates 5-chlorouracil in human atherosclerotic tissue: a potential pathway for somatic mutagenesis by macrophages. J Biol Chem. 2006;281:3096–3104. doi: 10.1074/jbc.M509236200. [DOI] [PubMed] [Google Scholar]
- Tarpey MM, Wink DA, Grisham MB. Methods for detection of reactive metabolites of oxygen and nitrogen: in vitro and in vivo considerations. Am J Physiol Regul Integr Comp Physiol. 2004;286:R431–R444. doi: 10.1152/ajpregu.00361.2003. [DOI] [PubMed] [Google Scholar]
- Thomas EL, Grisham MB, Jefferson MM. Preparation and characterization of chloramines. Methods Enzymol. 1986;132:569–585. doi: 10.1016/s0076-6879(86)32042-1. [DOI] [PubMed] [Google Scholar]
- Thukkani AK, Martinson BD, Albert CJ, Vogler GA, Ford DA. Neutrophil-mediated accumulation of 2-ClHDA during myocardial infarction: 2-ClHDA-mediated myocardial injury. Am J Physiol Heart Circ Physiol. 2005;288:H2955–H2964. doi: 10.1152/ajpheart.00834.2004. [DOI] [PubMed] [Google Scholar]
- Thukkani AK, McHowat J, Hsu FF, Brennan ML, Hazen SL, Ford DA. Identification of alpha-chloro fatty aldehydes and unsaturated lysophosphatidylcholine molecular species in human atherosclerotic lesions. Circulation. 2003;108:3128–3133. doi: 10.1161/01.CIR.0000104564.01539.6A. [DOI] [PubMed] [Google Scholar]
- Uetrecht J, Zahid N. N-chlorination of phenytoin by myeloperoxidase to a reactive metabolite. Chem Res Toxicol. 1988;1:148–151. doi: 10.1021/tx00003a004. [DOI] [PubMed] [Google Scholar]
- Uetrecht J, Zahid N, Tehim A, Fu JM, Rakhit S. Structural features associated with reactive metabolite formation in clozapine analogues. Chem Biol Interact. 1997;104:117–129. doi: 10.1016/s0009-2797(97)00017-3. [DOI] [PubMed] [Google Scholar]
- Uetrecht JP, Zahid N. N-Chlorination and oxidation of procainamide by myeloperoxidase: toxicological implications. Chem Res Toxicol. 1991;4:218–222. doi: 10.1021/tx00020a015. [DOI] [PubMed] [Google Scholar]
- Van der Korst JK, van de Stadt RJ, Muijsers AO, Ament HJ, Henrichs AM.Pharmacokinetics of D-penicillamine in rheumatoid arthritis Modulation of Autoimmunity and Disease 1981Praeger: New York; 165–171.In: Maini RN, Berry H (eds). [Google Scholar]
- van Zyl JM, Basson K, Kriegler A, van der Walt BJ. Activation of chlorpromazine by the myeloperoxidase system of the human neutrophil. Biochem Pharmacol. 1990;40:947–954. doi: 10.1016/0006-2952(90)90478-4. [DOI] [PubMed] [Google Scholar]
- van Zyl JM, Basson K, Uebel RA, van der Walt BJ. Isoniazid-mediated irreversible inhibition of the myeloperoxidase antimicrobial system of the human neutrophil and the effect of thyronines. Biochem Pharmacol. 1989a;38:2363–2373. doi: 10.1016/0006-2952(89)90477-2. [DOI] [PubMed] [Google Scholar]
- van Zyl JM, Basson K, van der Walt BJ. The inhibitory effect of acetaminophen on the myeloperoxidase-induced antimicrobial system of the polymorphonuclear leukocyte. Biochem Pharmacol. 1989b;38:161–165. doi: 10.1016/0006-2952(89)90163-9. [DOI] [PubMed] [Google Scholar]
- Van Zyl JM, van der Walt BJ. Thiacetazone acts as a scavenger of myeloperoxidase-generated hypochlorous acid and singlet oxygen. Med Sci Res. 1996;24:181–183. [Google Scholar]
- Weiss SJ, Klein R, Slivka A, Wei M. Chlorination of taurine by human neutrophils. Evidence for hypochlorous acid generation. J Clin Invest. 1982;70:598–607. doi: 10.1172/JCI110652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams DP, Pirmohamed M, Naisbitt DJ, Maggs JL, Park BK. Neutrophil cytotoxicity of the chemically reactive metabolite(s) of clozapine: possible role in agranolocytosis. J Pharmacol Exp Ther. 1997;283:1375–1382. [PubMed] [Google Scholar]
- Wolber G, Langer T. LigandScout: 3-D pharmacophores derived from protein-bound ligands and their use as virtual screening filters. J Chem Inf Model. 2005;45:160–169. doi: 10.1021/ci049885e. [DOI] [PubMed] [Google Scholar]
- Yamada M, Kurahashi K. Regulation of myeloperoxidase gene expression during differentiation of human myeloid leukemia HL-60 cells. J Biol Chem. 1984;259:3021–3025. [PubMed] [Google Scholar]
- Yang X-X, Hu Z-P, Chan SY, Zhou SF. Monitoring drug–protein interaction. Clin Chim Acta. 2006;365:9–29. doi: 10.1016/j.cca.2005.08.021. [DOI] [PubMed] [Google Scholar]
- Zederbauer M, Furtmüller PG, Bellei M, Stampler J, Jakopitsch C, Battistuzzi G, et al. Disruption of the aspartate to heme ester linkage in human myeloperoxidase: impact on ligand binding, redox chemistry and interconversion of redox intermediates. J Biol Chem. 2007a;282:17041–17052. doi: 10.1074/jbc.M610685200. [DOI] [PubMed] [Google Scholar]
- Zederbauer M, Furtmüller PG, Ganster B, Moguilevsky N, Obinger C. Manipulating the vinyl-sulfonium bond in human myeloperoxidase: impact on compound I formation and reduction by halides and thiocyanate. Biochem Biophys Res Commun. 2007b;356:450–456. doi: 10.1016/j.bbrc.2007.02.157. [DOI] [PubMed] [Google Scholar]
- Zederbauer M, Jantschko W, Neugschwandtner K, Jakopitsch C, Moguilevsky N, Obinger C, et al. The role of the covalent Glu242-heme linkage in the formation and reactivity of redox intermediates of human myeloperoxidase. Biochemistry. 2005;44:6482–6491. doi: 10.1021/bi0501737. [DOI] [PubMed] [Google Scholar]
- Zeng J, Fenna RE. X-ray crystal structure of canine myeloperoxidase at 3 Å resolution. J Mol Biol. 1992;226:185–207. doi: 10.1016/0022-2836(92)90133-5. [DOI] [PubMed] [Google Scholar]
- Zheng L, Nukuna B, Brennan ML, Sun M, Goormastic M, Settle M, et al. Apolipoprotein A-I is a selective target for myeloperoxidase-catalyzed oxidation and functional impairment in subjects with cardiovascular disease. J Clin Invest. 2004;114:529–541. doi: 10.1172/JCI21109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou T, Zhou SH, Qi SS, Shen XQ, Zeng GF, Zhou HN. The effect of atorvastatin on serum myeloperoxidase and CRP levels in patients with acute coronary syndrome. Clin Chim Acta. 2006;368:168–172. doi: 10.1016/j.cca.2005.12.040. [DOI] [PubMed] [Google Scholar]
- Zhu BZ, Carr AC, Frei B. Pyrrolidine dithiocarbamate is a potent antioxidant against hypochlorous acid-induced protein damage. FEBS Lett. 2002;532:80–84. doi: 10.1016/s0014-5793(02)03637-2. [DOI] [PubMed] [Google Scholar]
- Zuurbier KWM, Bakkenist ARJ, Fokkens RH, Nibbering NMM, Wever R, Muijsers AO. Inhibition of the chlorinating activity of myeloperoxidase by diclofenac and oxidation of diclofenac to dihydroxyazobenzene by myeloperoxidase. Biochem Pharmacol. 1990;40:1801–1808. doi: 10.1016/0006-2952(90)90359-s. [DOI] [PubMed] [Google Scholar]