

Abstract
Nkx2-5 is a homeobox containing transcription factor that is conserved and expressed in organisms that form hearts. Fruit flies lacking the gene (tinman) fail to form a dorsal vessel, mice that are homozygous null for Nkx2-5 form small, deformed hearts, and several human cardiac defects have been linked to dominant mutations in the Nkx2-5 gene. The Xenopus homologs (XNkx2-5) of two truncated forms of Nkx2-5 that have been identified in humans with congenital heart defects were used in the studies reported here. mRNAs encoding these mutations were injected into single cell Xenopus embryos, and heart development was monitored. Our results indicate that the introduction of truncated XNkx2-5 variants leads to three principle developmental defects. The atrial septum and the valve of the atrioventricular canal were both abnormal. In addition, video microscopic timing of heart contraction indicated that embryos injected with either mutant form of XNkx2-5 have conduction defects.
Keywords: Xenopus, Nkx2-5, heart development
INTRODUCTION
Many of the molecules used to direct and regulate heart development are conserved in different organisms. Among these common molecules are members of the NK-2 gene family. These genes encode homeodomain proteins that regulate the transcription of other genes required to execute particular developmental programs. The first member of this family reported to be involved in cardiac development was the Drosophila tinman gene, followed by the discovery of its homolog, the Nkx2-5 gene, in fish, birds, amphibians and mammals (Bodmer, 1993; Komuro and Izumo, 1993; Tonissen et al., 1994; Evans et al., 1995). Nkx2-5 is expressed early during embryogenesis, and although not exclusively found in embryonic regions destined to be heart, it helps define the cardiogenic field. In many organisms, its expression persists in the heart throughout development.
Tinman, Nkx2-5, and related proteins share several common domains (Newman and Krieg, 1998) that are critical for their function as transcription factors (Sepulveda et al., 1998, 2002; Belaguli et al., 2000; Kasahara et al., 2000). Starting at the N-terminus of the protein (and schematized for Xenopus Nkx2-5 in Fig. 1) are the TN domain, the homeodomain, the NK domain and finally the GIRAW domain at the carboxy terminus. Amino acid sequence identity between all the members of the family and tinman is just over 65% in the homeodomain and 70 – 80% for the NK2-specific domains (Newman and Krieg, 1998). Deletion analysis indicates that the N-terminal domains are essential for promoter activation (Sepulveda et al., 1998) and that the homeodomain mediates DNA binding.
Fig. 1.

Schematic of the Xenopus Nkx2-5 protein (XNkx2-5). The protein is 299 amino acids long and shares conserved domains with other members of the tinman-related family of transcription factors. These conserved domains include 10 amino acid TN (TPFSVKD*LN) shown in vertical stripes, a 60 amino acid homeodomain (diagonal stripes), a 20 amino acid NK-2 domain (horizontal stripes), and a C-terminal 5 amino acid sequence GIRAW (checked). Arrows indicate the location of the two truncation mutants used in this study, XNkx2-5 His156ter and Gln185ter.
Progressive deletion of the C-terminal domains enhances transcriptional activation by Nkx2-5, suggesting these sequences serve to modulate activity. The modulating activity of the C-terminus was lost when Nkx2-5 served as a transcriptional partner with GATA-4 (Sepulveda et al., 1998). Kasahara and colleagues (Kasahara et al., 2001a,b) were able to identify regions within Nkx2-5 important for interaction as a homodimer and for heterodimerization with Nkx2-3, Nkx2-6, and GATA-4. Protein:protein interactions could occur even when DNA binding capacity was lost.
The function of Nkx2-5 and its relatives has been examined in a variety of ways. For instance, the loss of the tinman gene in flies prevented the development of the dorsal vessel (the fly’s heart; Bodmer, 1993). Mice without functional Nkx2-5 formed small hearts that failed to loop, failed to septate, had underdeveloped ventricles and malformed atrioventricular canals (Lyons et al., 1995; Harvey, 1997). These null studies are complemented by overexpression studies carried out in Xenopus that showed that increased XNkx2-5 levels (Cleaver et al., 1996) led to enlarged hearts in early embryos.
Expression of a dominant-negative XNkx2-5 in frog embryos led to aberrant hearts of markedly reduced size (Fu et al., 1998; Grow and Krieg, 1998). In some cases, the heart was undetected (Grow and Krieg, 1998). These dominant-negative mutations were constructed in two ways and give some clues to how Nkx2-5 functions. Grow and Krieg (1998) introduced a single mutation into the third helix of Nkx2-5 homeodomain rendering the protein unable to bind to DNA. This sort of dominant-negative presumably interacts with its normal protein-binding partners, but fails to bind to appropriate regulatory sites on a promoter. Fu et al. (1998) fused the isolated DNA binding domain from Nkx2-5 to the engrailed repressor. This protein would bind to appropriate promoter sequences but be incapable of interacting with other protein partners and serve exclusively as a repressor. They reported similar results to those described by Grow and Krieg (Grow and Krieg, 1998). All these studies indicated that the expression of Nkx2-5 was critical for proper heart formation and that the regulatory role of Nkx2-5 is relatively cardiac-specific and dose-dependent.
Mutations in the Nkx2-5 gene have been reported in humans with congenital heart disease. Individuals were heterozygous for the mutations, indicating the mutations are either dominant or the result of haploinsufficiency. The phenotypes of patients were varied. Two specific Nkx2-5 mutations (Nkx2-5 Gln170ter and Gln198ter) usually led to problems that included atrial septal defects, atrioventricular heart block (conduction system defects), and, less commonly, tetralogy of Fallot, mitral valve defects, left ventricular hypertrophy, pulmonary atresia, and ventricular septal defects (Schott et al., 1998). Notably, these mutations do not lead to the severe phenotypes observed in the dominant-negative studies discussed above. These proteins both lack the C-terminal regions that encourage association with other transcription factors like GATA-4 and are truncated beyond the point where hyperactivation of test promoters was observed (Kasahara et al., 2000). The smaller of the two mutants (Nkx2-5 Gln170ter) fails to bind to DNA and fails to activate test promoters, whereas the larger mutant (Nkx2-5 Gln198ter) has both these activities when tested in tissue culture cells (Kasahara et al., 2000). When expressed in the presence of a normal copy of Nkx2-5 in humans, however, they give rise to similar phenotypes (Schott et al., 1998).
We have examined the effect of expressing the Xenopus Nkx2-5 proteins that correspond to the Nkx2-5 Gln170ter and Nkx2-5 Gln198ter human mutations. Because the frog proteins are slightly smaller, XNkx2-5 His156ter and Gln185ter are equivalent to the human point mutations Nkx2-5 Gln170ter and Nkx2-5 Gln198ter. Prior studies on dominant-negative XNkx2-5 resulted in very severe problems and were not amenable to inspection of more subtle changes, such as changes in internal cardiac structure or cardiac contractility. In this study, we have waited to analyze the effect of the treatments until hearts have looped, formed their atrial septum and atrioventricular valve and cardiac contraction could be simply timed and analyzed using video microscopy (Kolker et al., 2000; Bartlett et al., 2004). We present here our findings that early expression of XNkx2-5 truncation mutants alter heart morphology and physiology. We conclude that the transient expression of these Nkx2-5 mutations in Xenopus leads to phenotypes similar to those seen in mammals carrying the mutations. These studies suggest that haploinsufficiency does not account for the defects caused by the mutations and the sustained expression of the mutant protein is not needed to cause congenital defects.
RESULTS
Generation of XNkx2-5 Corresponding to the Human Point Mutations Gln170ter and Gln198ter
Among the first Nkx2-5 mutations linked to cardiovascular defects in humans were two point mutations that caused premature termination of the Nkx2-5 protein, specifically Gln170ter and Gln198ter (Schott et al., 1998). The alignment of the human Nkx2-5 protein with the Xenopus laevis Nkx2-5 protein reveals these two proteins are 56% identical and 67% similar. Not surprisingly, the DNA binding domain has conserved sequence of 61 of 64 amino acids. The Gln170ter mutation in the human is found 28 amino acids inside this region of high homology. The corresponding mutation in Xenopus ends the protein at the only nonsimilar residue in the region, a histidine at position 156 rather than a glutamine. The Gln198 in the human corresponds to the 185th amino acid in the Xenopus protein. Adjacent to the homeobox region is the 25 amino acid NK domain. Humans and frogs share 23 of 25 identical residues with 2 similar residues. This domain is thought to coordinate protein–protein interactions (Sepulveda et al., 1998; Kasahara et al., 2000) and is eliminated in both mutants tested. A schematic of the XNkx2-5 protein is shown in Figure 1. As noted in the material and methods section, plasmids were constructed to allow in vitro synthesis of mRNA encoding XNkx2-5 His156ter and XNkxGln185ter truncated proteins. Both DNA sequence analysis and in vitro translation of the mRNAs confirmed their ability to encode an appropriate protein.
Injection of mRNA Encoding Nkx2-5 or Two Different Carboxyl-Terminal Deletions of Nkx2-5 Resulted in Embryos With Enlarged Atria
We injected 1 to 2 ng of XNkx2-5 into one-cell embryos to study the persistent effect of the early expression of normal or either of the two mutant forms of XNkx2-5 on heart development. Although all mRNAs were degraded over time, there was no difference in the stability of full-length and mutant encoding XNkx transcripts. Approximately half the injected mRNA was degraded by 5 hr after injection. By 10 hr (stage 10.5), approximately 20% remained and by 20 hr (approximately stage 22) between 1 and 4% of input was recovered. In other studies (data not shown, Allen and Weeks, manuscript in preparation), we have examined the persistence of epitope-tagged full-length XNkx2-5 protein generated through mRNA injection into the one-cell stage. Epitope tagged XNkx2-5 was detectable by Western Blot analysis through stage 28, but not after stage 32. Thus, any changes in phenotype we observe have their origins in the early expression of the mutant protein.
Embryos injected with mRNA encoding full-length or truncated Nkx2-5 developed in a manner grossly indistinguishable from controls. This observation is consistent with findings reported by Cleaver et al. (1996). We chose to assay the effect on the morphology of the heart in stage 46 embryos. By stage 46, the heart has finished looping, the atria have assumed a more rostral position and septated, the atrioventricular valve has formed, and the outflow tract is well formed and bent to the left. In addition, blood flow is established and the ventricle is well trabeculated.
Visual inspection of overall embryonic development indicated no global effect due to the injection of XNkx2-5. Once the embryos had reached stage 46, they were fixed, placed on their backs and their hearts examined. What was immediately apparent in our studies was altered atrial size. Digital imaging and measurement of the cross-sectional area of the widest portion of the atria tested this subjective impression.
Atrial size from a representative trial is displayed in Table 1. We noted some absolute size variation with different mating pairs of frogs, however, Table 1 reflects the common trend we observed. The mean atrial area of embryos injected with XNkx2-5 mRNAs was larger than the controls at stage 46. Pairwise comparisons of mRNA-injected embryos with controls confirm the statistical significance of this increase. Measurements analyzed from three such trials indicate that the extent of atrial enlargement does not allow a clear distinction between XNkx2-5 FL and the two termination mutants.
TABLE 1.
Mean Atrial Area of Stage 46 Embryos Injected With Either Water or 1–2 ng of mRNA Encoding XNkx2-5 Full-length (FL) or Translation Termination Mutants Giving Rise to Carboxyl-End Truncated Proteins of 155 or 184 Amino Acids in Lengtha
| Treatment | n | Mean atrial area | Standard Deviation | Standard error of the mean | P value vs water |
|---|---|---|---|---|---|
| Water injected | 27 | 9.2 | 2.77 | 0.533 | |
| Nkx2-5 FL | 66 | 13.23 | 2.7 | 0.332 | <0.001 |
| Nkx2-5 His156ter | 50 | 15.95 | 2.11 | 0.298 | <0.001 |
| Nkx2-5 Gln185ter | 46 | 12.33 | 2.54 | 0.375 | <0.001 |
All embryos were derived from the same pair of adult frogs. Embryos were fixed in Dent’s fixative and stained by whole-mount reactions with anti-troponin T monoclonal antibody followed by reaction with anti-mouse Alexa 568. Measurements were made by focusing on the widest part of the atrial field with a Zeiss Axiophot fluorescence microscope and capturing that image with a SPOT camera. Digital images were exported to NIH image, and the area of the atrial field was determined.
To determine whether the differences in atrial size were due to increased atrial cell number, we counted the number of cells in the heart. Cleaver et al. (1996) reported that stage 31–33 embryos injected with Nkx2-5 mRNA had 1.6 times more myocardial cells than uninjected embryos, indicating that overexpression of Nkx2-5 increases the number of cells in the early heart. We looked to see if the mutant forms of Nkx2-5 had a similar effect by counting cardiac cells at two stages of development after injection of mRNA encoding mutant Nkx2-5. We compared the number of cells with the number in non-injected embryos. At stage 36, we compared 8 to 12 embryos expressing mutant Nkx2-5 to controls. In non-injected embryos, we counted approximately 1,400 total cardiac cells, 700 cells in the inflow region and approximately 600 cells in the outflow region of the embryonic heart. We found no significant difference in a pairwise analysis of hearts from embryos injected with either XNkx2-5 His156ter or Gln185ter compared with non-injected embryos. We also looked for variation in total atrial cell numbers in stage 46 embryos. Atrial cell numbers in all groups fell within the range noted for non-injected embryos (1,550 ± 400 cells) and were not statistically different. This finding suggests that the increased atrial size cannot be attributed to a transient early or a persistent increase in cell number.
Overexpression of Normal XNkx2-5 Can Be Distinguished From the Overexpression of XNkx2-5 His156ter and Gln185ter by Examination of the Inside of the Embryonic Heart
We have previously characterized normal heart development using confocal microscopy (Kolker et al., 2000) and have used this approach to compare internal cardiac structure from non-injected embryos with embryos randomly selected from each experimental group. Water-injected controls and non-injected controls in this study showed no differences in heart structure. However, the embryos injected with mutant XNkx2-5 transcripts often showed altered formation of both the atrial septum and the atrioventricular canal.
Representative phenotypes of the atrial septum are shown in Figure 2 and tabulated in Table 2. The atrial septum normally develops as a single, contiguous extension from the dorsal wall of the atria toward the atrioventricular canal. A normal atrial septum can be seen in Figure 2A,B,E, in pictures taken of a buffer injected control embryo (Fig. 2A,E) and an XNkx-FL mRNA injected embryo (Fig. 2B). The ventral view of sections compiled from the atrial region shows that the normal septum unevenly divides the right and left atria and centers on the atrioventricular canal. Histological section of a normal septum (buffer injected control embryo) is shown in Figure 2E. Figure 2C shows the atrial septum from an embryo injected with XNkx2-5 His156ter. The septum is noticeably thinner than those seen in either panels A or B. The second aberrant morphology seen was atrial septum with a frayed appearance (Fig. 2D, and in histological section in Fig. 2F) and deviates from the center of the atrioventricular canal. The aberrations of the septae (thinning as seen in Fig. 2C or frayed as seen in Fig. 2D) were not exclusive to one form of the mutant mRNA or the other. However, greater than 90% of embryos injected with XNkx2-5 His156ter and 75% of the embryos injected with XNkx2-5 Gln185ter had septal aberrations (Table 2).
Fig. 2.
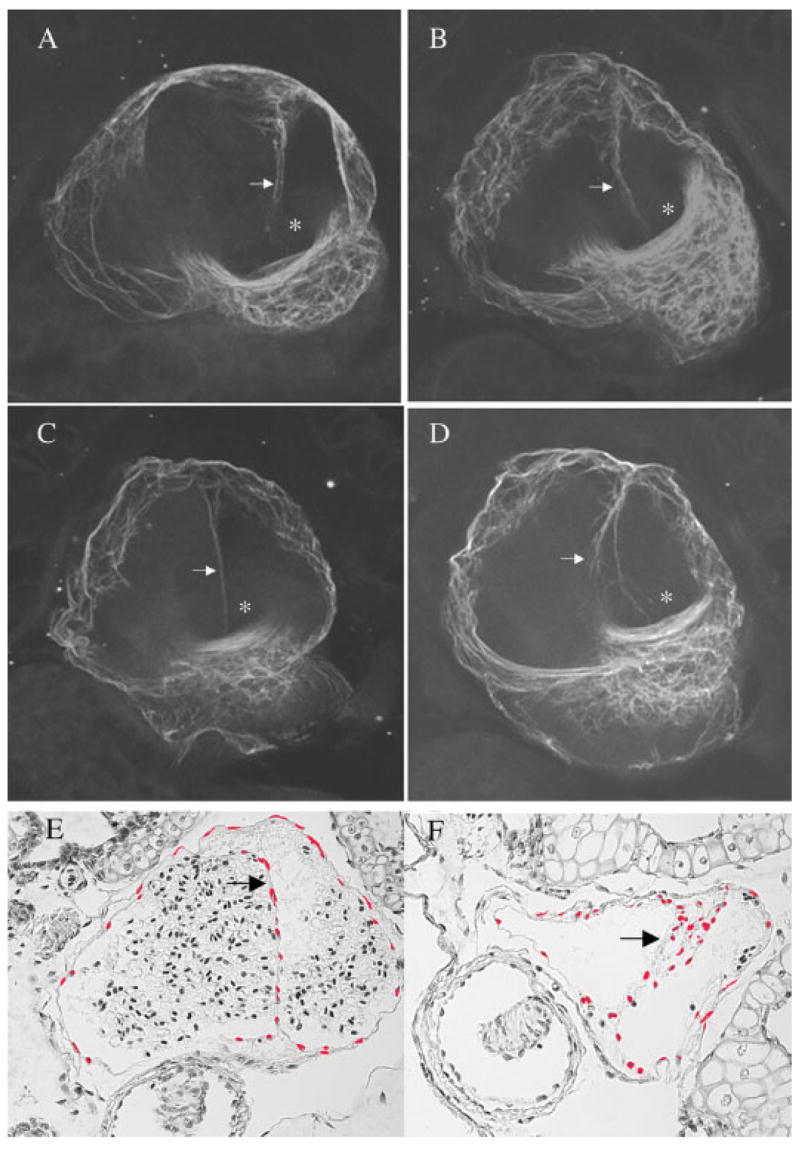
Atrial septum morphology at embryonic stage 46. The images are of 20 compiled confocal sections taken at 8-micron intervals. Embryos were fixed in Dent’s fixative and used for whole-mount immunostaining. The primary antibody used was anti-cardiac troponin T (CT3) monoclonal antibody followed by incubation with anti-mouse Alexa 568 conjugate. Arrowheads point to the atrial septum and the atrioventricular canal is marked with an asterisk. A–D: A normal septum seen in a heart from a water injected control (A), a normal septum seen in a heart from an embryo injected with the XNkx2-5 full length mRNA (B), thin septum seen in a heart from an embryo injected with XNkx2-5 His156ter (C), and a frayed and deviated septum seen in the heart of an embryo injected with XNkx2-5 Gln185ter (D). E,F: Histological sections of hearts from a buffer-injected embryo (E) or an embryo injected with the XNkx2-5 His156ter mRNA (F) allow an alternative comparison of normal and frayed septum. In panels E and F, atrial nuclei were pseudocolored red using Adobe Photoshop to provide a clear view of septal structure.
TABLE 2.
Atrial Septal Morphology in Embryos Injected With XNkx2-5 mRNAa
| Treatment | n | Normal atrial septum/mean atrial area | Thin atrial septum/atrial area | Frayed atrial septum/atrial area |
|---|---|---|---|---|
| Non-injected | 12 | 11/8.3 | 1/9 | 0 |
| Nkx2-5 FL | 28 | 18/11.8 | 10/14.8 | 0 |
| Nkx2-5 His156ter | 31 | 2/14.5 | 20/15.7 | 9/14.7 |
| Nkx2-5 Gln185ter | 37 | 9/11.8 | 10/13 | 18/11.8 |
None of the embryos injected with XNkx2-5 FL had the frayed phenotype, but approximately a third of the embryos injected with Nkx2-5 FL had thinned atrial septum. Because all treatment groups resulted in atrial enlargement, we sought to determine whether enlargement of the atrial area was a predictor of septal defects. As noted in Table 1, all groups had a range of atrial size. As listed in Table 2, we measured atrial size of those embryos with aberrant septa and those with normal septa. For embryos injected with XNkx2-5 FL, hearts with thin septa tended to have larger atrial areas. This trend was apparent for all treatments where thinning of the septa was observed. This correlation did not hold up when frayed septa were observed.
Both of the XNkx2-5 truncation mutants also gave rise to embryos with aberrant atrioventricular canals. This phenotype was less penetrant (from 20% of embryos to 75% of embryos depending on the trial) but striking. Examples of normal and aberrant atrioventricular canals are shown in Figure 3. The normally smooth and well opposed leaflets of the valve seen in control (Fig. 3A) or XNkx2-5 FL-injected embryos (Fig. 3B) were replaced by a dysplastic valve with a smaller annulus and poor leaflet coaptation. The less penetrant phenotype seen for atrioventricular canal formation may be influenced by the genetic composition of the mating pair of adults used to generate embryos. We did not observe atrioventricular canal defects in control or XNkx2-5 FL-injected embryos and noted no other morphological defects other than the anomalies discussed above.
Fig. 3.
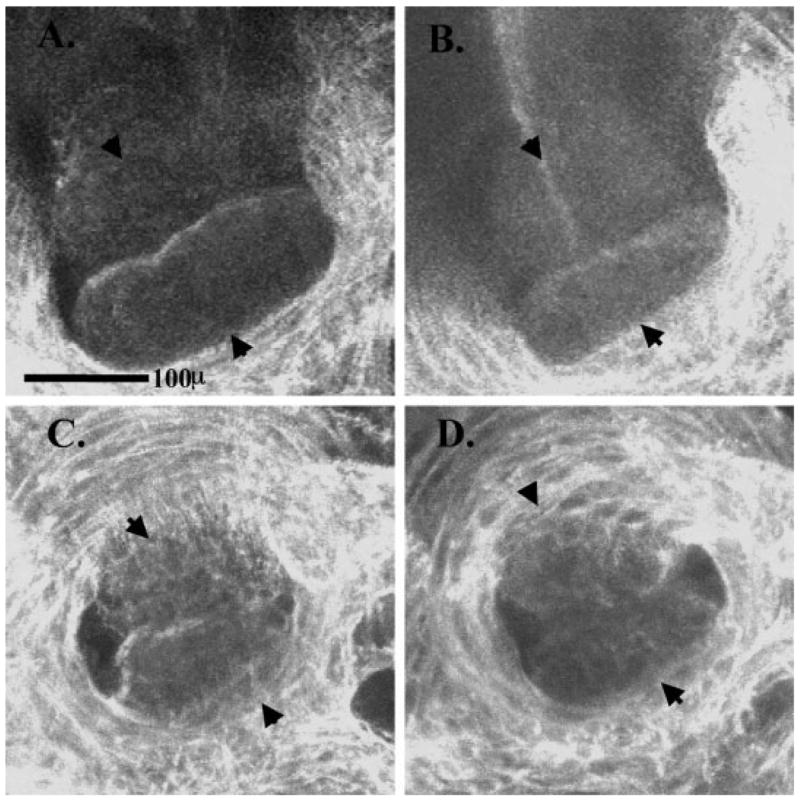
Altered morphology of the atrioventricular canal in embryos injected with XNkx2-5 encoding truncated protein. Embryos were fixed in Dent’s fixative and used for whole-mount immunostaining using JB3 (anti-fibrillin) antibody followed by a Cy5 secondary antibody and viewed using a confocal microscope. Images show the atrioventricular canal viewed from the atrial side of the valve and include arrowheads to denote the anterior and posterior leaflets of the atrioventricular valve. A: Non-injected embryo. B: XNkx2-5 FL-injected embryo. C: XNkx2-5 His156ter-injected embryo. D: XNkx2-5 Gln185ter-injected embryo. Note the dysplastic appearance and poor coaptation of the valve leaflets in the embryos injected with mutant XNkx2-5 (C,D).
Overexpression of XNkx2-5 His156ter and Gln185ter Can Be Distinguished From the Overexpression of Normal XNkx2-5 by Examination of the Cardiac Conduction Intervals
Humans carrying either of the two Nkx2-5 mutations studied here have defects in the conduction system of the heart. The size of the Xenopus early embryonic heart (approximately 0.3 mm in diameter) presents a challenge in making assessments on the conduction system. Recently we have correlated electrocardiograms and video microscopic measurements taken from stage 46 tadpole hearts and established the baseline information needed to examine the effect the truncation mutants on cardiac contraction (Bartlett et al., 2004). We examined the cardiac contraction intervals of stage 46 embryos injected with XNkx2-5 His156ter or Gln185ter and compared them with embryos injected with normal XNkx2-5, water, or non-injected (Table 3). Contraction intervals measured include the AA interval, the VV interval, and the AV interval. The AA interval, the time between sequential atrial contractions, assesses the function of the cardiac pacemaker. The VV interval, the time between sequential ventricular contractions, measures the functional heart rate. The AV interval assesses atrioventricular conduction by measuring the time delay between atrial and ventricular contractions. This evaluates the coordination of the sequential nature of atrial and ventricular contractions and detects heart block. Embryos injected with XNkx2-5 His156ter had significantly prolonged AV intervals, averaging 237 ± 12 msec, 30% longer than the other treatment groups. This finding was not seen in any of the four other treatment groups, including those injected XNkx2-5 Gln185ter. Embryos injected with XNkx2-5 Gln185ter were tachycardic, however, with significantly shorter AA and VV intervals (25% increase in heart rate), measuring 404 ± 26 msec, compared with the four other treatment groups.
TABLE 3.
Timing of Stage 46 Embryonic Heart Contraction by Video Microscopya
| Treatment | n | AA interval (msec) | VV interval (msec) | AV interval (msec) |
|---|---|---|---|---|
| Non-injected | 31 | 508 ± 17* | 508 ± 17* | 169 ± 8** |
| Water | 37 | 523 ± 21* | 522 ± 21* | 186 ± 10** |
| Wild-type | 16 | 516 ± 32* | 516 ± 32* | 177 ± 15** |
| His156ter | 27 | 526 ± 26* | 527 ± 26* | 237 ± 12 |
| Gln185ter | 23 | 404 ± 26 | 405 ± 26 | 174 ± 12** |
The mean atrial cycle length (the AA interval), the ventricular cycle length (the VV interval), and the delay between onset of the atrial and ventricular contractions (AV interval) and the standard error of the mean (SEM) by treatment group are listed. Embryos injected with the His156ter mutant had prolongation of the AV interval. Alternately, the Gln185ter mutant was associated with tachycardia demonstrated by shortening of AA and VV intervals.
Significantly higher than Gln185ter (P < 0.005 for water; P < 0.01 for His156ter; P < 0.05 for wild-type; P < 0.05 for non-injected).
Significantly lower than His 156ter (P < 0.0001 for non-injected; P < 0.0025 for Gln185ter; P < 0.025 for wild-type; P < 0.01 for water).
DISCUSSION
The power of analyzing null or dominant-negative mutations of developmentally important genes is undeniable. However, sometimes these approaches may have too broad an effect to tease out more subtle developmental influences of different regulators. The studies here concentrate on two mutant forms of XNkx2-5. The truncation mutations at amino acid 156 and 185 match the human mutations found at amino acid 170 and 196. These mutants are not embryonic lethal but nonetheless cause developmental abnormalities in a dominant manner.
Our studies focused on detecting morphological and functional changes in the heart at embryonic stage 46. By stage 46, most of the landmark features of cardiac development have been completed; the heart has looped; chambers, septae, and valves have formed; and the conduction system is operational. The average atrial size of embryos that were injected with XNkx2-5 mRNA (full-length or truncated) was larger than controls. In younger embryos, the transient expression of full-length XNkx2-5 increases both heart cell and overall cell number (Cleaver et al., 1996) but the almost doubling of cross-sectional area of the heart seen in stages 31–33 diminishes as the embryo ages, and by stage 46, it is more difficult to detect a size change (P. Krieg, personal communication). We observed no statistical change in stage 46 ventricle size and a more modest 40% change in atrial cross-sectional area when full-length Nkx2-5 mRNA was injected, consistent with the observations of the Krieg lab. The increase in cross-sectional area of the atria was not accompanied by an increase in atrial cell number whether the embryos were injected with full-length or with the mutant forms of Nkx2-5, suggesting that the increase in cross-sectional area is likely due to hypertrophy of the cells. By stage 46, cardiac size alone and more specifically atrial size did not distinguish the embryos injected with full-length or truncated Nkx2-5.
Internal inspection of the hearts using confocal microscopy uncovered a high frequency of atrial septal malformation in embryos injected with mutant encoding mRNA. One of the phenotypes encountered, atrial septum thinning, was also found in XNkx2-5 full-length mRNA-injected embryos, although this thinning was less common. Thinned septa correlated with those embryos with the largest atria, so perhaps this phenotype really reflects the process of having the same number of septal cells having to span a greater distance. The second atrial phenotype was typified by both atrial septum misplacement and fraying of the septum. This phenotype was only noted in embryos injected with mRNA encoding the truncated forms of XNkx2-5. In frogs, the atrial septum normally develops as a single, contiguous extension from the dorsal wall of the atria toward the atrioventricular junction. The timing is coincident with formation of the atrioventricular and spiral valves. The abnormal atrial septal phenotype reported in our experiments is not seen at any stage of normal development and does not correspond to a general developmental delay. Thus, we suggest that the early regulation of septal formation is disrupted in tadpoles, an anomaly consistent with defects seen in humans.
We also noted significant changes in the atrioventricular canal. This phenotype had variable penetrance, but was only seen in embryos injected with the truncated XNkx2-5 mRNAs. The annulus of the atrioventricular canal was smaller and the atrioventricular valve was dysplastic with poor leaflet coaptation. Although a strictly analogous phenotype has not been reported in humans, a range of defects has been noted that include atrioventricular valve defects (Schott et al., 1998, Benson et al., 1999).
Both truncation mutants in humans correlate with conduction system defects. We recently described methods to assess cardiac conduction in embryonic hearts (Bartlett et al., 2004) using video microscopy. Injection of truncated XNkx2-5 encoding mRNA leads to conduction defects in embryos. Both mutations lead to statistically significant changes in the conduction system physiology. For the smaller truncation (XNkx2-5 His156ter), the defect is manifested in a delay in the AV interval, a phenotype noted in humans carrying the human form of this mutation (Schott et al., 1998). For the larger truncation, we noted distinct tachycardia. Atrial arrhythmias have been seen in human disease (Benson et al., 1999) and some mouse models testing the role of a different Nkx2-5 mutation (Wakimoto et al., 2002). At this point, we cannot differentiate between XNkx2-5 being directly involved in the changes we see (by means of, for instance, alteration in specific gene expression) or whether some of the phenotypes seen are secondary effects due to hemodynamic changes in cardiovascular system. However, studies to look at changes of early cardiac gene expression are under way.
Several previous studies on the embryonic effects of mutations in Nkx2-5 have been carried out. For example, studies in both Xenopus and mouse replace a conserved leucine residue between the second and third helices of the homeodomain with a proline, making the protein unable to bind DNA (Grow and Krieg, 1998; Wakimoto et al., 2002) but leaving intact the carboxyl-terminus of the protein. These mutations lead to a much more severe phenotype than the truncation mutants we used. In Xenopus, the mutant protein leads to embryos with very small hearts. In mouse, the same mutation expressed under the control of the β-myosin heavy chain promoter resulted in death shortly after birth (Kasahara et al., 2000; Wakimoto et al., 2002) due to severe conduction system defects. However, delaying expression of the mutant postnatally (using the alpha-myosin heavy chain promoter) delayed overt abnormalities. Our observations that early expression of mutated Nkx2-5 can lead to irreversible cardiac defects is consistent with these previous studies.
Cell culture and in vitro studies have looked at the human mutations Nkx170ter and Nkx198ter that we used to model XNkx2-5 His156ter and Gln185ter (Sepulveda et al., 1998; Kasahara et al., 2000). These studies describe functional differences in the two truncations. For example, Nkx2-5 Gln198ter bound to DNA was deficient in coordinated control of genes coregulated with GATA-4 but still served to activate genes. Nkx2-5 Gln170ter did not bind DNA nor activate genes. However, there was little to distinguish the morphological consequences of these two mutations in humans (Schott et al., 1998). In humans, we presume that the mutant protein is expressed at the developmentally appropriate time and that expression persists. In our experiments, the mutant protein is expressed earlier than normal and the capacity to continue to make the protein disappears as the injected message is degraded. It is also true that, in Xenopus, another protein, Nkx2-3, is expressed at nearly the same time and the same regions expressing Nkx2-5 (Evans et al., 1995; Newman and Krieg, 1998). Both the overexpression of Nkx2-3 (Cleaver et al., 1996) or the overexpression of severe dominant-negative Nkx2-3 proteins had similar consequences for the embryo (Fu et al., 1998, Grow and Krieg, 1998). Nonetheless, it is apparent from our studies, and consistent with those reported by others, that even early and transient changes in protein expression can lead to congenital damage to the heart (Cleaver et al., 1996; Fu et al., 1998; Grow and Krieg, 1998; Dagle et al., 2003).
The mutations in Nkx2-5 that are especially relevant to human health are not embryonic lethal. We show here that two of the mutants that lead to congenital heart defects in human adversely affect heart development in Xenopus. These mutations lead to very different activities with respect to DNA binding or protein interaction, yet both mutants altered the normal development of the atrial septum, AV valve, and the establishment of conduction system. However, the defects in the conduction system were specific to each mutant. Thus, the studies of these mutations may provide an opportunity to focus on disruption of specific events in cardiac developmental.
EXPERIMENTAL PROCEDURES
Plasmids and RNA
The wild-type Xenopus Nkx2-5 cDNA clone (pXNkx2-5) used for this study was a gift of Dr. Paul Krieg (University of Arizona). The two variants were generated by a polymerase chain reaction-based approach. The His156ter mutant used oligonucleotides complementary to the cDNA from the 146th to the 155th codon, and included the complement to the stop codon TAG at the 156th codon. A BglII site was added to the 5′-end of the oligo for cloning purposes (GACAGATCTCTAGACCCTCTCAGGGGCTGACAGATA). The Gln185ter mutant used oligonucleotides complementary to the cDNA from the 178th to the 184th codon, and included the complement to the stop codon TAG at the 185th codon. A BglII site was added to the 5′-end of the oligo for cloning purposes (GACAGATCTCTACCTCTGCCGTTTGCATTTGTACCT). These two oligonucleotides were used with an oligonucleotide containing the XNkx2-5 start site region (GACGTCGACATGTTTGCCAGTCCTGTGACATCACT) and a SalI site. The His156ter fragment was cloned into a variant of pGem 3Z (Promega) that contained the last 30 nucleotides (nt) of the 3′-untranslated region of XNkx2-5 as well as a 30-nt polyA tail. The Gln185ter was cloned into a modified pSP73 (Promega) containing a 30-nt polyA stretch 3′ of the insertion site. A polyA stretch was also added to the original pXNkx2-5 plasmid to allow the synthesis of a polyadenylated wild-type transcript. The identity of all clones was verified by DNA sequence analysis. RNA was transcribed using the mMessage Machine kit (Ambion) to produce capped RNAs. An aliquot of the RNA was inspected before use by formaldehyde gel electrophoresis and quantitated by measuring the absorbance ratio at 260 nm and 280 nm. The relative stability of the mRNAs injected was assayed using 32P-labeled, capped transcripts. Labeled transcripts were injected into one-cell embryos, and total RNA was isolated from sibling embryos using methods previously described. Triplicates of 10 embryos were harvested at 0 hr, 2.5 hr, 5 hr, 10 hr, and 20 hr after injection. RNA isolated from equal numbers of embryos was separated by formaldehyde gel electrophoresis, the gels were dried, and radioactive RNA was quantified using a Packard instant imager area scintillation detector.
Animals and Injections
Frogs were purchased from both Xenopus I and Nasco for these experiments. Females were induced to lay eggs by injection with human chorionic gonadotropin (500–1,000 units, Sigma Chemical, St. Louis, MO). Eggs were collected in plastic Petri plates just before fertilization. Testes were removed from male frogs that had been killed with 1 ml of a 10-mg/ml solution of Tricaine (Sigma Chemical). A small piece of testes was crushed in 0.1× MMR (1× MMR = 100 mM NaCl, 2 mM KCl, 1 mM MgCl2, 2 mM CaCl2, 5 mM Hepes [pH 7.4]) and added to the eggs. The egg/sperm mix was flooded with 0.1× MMR, and the embryos were allowed to develop at room temperature. Fertilized eggs were dejellied for 10–15 min in 2% cysteine, pH 7.0. The eggs were then rinsed and placed in 1× MMR, 1% Ficoll-400 with penicillin (25 U/ml) and streptomycin (25 μg/ml) before injection. Then, 10 nl of RNA (0.1 to 0.2 ng/nl) was injected into each embryo before first cleavage. Embryonic stages were identified by the criteria describe in Nieuwkoop and Faber (Nieuwkoop and Faber, 1967).
Fixation and Immunohistochemistry
Fixation and immunohistochemistry were as described in Kolker et al. (2000). Briefly, tadpoles were fixed in Dent’s fixative (80% methanol, 20% dimethyl sulfoxide [DMSO]) and stored at −20°C. Except where noted, subsequent treatments and rinses were carried out on samples in 24-well plates on an orbital shaker. Before immunolabeling, the ventral dermal layer overlying the heart of stage 46 tadpoles was manually removed to help antibody penetration and ensure optimal imaging of the hearts.
Embryos were rehydrated to phosphate buffered saline (PBS) in a methanol-PBS series that included 1% DMSO at each step. Embryos were then treated with bovine testicular hyaluronidase (1 mg/ml in 50 mM acetate buffer) for 30 – 45 min at room temperature, rinsed in PBS-TD (PBS, 1% Tween-20, 1% DMSO, 0.02% NaN3) and blocked for 4 hr at room temperature or overnight at 4°C in block solution (PBS-TD containing 0.1 M glycine, 2% powdered milk, and 5% serum from the host species of the secondary antibody or 1% bovine serum albumin). Primary antibodies, mouse anti-bovine cardiac troponin T (CT3, Developmental Studies Hybridoma Bank [DSHB], Univ. of Iowa) and mouse anti-chicken fibrillin (JB3, gift from Dr. Charlie Little, Kansas University) were diluted 1:50 with block solution. Incubation with primary antibody was carried out overnight at 4°C. Samples were then rinsed 6 – 8 times over 8–10 hr with PBS-TD. Secondary antibodies, anti-mouse conjugated to Alexa 568 (from Molecular Probes) and anti-mouse conjugated to Cy5 (from Jackson ImmunoResearch), were diluted 1:200 in blocking solution and incubated with the sample overnight at 4°C. Samples were rinsed as described for primary antibody treatment and subsequently dehydrated in an ethanol series.
Embryos were mounted for viewing in constructed “mounting wells.” Depressions of concave slides were filled with Sylgard 184 silicone elastomer (Dow Corning) working solution and allowed to polymerize at 55°C overnight. Wells to accommodate the embryos were carved out of the elastomer with a scalpel. Embryos were positioned in the mounting wells and excess EtOH was removed. The specimens were held down with forceps as the clearing agent BABB (benzyl alcohol: benzyl benzoate, 1:2, adjusted to pH 7.0) was added, because the embryos became buoyant in the dense BABB and were difficult to reposition after clearing. Embryos were covered with a coverslip and viewed on a Nikon optiphot/Bio-Rad MRC-1024 confocal microscope equipped with a krypton/argon laser. Images were initially saved at a resolution of 1,024 × 1,024 pixels/inch. In some cases, the resolution was reduced to accommodate memory capacity of computers and/or printers. Confocal Assistant 4.02 and Adobe Photoshop 5.0 were used for image processing.
The number of cells in the hearts of stage 36 embryos was determined as follows. After injection, embryos were allowed to develop until stage 36 and then were placed in vials barely covered with 0.1 MMR. Embryos were quick frozen in a dry ice/ethanol bath for 30 min then stored at −80°C. The frozen embryos were defrosted in a solution of 80% Dent’s and 20% glycerol at 4°C for at least 2 hr then washed twice for 30 min in 100% methanol and stored in 100% methanol at −20°C. The stage 36 embryos were stained for 5 hr at room temperature with 0.53 μM phalloidin–fluorescein isothiocyanate (Sigma-Aldrich) in methanol. After staining, the embryos were rinsed twice for 20 min in 100% methanol. The embryos were then stained with TO-PRO-3 at 1:100 in methanol for 2 hr at room temperature, followed by three washes for 1 hr each in 100% methanol. Embryos were placed in mounting wells as described above in a lateral position and imaged on a Zeiss Axiophot microscope in 10-μm sections at ×20 magnification. Using ImageJ analysis software (Wayne Rasband, National Institutes of Health, Bethesda, MD, http://rsb.info.nih.gov/ij/) the cardiac area was traced and measured for each image and cardiac volumes were calculated. Additionally, the brightness and contrast of the images were adjusted to optimize visualization and the stained nuclei were counted manually.
For stage 46 embryos, histological sections of treated and control stage 46 hearts were obtained after mounting embryos in paraffin blocks. Ten-micron transverse sections of the thorax were stained with hematoxylin to allow visualization of nuclei. Sections were imaged at 10× magnification using a Zeiss Axiophot microscope, and atrial nuclei were manually counted by an observer masked to the treatment group.
Data Analysis
Atrial circumferences were traced using digital images captured with a SPOT camera mounted on a Zeiss Axiophot microscope with fluorescence. The camera was focused on the widest possible atrial region before digital capture of the image. Atrial areas were traced using the Scion Image program (NIH image), and areas were statistically analyzed using Microsoft Excel.
Contraction Interval Assessment
Analysis of cardiac contraction used methods described in Bartlett et al. (2004). Briefly, live stage 46 Xenopus laevis embryos were paralyzed by being placed in a 3-ml room temperature (22°C) bath of 0.1× MMR containing cisatracurium (Abbott) 0.33 mg/ml for 15 min. Embryos were positioned in a right anterior oblique position to optimize visualization of the posteriorly positioned atria. Using a Zeiss dissecting microscope attached to a Panasonic CCD video camera, video images of the heart were acquired at a rate of 29.97 frames per second (33.3 msec per frame) as a Quicktime movie format (See Supplementary Movie S1, which can be viewed at http://www.interscience.wiley.com/jpages/1058-8388/suppmat). The movies were imported into ImageJ analysis software as 8-bit grayscale. Circular areas of approximately 40 pixels were designated in the posterior atria and the anterior ventricle just inferior to the outflow tract. The average grayscale value of the pixels within the designated areas was measured in each video frame. The grayscale value was plotted relative to the movie frame to obtain waveforms depicting filling and emptying of the cardiac chambers. The atrial cycle length (the AA interval), the ventricular cycle length (the VV interval), and the delay between onset of the atrial and ventricular contractions (AV interval) were measured. The measurements were made for each of 10 cardiac cycles for each embryo. Cardiac conduction intervals between groups were compared by fitting a general linear model using the mixed procedure in SAS (version 9.1). The 1,310 measurements obtained from analyzing the targeted number of 10 cycles for each of the 131 embryos were included in the analysis. Because of the unbalanced nature of the data, Satterthwaite’s approximation method was used to determine the appropriate degrees of freedom for hypothesis testing. The significance level for testing the overall null hypothesis of no difference among the five treatment group-specific means for each measure was set at 0.05. If the null hypothesis was rejected, follow-up pairwise comparisons were made among the treatment group-specific means using Tukey’s approach to control the experiment-wide error rate at 0.05.
Supplementary Material
The Supplementary Material referred to in this article can be found at http://www.interscience.wiley.com/jpages/1058-8388/suppmat
Acknowledgments
The authors thank John Dagle, Tom Scholz, and Fred Lamb of the University of Iowa Department of Pediatrics for useful discussion and technical advice; Trudy Burns of the College of Public Health at the University of Iowa for help on statistical analysis; and Bobby Thompson of the Medical University of South Carolina for thoughtful comments during the course of this work. The authors thank Paul Krieg for the original XNkx2-5 clone and insightful discussions about the effects of Nkx2-5 mRNA injection, and Charlie Little for the JB3 antibody. The authors recognize the assistance of the University of Iowa Central Microscopy Research Facility generating histological sections. CH1 and CT3 antibodies, developed by Dr. Jim Lin, were obtained from the Developmental Studies Hybridoma Bank maintained by the University of Iowa, Department of Biological Sciences, Iowa City, IA 52242. Technical assistance and discussions with Bryan Allen, Jen Littig, Chip Greaves, Shweta Padmanabha, and Abi Struck-Marcell are acknowledged. This work was supported by a SCOR on congenital heart disease from NIHLB (D.L.W.), and also by NIH sponsored Pediatric Cardiology Fellowships (U.T. and H.L.B.). L.S., S.K., U.T., and V.D. contributed to this work while members of the Weeks lab at the Department of Biochemistry at the University of Iowa. The contents of this study are solely the responsibility of the authors and do not necessarily represent the official views of the NIH.
Grant sponsor: NIHLB; Grant number: GM069944; Grant number: HL062178; Grant sponsor: NIH.
References
- Bartlett HL, Scholz TD, Lamb FS, Weeks DL. Characterization of embryonic cardiac pacemaker and atrioventricular conduction physiology in Xenopus laevis using noninvasive imaging. Am J Physiol Heart Circ Physiol. 2004;286:H2035–H2041. doi: 10.1152/ajpheart.00807.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belaguli NS, Sepulveda JL, Nigam V, Charron F, Nemer M, Schwartz RJ. Cardiac tissue enriched factors serum response factor and GATA-4 are mutual coregulators. Mol Cell Biol. 2000;20:7550–7558. doi: 10.1128/mcb.20.20.7550-7558.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benson DW, Silberbach GM, Kavanaugh-McHugh A, Cottrill C, Zhang Y, Riggs S, Smalls O, Johnson MC, Watson MS, Seidman JG, Seidman CE, Plowden J, Kugler JD. Mutations in the cardiac transcription factor NKX2.5 affect diverse cardiac developmental pathways. J Clin Invest. 1999;104:1567–1573. doi: 10.1172/JCI8154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodmer R. The gene tinman is required for specification of the heart and visceral muscles in Drosophila. Development. 1993;118:719–729. doi: 10.1242/dev.118.3.719. [DOI] [PubMed] [Google Scholar]
- Cleaver OD, Patterson KD, Krieg PA. Overexpression of the tinman-related genes XNkx-2.5 and XNkx-2.3 in Xenopus embryos results in myocardialhyperplasia. Development. 1996;122:3549–3556. doi: 10.1242/dev.122.11.3549. [DOI] [PubMed] [Google Scholar]
- Dagle JM, Sabel JL, Littig JL, Sutherland LB, Kolker SJ, Weeks DL. Pitx2c attenuation results in cardiac defects and abnormalities of intestinal orientation in developing xenopus laevis. Dev Biol. 2003;262:268–281. doi: 10.1016/s0012-1606(03)00389-0. [DOI] [PubMed] [Google Scholar]
- Evans SM, Yan W, Murillo MP, Ponce J, Papalopulu N. tinman, a Drosophila homeobox gene required for heart and visceral mesoderm specification, may be represented by a family of gene in vertebrates:XNkx-2.3, a second vertebrate homologue of tinman. Development. 1995;121:3889–3899. doi: 10.1242/dev.121.11.3889. [DOI] [PubMed] [Google Scholar]
- Fu Y, Yan W, Mohun TJ, Evans SM. Vertebrate tinman homologues XNkx2–3 and XNkx2–5 are required for heart formation in a functionally redundant manner. Development. 1998;125:4439–4449. doi: 10.1242/dev.125.22.4439. [DOI] [PubMed] [Google Scholar]
- Grow MW, Krieg PA. Tinman function is essential for vertebrate heart development: elimination of cardiac differentiation by dominant inhibitory mutants of the tinman-related genes, XNkx2–3 and XNkx2–5. Dev Biol. 1998;204:187–196. doi: 10.1006/dbio.1998.9080. [DOI] [PubMed] [Google Scholar]
- Harvey RP. Nkx2.5 controls myogenesis and morphogenesis in the developing mammalian heart. In: Harvey RP, Olsen EN, Schulz RA, Altman JS, editors. Genetic control of heart development. Strasbourg: Human Frontier Science Program; 1997. pp. 55–62. [Google Scholar]
- Kasahara H, Lee B, Schott JJ, Benson DW, Seidman JG, Seidman CE, Izumo S. Loss of function and inhibitory effects of human CSX/NKX2.5 homeoprotein mutations associated with congenital heart disease. J Clin Invest. 2000;106:299–308. doi: 10.1172/JCI9860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasahara H, Usheva A, Ueyama T, Aoki H, Horikoshi N, Izumo S. Characterization of homo- and heterodimerization of cardiac Csx/Nkx2.5 homeoprotein. J Biol Chem. 2001a;276:4570–4580. doi: 10.1074/jbc.M004995200. [DOI] [PubMed] [Google Scholar]
- Kasahara H, Wakimoto H, Liu M, Maguire CT, Converso KL, Shioi T, Huang WY, Manning WJ, Paul D, Lawitts J, Berul CI, Izumo S. Progressive atrioventricular conduction defects and heart failure in mice expressing a mutant Csx/Nkx2.5 homeoprotein. J Clin Invest. 2001b;108:189–201. doi: 10.1172/JCI12694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolker SJ, Tajchman U, Weeks DL. Confocal imaging of early heart development in Xenopus laevis. Dev Biol. 2000;218:64–73. doi: 10.1006/dbio.1999.9558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komuro I, Izumo S. Csx: a murine homeobox-containing gene specifically expressed in the heart. Proc Natl Acad Sci U S A. 1993;90:8195–8199. doi: 10.1073/pnas.90.17.8145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyons I, Parsons LM, Hartley L, Li R, Andrews JE, Robb L, Harvey RP. Myogenic and morphogenetic defects in the heart tubes of murine embryos lacking the homeo box gene Nkx2–5. Genes Dev. 1995;9:1654–1666. doi: 10.1101/gad.9.13.1654. [DOI] [PubMed] [Google Scholar]
- Newman CS, Krieg PA. tinman-related genes expressed during heart development in Xenopus. Dev Genet. 1998;22:230–238. doi: 10.1002/(SICI)1520-6408(1998)22:3<230::AID-DVG5>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- Nieuwkoop PD, Faber J. Normal table of Xenopus laevis (Daudin) Amsterdam: North Holland publishing Company; 1967. [Google Scholar]
- Schott JJ, Benson DW, Basson CT, Pease W, Silberbach GM, Moak JP, Maron BJ, Seidman CE, Seidman JG. Congenital heart disease caused by mutations in the transcription factor NKX2–5. Science. 1998;281:108–111. doi: 10.1126/science.281.5373.108. [DOI] [PubMed] [Google Scholar]
- Sepulveda JL, Belaguli N, Nigam V, Chen CY, Nemer M, Schwartz RJ. GATA-4 and Nkx-2.5 coactivate Nkx-2 DNA binding targets: role for regulating early cardiac gene expression. Mol Cell Biol. 1998;18:3405–3415. doi: 10.1128/mcb.18.6.3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sepulveda JL, Vlahopoulos S, Iyer D, Belaguli N, Schwartz RJ. Combinatorial expression of GATA4, Nkx2-5, and serum response factor directs early cardiac gene activity. J Biol Chem. 2002;277:25775–25782. doi: 10.1074/jbc.M203122200. [DOI] [PubMed] [Google Scholar]
- Tonissen K, Drysdale T, Lints TJ, Harvey RP, Krieg PA. XNkx-2.5, a Xenopus gene related to Nkx-2.5 and tinman: evidence for a conserved role in cardiac development. Dev Biol. 1994;162:325–328. doi: 10.1006/dbio.1994.1089. [DOI] [PubMed] [Google Scholar]
- Wakimoto H, Kasahara H, Maguire CT, Izumo S, Berul CI. Developmentally modulated cardiac conduction failure in transgenic mice with fetal or postnatal overexpression of DNA nonbinding mutant Nkx2.5. J Cardiovasc Electrophysiol. 2002;13:682–688. doi: 10.1046/j.1540-8167.2002.00682.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The Supplementary Material referred to in this article can be found at http://www.interscience.wiley.com/jpages/1058-8388/suppmat