

Abstract
The herpesvirus proteases are an example in which allosteric regulation of an enzyme activity is achieved through the formation of quaternary structure. Here, we report a 1.7 Å resolution structure of Kaposi’s Sarcoma herpesvirus protease in complex with a hexapeptide transition state analogue that stabilizes the dimeric state of the enzyme. Extended substrate binding sites are induced upon peptide binding. In particular, 104 Å2 of surface are buried in the newly formed S4 pocket when tyrosine binds at this site. The peptide inhibitor also induces a rearrangement of residues that stabilize the oxyanion hole and the dimer interface. Concomitant with the structural changes, an increase in catalytic efficiency of the enzyme results upon extended substrate binding. A nearly 20-fold increase in kcat/KM results upon extending the peptide substrate from a tetrapeptide to a hexapeptide exclusively due to a KM effect. This suggests that the mechanism by which herpesvirus proteases achieve their high specificity is by using extended substrates to modulate both the structure and activity of the enzyme.
INTRODUCTION
The herpesvirus family encodes a protease whose activity is regulated by a unique oligomerization dependent allosteric mechanism 1–6. The inactive monomeric protease is expressed fused to an assembly protein in the cytosol and subsequently imported into the nucleus 7–11. Concentration dependent dimerization within the nuclear localized immature capsids activates the protease 1,2,12. The active protease is released from the assembly protein by autoprocessing at the release (R) site 10,11,13–15. Protease processing of the assembly protein, a major scaffold protein, at the maturation (M) site leads to capsid maturation and virion formation 11,14,16,17.
Recent studies on the Kaposi’s Sarcoma herpesvirus (KSHV) protease suggest an induced structure upon dimerization as a concentration dependent activation of the enzyme 4–6. This induced structure model differs from canonical induced fit in that the free energy of subunit dimerization is used to organize the active site and provide a unique mechanism for activating proteolytic activity. In this model, dimerization induced folding of helix 5, the major contributor to the dimer interface, and helix 6, which is crucial for stabilization of the oxyanion hole, stabilizes elements that are essential for catalytic activity.
Similarly, structural studies of the related human cytomegalovirus (HCMV) protease 3, reveal loss of catalytic activity of a dimer interface mutant due to disordering of both the C-terminal helix G and the L10 loop (H6 and L10 in KSHV protease) that support the catalytic machinery. In addition, the ordering of these structural elements, in the HCMV protease variant, can be induced by binding of a peptidomimetic inhibitor. Finally, upon binding of a peptide phosphonate inhibitor at the active site of KSHV protease, the equilibrium is shifted toward the dimeric enzyme, suggesting communication between the active site and the dimer interface4.
We sought to understand how substrate binding and its effect on the disorder to order transition could regulate catalytic activity in KSHV protease. To understand the structural basis of the stabilized oligomer state we have used the hexapeptide diphenylphosphonate transition state analog previously shown to dramatically shift the equilibrium to the dimeric form of the enzyme. We have crystallized KSHV protease bound to this inhibitor and determined the structure of the complex. The structure elucidates atomic details of substrate binding pockets, which are composed of loops previously disordered in the apo structure of KSHV protease. In addition, an induced fit to the extended substrate binding pockets of the protease is revealed, which is most pronounced at S4. The structural evidence for protease extended substrate specificity that leads to enhanced catalytic efficiency is revealed. This suggests that substrate binding could provide another level of regulation of protease activity in this enzyme beyond the initial concentration dependent activation mechanism described previously.
RESULTS
Extended substrate binding improves catalytic efficiency
The positional scanning synthetic combinatorial libraries (PS-SCL) previously used to determine the KSHV non-prime side substrate specificity contained only tetrameric substrates 4. To determine the extent of the extended substrate specificity for KSHV protease we synthesized five nested sets of substrates with the trimeric tButyl-Gln-Ala being the shortest and the heptameric Ser-Pro-Val-Tyr-tButyl-Gln-Ala substrate being the longest. All substrates were acetylated on their N-termini and included the fluorogenic reporter group 7-amino-4-carbamoyl-methylcoumarin to monitor enzyme activity spectroscopically. The amino acid sequence of the tri-, tetra-, penta- and hexameric substrates corresponds to the sequence of the hexapeptidyl phosphonate inhibitor 4, while in the heptapetide, Ser was incorporated at the P7 position in accordance with the natural R site sequence.
Kinetic analysis of the peptide substrates (Table 1) reveals an increase in catalytic efficiency for the hydrolysis of longer substrates with an 18.2-fold increase for heptameric compared to tetrameric substrates. The improvement in catalytic efficiency for longer substrates comes solely from improved KM values, whereas the turnover rate, kcat remains virtually unchanged.
Table 1.
Hydrolysis rates of the extended substrates for KSHV protease
| kcat (s−1) | KM (μM) | kcat/KM (M−1s−1) | Surface area (Å2) | ΔΔG kcal/mol | ||
|---|---|---|---|---|---|---|
| P3-P1 | Ac-tQA-ACC | N.D. | N.D. | N.D. | ||
| P4-P1 | Ac-YtQA-ACC | 0.0084 ± 0.0003 | 80.0 ± 8.8 | 105 | 359 | 5.7 |
| P5-P1 | Ac-VYtQA-ACC | 0.0075 ± 0.0003 | 15.9 ± 2.5 | 472 | 408 | 6.5 |
| P6-P1 | Ac-PVYtQA-ACC | 0.0083 ± 0.0002 | 8.5 ± 0.8 | 976 | 446 | 7.1 |
| P7-P1 | Ac-SPVYtQA-ACC | 0.0088 ± 0.0003 | 4.6 ± 0.7 | 1913 | N/A | N/A |
Surface area that substrate buries upon binding is calculated from the structure of complexed KSHV protease. The difference in Gibbs free energy for binding of the each residue was determined based on the burial of hydrophobic solvent accessible surface area of protease upon substrate binding.
Using the high resolution three-dimensional structure of the enzyme inhibitor complex (see below) we calculate that binding of the hexapeptide inhibitor buries 450 Å2 of hydrophobic surface area (Table 1). Binding of the tetrapeptide inhibitor would bury 360 Å2. This difference of 90 Å2 between hexapeptide and tetrapeptide binding contributes 1.44 kcal/mol to substrate binding energy. This is in agreement with the 1.40 kcal/mol difference in Gibbs free energy calculated for binding of these two substrates from their kcat/KM values.
Ordering of unstructured regions of KSHV protease
The crystal structure of the KSHV protease complexed with a hexapeptide transition state analogue was determined at a resolution of 1.7Å (Figure 1b). The overall fold of the KSHV protease-inhibitor structure is similar to the reported apo protease structure 18 (Figure 1b). However, several previously disordered regions are present in our structure accounting for new secondary structure elements, namely, 3 loops and 5 helices (Figure 1c). The ordered loops L0a, L0b and L9 encompass the following residues: 15–20, 24–26, and 125–129, respectively. To preserve previous helix nomenclature we have kept the same numbering for the helices and we introduce a letter following the already assigned corresponding helix number to annotate new helices. Also, we annotated a small helix encompassing residues 88–91, that was left out from the nomenclature in the previous structure of apo KSHV protease. Thus, we define helices as follows: α0: residues 21–23, α0a: residues 27–33, α1: residues 74–84, α1a: residues 88–91, α2: residues 100–110, α2a: residues 120–124, α3: residues 154–159, α4: residues 166–179, α4a: residues 181–183, α5: residues 193–203 and α6: residues 209–220.
Figure 1.
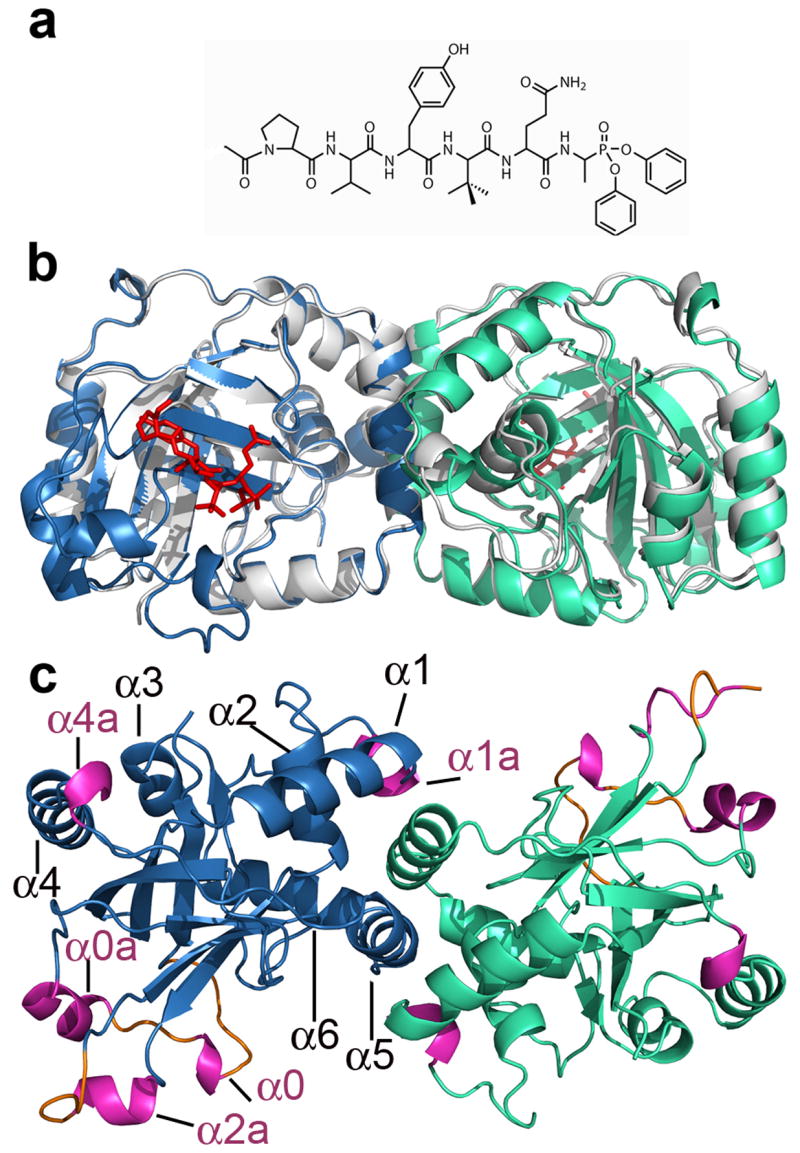
The crystal structure of the KSHV protease in complex with the hexapeptide transition state analogue. (a) Chemical structure of a Ac-Pro-Val-Tyr-tBut-Gln-Ala-(OPh)2 inhibitor. (b) The overlay of the apo KSHV protease structure (pdb code: 1FL1) shown in grey and complexed protease with monomer A shown in skyblue and monomer B in greencyan. The hexapeptidyl phosphonate inhibitor is shown in red. The same color scheme is used throughout paper. (c) The structure of KSHV protease:inhibitor complex is rotated 90º around the horizontal axis. New secondary structure elements helices and loops are highlighted in pink and orange, respectively.
Newly ordered regions (Figure 1c) encompass residues involved in substrate binding that belong to the extended substrate binding pockets. The helix 2a residues Pro120, Arg 121 and Glu 122 contribute to the formation of P3, P5 and P6 binding pockets respectively, while Ser 128 and Gly 129 residues further delineate the P6 binding pocket. The structure reveals new evidence for helix 0 (21–23) involvement in electrostatic stabilization of the oxyanion hole by residue Glu 22.
Hexapeptide transition-state analogue binding specificity
Six structures of HCMV have been reported complexed to peptidomimetic inhibitors with amino acids different from the substrate recognition sequences 3,19,20. These structures were quite informative and compelled us to determine a high resolution structure of KSHV protease complexed with a peptide transition state analogue that contained a peptide sequence optimized for substrate binding. To design an optimal peptide sequence for this inhibitor we incorporated data from our substrate specificity profile of KSHV protease together with its natural R site sequence 4. T-butyl glycine, as opposed to the endogenously occurring valine, was incorporated into the substrate at P3 to increase the Kcat/KM (unpublished results) as has been reported for HCMV Pr as well 21.
The atomic details of interactions of both phosphonate and peptide moieties of the inhibitor with the protease are revealed in the structure. The phosphonate group of the tetrahedral intermediate is covalently bound to the catalytic Ser 114 and is further stabilized by a hydrogen bond to the Nε2 nitrogen of His 46. The electron density surrounding the phosphonate group shows that the inhibitor exists in an “aged” form, corresponding to the release of the phenoxy groups. The presence of the aged complex was expected due to the slow crystallization time of the complex22. The phosphonate O1B occupies the oxyanion hole and is held in place by hydrogen bonds to the main chain amide of Arg 142 and water 16. A water molecule occupying an equivalent position has been described in previous structures of herpesvirus proteases 20,23–27. This water molecule is further stabilized by a hydrogen bond to Thr 146 Oγ1, the backbone amide of Arg 143, and to the carbonyl oxygen of Leu 113. The side chain guanidino group of Arg 142 further stabilizes the carbonyl oxygen of the P2 Gln residue, which is consistent with HCMV protease structures complexed with peptidomimetic inhibitors 3,19,20.
The peptide moiety of the inhibitor is bound in an extended β-strand conformation, in agreement with previously reported structures of shorter peptides in complex with other herpesvirus proteases 3,19,20,28 (Figure 2a). In the peptide moiety of the inhibitor, all residues make both hydrogen bonds and hydrophobic contacts with the protease. The backbone atoms of the residues P2–P4 form 4 hydrogen bonds with the backbone atoms of the protease β-strand encompassing residues 113–120, which includes the catalytic Ser 114 (Figure 2b). The side chains of P2 Gln and P4 Tyr make additional hydrogen bonds with the protease interacting with Glu 133 (through a water molecule) and Pro130, respectively. Pro at P6 is stabilized by 4 hydrophobic contacts formed via its side chain atoms. These additional interactions may contribute to the improved binding affinity (lower KM) that we observe for the longest heptapeptide substrate.
Figure 2.
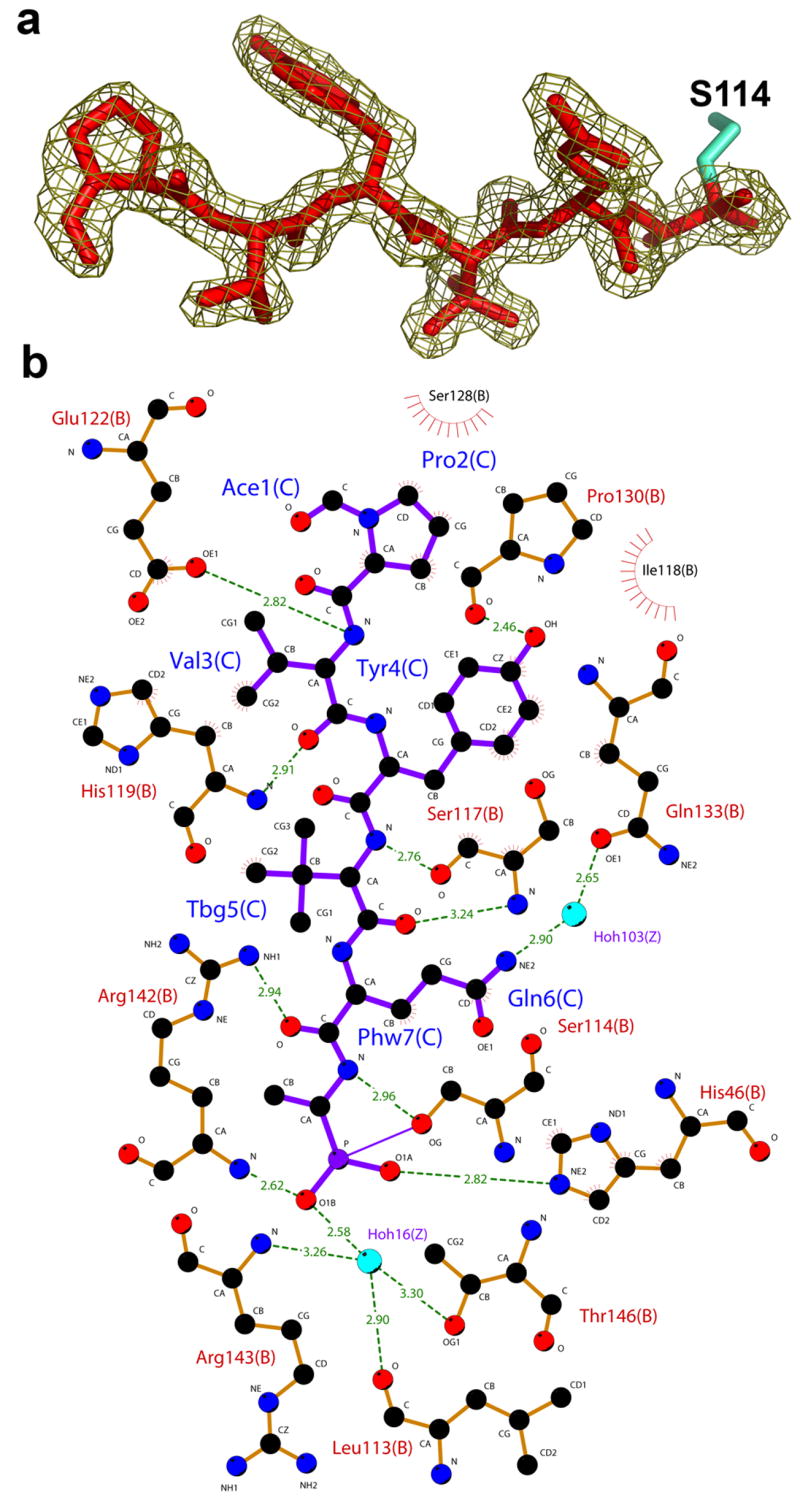
Interactions of the hexapeptide transition state analogue with KSHV protease. (a) The 2fo–fc electron density map of the inhibitor at contour level of 1σ. (b) A Ligplot 50 representation of the interactions between inhibitor (purple) and protease residues (orange). Water molecules are colored in blue, protease residues that are involved in hydrogen bonds are shown in ball and stick representation and hydrogen bonds are represented with dashed green lines. Residues that are involved in hydrophobic contacts are labeled with radial lines facing their contact residue.
Induced fit in the extended substrate binding pocket
Previous structures of herpesvirus proteases suggested that conformational changes occur in the substrate binding sites upon binding of an inhibitor 3,20,26,29. With the optimized peptide inhibitor of KSHV protease we were able to assess the capacity of the enzyme to accommodate its substrates through induced conformational rearrangements. In KSHV protease, the observed induced fit 30 encompasses both small changes at the active site with movement of the side chains of Arg 142 and Arg 143 as well as more dramatic rearrangements of the residues in the extended substrate binding pockets (Figure 3b).
Figure 3.
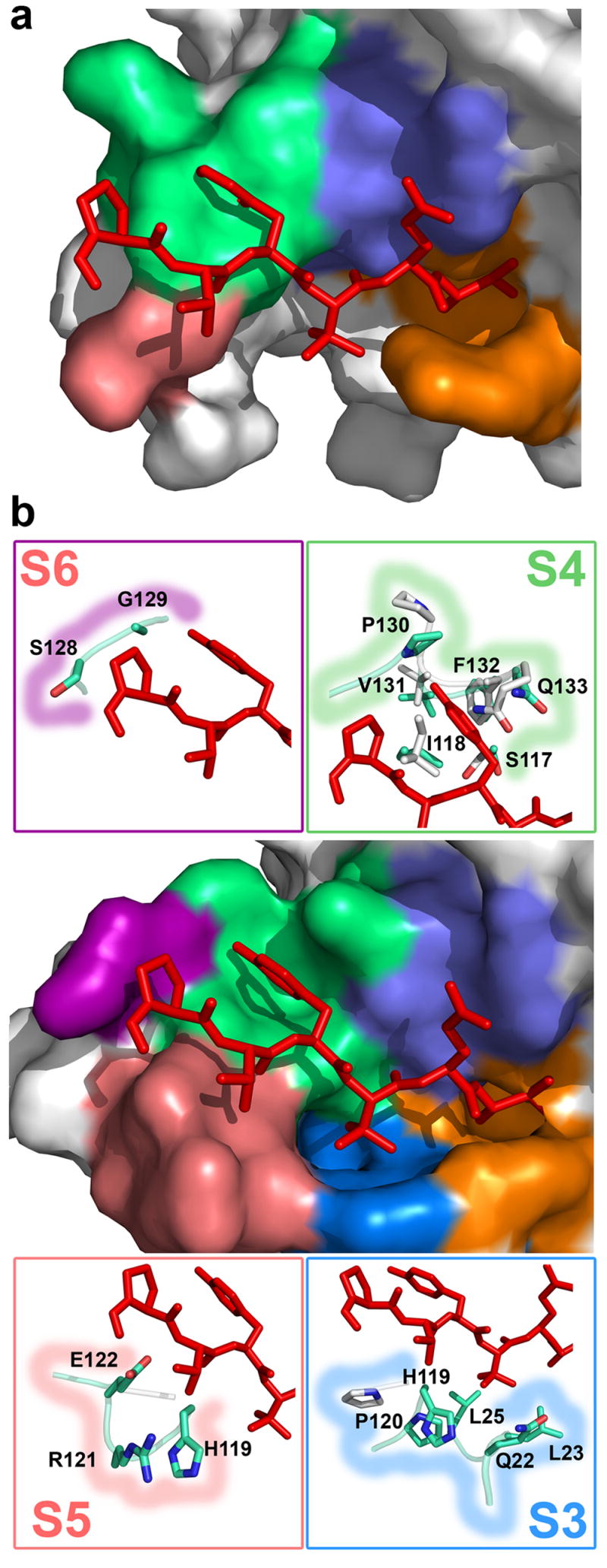
Induced fit and formation of the substrate binding pockets. (a) The molecular surface representation of the apo KSHV protease structure close to the active site shows mostly disordered extended binding sites. Inhibitor (stick representation, red) from complexed KSHV protease structure is superimposed for reference. (b) The molecular surface representation of the complexed KSHV protease shows that upon inhibitor binding (stick model, red), the extended substrate binding sites (S1, orange; S2, slate; S3, deepblue; S4, green; S5, salmon; and S6, purple) are ordered and a deep cavity is induced in the S3 and S4 sites. Stick models in boxes represent the residues in corresponding S3–S6 binding sites that either undergo conformational changes or are ordered upon inhibitor binding.
A shallow S2 site, compared to other pockets, corresponds to lower protease selectivity at the P2 position due to the smaller buried surface area upon substrate binding. Similarly, the pockets for P5 and P6 residues are very shallow. In contrast to HCMV protease, the S3 site appears to be well-defined as a deep and narrow hydrophobic pocket designed to accommodate amino acids not larger than valine. Indeed, this protease has a strong in vitro preference for Val and somewhat less for Ile at this position 4, although both protease natural substrate sequence sites R and M have Leu at this position.
The most remarkable finding is the shape of the S4 site. A deep pocket is induced upon inhibitor binding to accommodate a large Tyr residue (Figure 3b), which buries 104 Å2. Moreover, it reveals the molecular basis of the previously reported strong preference for the large hydrophobic residues (Tyr, Trp, and Phe) at position P4 4. The conformational change induced by the transition state analog involves residues in loop 9 (residues 125–133) (Figure 3b). The side chain amide of Gln 133 moves 4.3 Å to allow binding of P4 Tyr, while Pro 130 nitrogen moves 4.6 Å to accommodate binding of the Tyr hydroxyl group. Previously disordered regions in the extended binding sites are present in our structure and in addition to S4, provide a complete description of the S3, S5 and S6 sub-sites (Figure 3b). The S3 site is mostly formed by residues in the new helix 0, Gln 22 and Leu 23 and is further delineated by Leu 25, His119 and Pro 120. Compared to the apo KSHV structure, the S3 site is also formed by an induced fit, revealed by a 4.66 Å shift in the position of His 119 Cδ and a 3.73 Å shift for Pro 120 N. The shallow S5 site is formed by His 119 and newly determined helix 2a residues Arg 121 and Glu 122. Inhibitor binding also induces ordering of two residues that form the S6 site, Ser 128 and Gly 129. Our structure suggests that KSHV protease can accommodate binding of substrates up to P6 and for the longer substrate presumably the P7 amino acid may contact the protease. Kinetic data are consistent with the ordering of the P7 binding site showing a 1.8-fold improvement in KM of the Ser-Pro-Val-Tyr-tButyl-Gln-Ala heptapeptide compared to the Pro-Val-Tyr-tButyl-Gln-Ala hexapeptide substrate.
Conformational changes occur at the active site and dimer interface which stabilize the dimer
Efficient proteolysis requires stabilization of the tetrahedral intermediate. This is partially accomplished in the herpesvirus protease by Arg 142 and Arg 143 stabilization of the oxyanion in the tetrahedral intermediate. Binding in the oxyanion hole appears to be sufficient for protease activation and dimer stabilization as phosphonylation of the catalytic serine by both diisopropyl fluorophosphonate (DFP) and peptidyl phosphonate inhibitors are known to stabilize the dimeric form of KSHV and EBV proteases 4,26. However based on the Epstein-Barr virus (EBV) protease structure inhibited with DFP 26, phosphonylation alone is incapable of inducing the conformational changes that we observe at the extended substrate binding sites.
Our structure reveals that binding of the peptide phosphonate inhibitor induces dimer stabilization through the network of interactions established by induced conformational changes both at the active site and the dimer interface (Figure 4a). Structural changes at the active site involve residues Arg 142 and Arg 143 in the oxyanion hole loop L10, which undergo conformational changes to stabilize the oxyanion hole. In contrast to other herpesvirus proteases a striking conformational change occurs in the side chain of Arg 143 to further support the formation of the oxyanion hole by interaction with residues in the newly formed helix 0. The side chain guanidino group of Arg 143 forms 6 hydrogen bonds, two directly to Leu 23 in the helix 0 and the rest indirectly through 3 water molecules to Val 11, Glu 22, Leu115, and Ser 117. This dense network of interactions explains why this residue is strongly conserved and why its mutation to Ala in HCMV protease reduces protease activity by 1500-fold 31. In addition, the side chain of Arg 142 rearranges to interact with Gln 22 from the same helix. This fixed conformation of the oxyanion hole loop in turn provides a rigid scaffold to stabilize helix 6 through a salt bridge between Arg 144 and Asp 216. Moreover, our structure reveals additional helix 6 interactions including the carbonyl oxygen of Val 219 (helix 6) and His 63 Nε2, which is further stabilized by an indirect hydrogen bond interaction to the side chain hydroxyl group of Ser 15. We hypothesize that interaction of the oxyanion hole loop and helix 0 positions the loop L0a in close proximity to helix 6 to support movement of His 63 (3.5 Å measured from the center of the side chain imidazole), which stabilize helix 6. The stabilized helix 6 is thought to be important for stabilization of helix 5 at the dimer interface 5.
Figure 4.

Stabilization of the oxyanion hole and the dimer interface. (a) The residues that undergo conformational change and are involved in stabilization of the oxyanion hole loop and helix 6 in the structures of apo (white) and inhibited (skyblue) KSHV protease are shown as a stick model. The oxyanion hole loop, helices 0 and 6, and the loop L0a are shown in a ribbon diagram. (b) Helix 5 in a monomer A (skyblue) and the helix 1A in a monomer B (greencyan) in the complexed protease structure are shown in ribbon diagram and key residues involved in stabilization of the dimer interface are shown in stick model. Corresponding residues and the secondary structure elements in the apo protease structure are shown in white. The rotation of the His 88 and the movement of Val 89 are emphasized by an arrow. (c) The molecular surface of the intersection of the dimer interface in the KSHV apo structure (left) and the complex structure (right) is shown for monomer A in white and the helix 5 is highlighted in skyblue. The helix 5 of the adjacent monomer B is drawn in the cartoon diagram in greencyan. The surface of the His 88 and Val 89 are colored in limegreen and red respectively.
Binding of the peptide phosphonate inhibitor at the active site also induces a significant conformational change at the KSHV protease dimer interface. The structure reveals unwinding of a single turn of helix 1a whose residues undergo a major conformational change to establish additional interactions at the dimer interface with an adjacent monomer’s helix 6 (Figure 4b, c). This conformational change involves rotation of His 88 in molecule A and a 5.2 Å movement of the imidazole ring, to allow interaction with helix 5 residues Asp 202 and Lys 199 in molecule B. A 4.7 Å movement of Val 89A Cβ results in further intermolecular interactions with helix 5 through hydrophobic contacts with Phe 205B and Thr 108B.
DISCUSSION
KSHV protease exhibits multiple levels of regulation and is a highly unique serine protease
To achieve controlled spatial and temporal activation of protease activity, the herpesvirus family has evolved a unique mechanism of oligomerization and substrate-dependent regulation of enzyme activity. This mechanism requires acquisition of quaternary structure and multiple conformational changes in protease tertiary structure prior to activation. Our current model depicted in Figure 5 consists of the following steps: i) concentration dependent dimerization leads to induced protease structure through folding of helices 5 and 6; ii) subsequent positioning of the oxyanion hole loop required for catalysis; iii) a substrate induced conformational change in the substrate binding pocket, and iv) substrate induced improvement in catalytic efficiency. Here we have used crystallography and kinetic studies to obtain a snapshot of the inhibitor stabilized quaternary structure of a herpesvirus protease. Furthermore, we have provided structural evidence for extended substrate specificity for a herpesvirus protease and induced fit for substrate binding particularly at the S4 site.
Figure 5.

The model for allosteric regulation of herpesvirus proteases. Herpesvirus proteases evolved structurally flexible molecule to allosterically regulate and modulate protease activity. Herpesvirus proteases are expressed as inactive monomers fused to an assembly protein. The concentration dependent dimerization activates protease through an induced structure that leads to the formation of the catalytic oxyanion hole. Protease activity can be further modulated by the binding of the extended substrates through an induced fit at extended binding sites.
Compared to classic serine proteases such as trypsin the catalytic efficiency of a herpesvirus protease is several orders of magnitude lower 32. Available structures of trypsin provide evidence for substrate specificity in predefined sub-sites, particularly at S1 and somewhat less for S2 and S3 32–34. In contrast, herpesvirus proteases have shallow substrate binding pockets. The decreased substrate binding affinity in KSHV protease is offset by extended substrate specificity reflected by a 20-fold decrease in KM between the tretrameric and heptameric substrate. Similarly, it has been reported for herpes simplex virus and EBV proteases that when comparing hydrolysis rates of peptides, whose prime side were fixed and non prime sides varied, P9-P8′ and P10-P10′ substrates are cleaved with higher efficiency than substrates with only four residues at the N-terminal side. 14,35. Our data suggest that the binding energy of an extended peptide substrate is sufficient to improve KM values for KSHV protease. In contrast, the proteolytic activity of trypsin increases dramatically (>104-fold) when the length of the non prime side of the synthetic peptides is moderately increased from P1-P′4 to P4-P′4 36. In this case, the increase in kcat is more pronounced than the decrease in KM. In canonical serine proteases, which bind substrates in a lock and key fashion using pre-formed sub-sites, the longer substrates appear to have a more favorable conformation and undergo acylation more readily, which is reflected in the increased turnover rate 32. On the other hand in herpesvirus proteases, substrate binding requires formation of the binding sites through an induced fit and, hence, they require extended substrates to achieve specificity and increase substrate affinity for efficient proteolysis.
Substrate induced fit can be used to sequentially regulate protease activity
Structural flexibility of a macromolecule offers a simple and effective way for the regulation of enzyme activity. Factor D is another example of a serine protease whose extremely restricted proteolytic activity, in the alternate complement pathway, is regulated by a substrate induced reversible transition from its zymogen-like form to the corresponding catalytically active protease 37,38. Binding of its natural substrate, C3b-bound factor B, induces conformational rearrangements to form an active catalytic triad conformation. In the herpesvirus protease family, activation is regulated by a concentration dependent dimerization that induces conformational changes at the active site to form an oxyanion hole, but not the catalytic triad. Substrate or inhibitor binding then provides additional stabilization of the oxyanion hole. This is supported by the ability of a peptidomimetic inhibitor to induce the ordering of the oxyanion hole loop in an HCMV dimer interface mutant 3.
Herpesvirus proteases show further structural flexibility to accommodate binding of their substrates. Our structure reveals atomic details of the substrate induced fit in the extended sub-sites. In addition, the reported structure of the EBV protease complexed to DFP 26 has all the secondary structure elements described in our structure and allows us to directly compare the effects of the phosphonate and the peptide moieties on the induced fit. Both DFP and the hexapapetide phosphonate inhibitor are capable of inducing changes in the active site to stabilize the oxyanion loop. However, when the hexapeptide phosphonate inhibitor is modeled on the DFP complexed structure of EVB protease, the S3 substrate binding site is open and not formed. Similarly, P4 Tyr is completely buried in the surface of the modeled EBV structure. This suggests that sub-sites S3 and S4 are induced by the binding of the peptide moiety of the inhibitor and not the phosphonate group.
Why did herpesvirus proteases evolve such a mechanism for substrate recognition and binding? The in vivo role of the protease in capsid assembly and maturation is to process assembly protein at the M and R sites. One of the two genes that encode the assembly protein is transcribed into a larger mRNA encoding for both the protease and assembly protein. Different expression levels result in a 10-fold difference between the protease and its assembly protein substrate. It has been estimated that the total protease concentration in the immature capsids is about 100 μM 5, which would correspond to the substrate concentration for protease autoprocessing at the R site. This concentration is on the order of KM for the tetrapeptide substrate (80 ±9 μM).
For efficient processing, substrate concentrations have to be several folds higher than the KM. To overcome the problem of low substrate concentration, the herpesvirus protease appears to have employed extended substrate specificity to lower the KM and improve the catalytic efficiency for autoprocessing at the R-site. In addition, the only strongly conserved residue in amino acid sequences of natural R sites across the herpesvirus protease family is Tyr at P4. Our structure reveals that binding of Tyr buries 104 Å2 surface area that contributes 2.1 kcal/mol of binding energy. It has been shown that in vitro processing of HCMV protease R-site peptides is 16 times slower than that of the M-site peptide and that P4 substitution of Val to Tyr in the M site sequence results in a 45% enhancement of the cleavage rate 28. In agreement with substrate profiling data our structure shows that the enzyme can easily accommodate Tyr at P4. The strict conservation of Tyr at P4 at the R but not M cleavage site may reflect another level of regulation and substrate differentiation. Recently it has been reported that HCMV protease fused to the assembly protein is active in the presence of a dimerization inducing kosmotrope 11. However, an earlier study on an EVB protease that included R-site and only 4 additional amino acids of the assembly protein reported that the resultant protease was inactive 26. Altogether, these results suggest a model where the protease fused to the intact assembly protein is able to dimerize at the capsid scaffold at high concentrations and preferentially process assembly protein at the M-site, prior to release by cleavage at the R-site. Clearly both substrate specificity and the conformation of assembly protein appear to have a role in the regulation of substrate processing and ultimately viral assembly. Such a highly regulated process provides unique opportunities for intervention at the therapeutic level.
Conclusions and implications
The herpesvirus proteases initially use dimerization as the activity on/off switch and then substrate induced fit to further finely control the catalytic efficiency. The dependence of the enzyme on using conformational flexibility to regulate its activity at multiple steps provides an opportunity to design a new generation of drugs for treatment of diseases caused by herpesviruses. Since the viral assembly process is exquisitely timed and proper proteolytic activity is essential, a small perturbation of the enzyme activity can result in catastrophic effects in viral replication. Structural studies reveal the importance of extended substrate specificity and deep S3 and S4 pockets suggesting a new target site on the herpesvirus protease. This site is more than 10Å away from the active site serine and plays an important role in substrate binding. Moreover, it offers deep hydrophobic cavities that are more suited for small molecule binding than sub-sites S2 and S1. The flexibility of this site may pose difficulties for design of tight binding inhibitors as part of the gain of free binding energy may be used to form the binding site. Efforts that take advantage of these sites may aid in the search for competitive inhibitors directed to the extended active site of KSHV protease.
MATERIALS AND METHODS
Recombinant expression and purification of KSHV Pr
A protease variant S204G resistant to autolysis was recombinantly expressed in E.coli and purified as previously described 39. Purified protease was stored in assay buffer: 25mM potassium phosphate pH 8.0, 150mM KCl, 100μM EDTA, 1mM β-mercaptoethanol.
Crystallization
Purified KSHV protease (26μM) was incubated with Ac-Pro-Val-Tyr-tBut-Gln-Ala-P(OPh)2 (50 μM) 4 for 24h at room temperature, followed by one more addition of the same inhibitor (50μM) and incubated for additional 48h. Following complete inhibition, the protease complex was purified over a Superdex-75 column and concentrated to 5.2 mg/ml in assay buffer. Crystallization was performed using the hanging drop method against: 0.8M NaFormate, 0.1 M NaAcetate, 5% PEG 3350. Crystals grew over the course of 1–2 months at 20°C as either small plates or rods dimensions 0.3 mm × 0.05 mm × 0.05 mm. Rod shaped crystals contained one dimer in the asymmetric unit with space group P21.
Data Collection and Refinement
A 1.73 Å data set was collected at the Advance Light Source at the Berkeley National Laboratory, beamline 8.3.1. using a crystal flash cooled to 100 K in mother liquor with 20% glycerol as the cryoprotectant. Diffraction data was integrated using Mosflm 40 and scaled and merged using Scala 41. The structure was solved by molecular replacement using Molrep 42 with the apo KSHV protease structure as the search model (PDB cokde:1FL1 18). Manual cycles of rebuilding were performed using the COOT graphical interface 43, followed by the refinement in Refmac5 44 In the final rounds of refinement 4 TLS 45 groups were defined one each per protein monomer and one each per inhibitor and refined in Refmac 5 44.
Our current model of KSHV protease dimer in complex with phosphonate inhibitor contains the complete sequence (3–230 amino acids) in molecule A, while in molecule B amino acid 126 is disordered. The crystallographic information is summarized in Table 2. The structure coordinates are deposited at the protein data bank under the code 2PBK.
Table 2.
Data collection and refinement statistics
| Data Collection | |
| Resolution limit (Å) | 1.73–20.00 |
| Unique Reflections | 47867 |
| Completeness (%) | 93.9 (61.8)b |
| Redundancy | 3.8 (2.6)b |
| I/σ<I> | 13.2 (4.8)b |
| Rmerge(%) | 0.048 (0.205)b |
| Refinement statistics | |
| Rcrys t (%) | 0.16 (0.23)a |
| Rfree (%) | 0.20 (0.26)a |
| Number of atoms | 4182 |
| Protein | 3538 |
| Ligand | 102 |
| Solvent | 542 |
| Average B-factor (Å2) | 30.9 |
| Protein | 29.2 |
| Ligand | 29.3 |
| Solvent | 42.3 |
| R.m.s. deviation | |
| Bond lengths (Å) | 0.015 |
| Angles (°) | 1.49 |
| Ramachandran (%) | |
| Most favored | 91.4 |
| Allowed | 8.6 |
Numbers in parenthesis are for the highest resolution shell: a (1.78–1.73) and b (1.82–1.73)
Solvent Accessible surface area and ΔG calculation
The buried solvent accessible surface area at the substrate binding pockets was calculated using the Protein interfaces, surfaces and assemblies service PISA at European Bioinformatics Institute (http://www.ebi.ac.uk/msd-srv/prot_int/pistart.html), authored by E. Krissinel and K. Henrick 46. The difference in Gibbs free energy (ΔΔG) was calculated with the assumption that burial of 1Å2 of solvent accessible hydrophobic surface contributes 16 cal/mol binding energy 47. The ΔΔG was calculated from kcat/KM values using following formula48:
where A is a longer substrate (hexapeptide) and B a shorter substrate (tetrapeptide).
Synthesis and Kinetic Analysis of peptide substrates
Individual peptide 7-amino-4-carbamoyl-methylcoumarin (ACC) substrates: P3-P1 (Ac-tBut-Gln-Ala-ACC), P4-P1 (Ac-Y-tBut-Gln-Ala-ACC), P5-P1-ACC (Ac-V-Y-tBut-Gln-Ala-ACC), P6-P1-ACC (Ac-P-V-Y-tBut-Gln-Ala-ACC), P7-P1-ACC (Ac-S-P-V-Y-tBut-Gln-Ala-ACC) were synthesized and purified as described 49. Concentrated KSHV protease was diluted to 1μM and incubated at 30ºC for 1 hour until monomer-dimer equilibrium was established. The 98μl of 1μM KSHV protease were aliquoted in individual wells on a 96-well plate. Peptide stocks were prepared in DMSO (0.6–30mM) and 2μl were aliquoted in the protease containing wells to a final substrate concentration 6–300μM. Reaction rate of substrate hydrolysis was monitored as an increase in fluorescence of the ACC leaving group for 60 minutes at 30ºC as described previously 4.
Supplementary Material
Acknowledgments
We thank Dr. Mark David Lim and Brandon Tavshanjian for assistance with peptide synthesis. This research was funded by a US National Institute of Health Grant GM56531 (C.S.C) and a University of California President’s dissertation year award (A.B.M).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Darke PL, Cole JL, Waxman L, Hall DL, Sardana MK, Kuo LC. Active human cytomegalovirus protease is a dimer. J Biol Chem. 1996;271:7445–9. doi: 10.1074/jbc.271.13.7445. [DOI] [PubMed] [Google Scholar]
- 2.Schmidt U, Darke PL. Dimerization and activation of the herpes simplex virus type 1 protease. J Biol Chem. 1997;272:7732–5. doi: 10.1074/jbc.272.12.7732. [DOI] [PubMed] [Google Scholar]
- 3.Batra R, Khayat R, Tong L. Molecular mechanism for dimerization to regulate the catalytic activity of human cytomegalovirus protease. Nat Struct Biol. 2001;8:810–7. doi: 10.1038/nsb0901-810. [DOI] [PubMed] [Google Scholar]
- 4.Marnett AB, Nomura AM, Shimba N, Ortiz de Montellano PR, Craik CS. Communication between the active sites and dimer interface of a herpesvirus protease revealed by a transition-state inhibitor. Proc Natl Acad Sci U S A. 2004;101:6870–5. doi: 10.1073/pnas.0401613101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nomura AM, Marnett AB, Shimba N, Dotsch V, Craik CS. Induced structure of a helical switch as a mechanism to regulate enzymatic activity. Nat Struct Mol Biol. 2005 doi: 10.1038/nsmb1006. [DOI] [PubMed] [Google Scholar]
- 6.Nomura AM, Marnett AB, Shimba N, Dotsch V, Craik CS. One functional switch mediates reversible and irreversible inactivation of a herpesvirus protease. Biochemistry. 2006;45:3572–9. doi: 10.1021/bi0523658. [DOI] [PubMed] [Google Scholar]
- 7.Weinheimer SP, McCann PJ, 3rd, O'Boyle DR, 2nd, Stevens JT, Boyd BA, Drier DA, Yamanaka GA, Dilanni CL, Deckman IC, Cordingley MG. Autoproteolysis of herpes simplex virus type 1 protease releases an active catalytic domain found in intermediate capsid particles. J Virol. 1993;67:5813–22. doi: 10.1128/jvi.67.10.5813-5822.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Donaghy G, Jupp R. Characterization of the Epstein-Barr virus proteinase and comparison with the human cytomegalovirus proteinase. J Virol. 1995;69:1265–70. doi: 10.1128/jvi.69.2.1265-1270.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gao M, Matusick-Kumar L, Hurlburt W, DiTusa SF, Newcomb WW, Brown JC, McCann PJ, 3rd, Deckman I, Colonno RJ. The protease of herpes simplex virus type 1 is essential for functional capsid formation and viral growth. J Virol. 1994;68:3702–12. doi: 10.1128/jvi.68.6.3702-3712.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Unal A, Pray TR, Lagunoff M, Pennington MW, Ganem D, Craik CS. The protease and the assembly protein of Kaposi's sarcoma-associated herpesvirus (human herpesvirus 8) J Virol. 1997;71:7030–8. doi: 10.1128/jvi.71.9.7030-7038.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brignole EJ, Gibson W. Enzymatic activities of human cytomegalovirus maturational protease assemblin and its precursor (pPR, pUL80a) are comparable: [corrected] maximal activity of pPR requires self-interaction through its scaffolding domain. J Virol. 2007;81:4091–103. doi: 10.1128/JVI.02821-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pray TR, Reiling KK, Demirjian BG, Craik CS. Conformational change coupling the dimerization and activation of KSHV protease. Biochemistry. 2002;41:1474–82. doi: 10.1021/bi011753g. [DOI] [PubMed] [Google Scholar]
- 13.Robertson BJ, McCann PJ, 3rd, Matusick-Kumar L, Newcomb WW, Brown JC, Colonno RJ, Gao M. Separate functional domains of the herpes simplex virus type 1 protease: evidence for cleavage inside capsids. J Virol. 1996;70:4317–28. doi: 10.1128/jvi.70.7.4317-4328.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dilanni CL, Drier DA, Deckman IC, McCann PJ, 3rd, Liu F, Roizman B, Colonno RJ, Cordingley MG. Identification of the herpes simplex virus-1 protease cleavage sites by direct sequence analysis of autoproteolytic cleavage products. J Biol Chem. 1993;268:2048–51. [PubMed] [Google Scholar]
- 15.Baum EZ, Bebernitz GA, Hulmes JD, Muzithras VP, Jones TR, Gluzman Y. Expression and analysis of the human cytomegalovirus UL80-encoded protease: identification of autoproteolytic sites. J Virol. 1993;67:497–506. doi: 10.1128/jvi.67.1.497-506.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Preston VG, Rixon FJ, McDougall IM, McGregor M, al Kobaisi MF. Processing of the herpes simplex virus assembly protein ICP35 near its carboxy terminal end requires the product of the whole of the UL26 reading frame. Virology. 1992;186:87–98. doi: 10.1016/0042-6822(92)90063-u. [DOI] [PubMed] [Google Scholar]
- 17.Kennard J, Rixon FJ, McDougall IM, Tatman JD, Preston VG. The 25 amino acid residues at the carboxy terminus of the herpes simplex virus type 1 UL26.5 protein are required for the formation of the capsid shell around the scaffold. J Gen Virol. 1995;76(Pt 7):1611–21. doi: 10.1099/0022-1317-76-7-1611. [DOI] [PubMed] [Google Scholar]
- 18.Reiling KK, Pray TR, Craik CS, Stroud RM. Functional consequences of the Kaposi's sarcoma-associated herpesvirus protease structure: regulation of activity and dimerization by conserved structural elements. Biochemistry. 2000;39:12796–803. doi: 10.1021/bi001019h. [DOI] [PubMed] [Google Scholar]
- 19.Tong L, Qian C, Massariol MJ, Deziel R, Yoakim C, Lagace L. Conserved mode of peptidomimetic inhibition and substrate recognition of human cytomegalovirus protease. Nat Struct Biol. 1998;5:819–26. doi: 10.1038/1860. [DOI] [PubMed] [Google Scholar]
- 20.Khayat R, Batra R, Qian C, Halmos T, Bailey M, Tong L. Structural and biochemical studies of inhibitor binding to human cytomegalovirus protease. Biochemistry. 2003;42:885–91. doi: 10.1021/bi027045s. [DOI] [PubMed] [Google Scholar]
- 21.Ogilvie W, Bailey M, Poupart MA, Abraham A, Bhavsar A, Bonneau P, Bordeleau J, Bousquet Y, Chabot C, Duceppe JS, Fazal G, Goulet S, Grand-Maître C, Guse I, Halmos T, Lavallée P, Leach M, Malenfant E, O'Meara J, Plante R, Plouffe C, Poirier M, Soucy F, Yoakim C, Déziel R. Peptidomimetic inhibitors of the human cytomegalovirus protease. Journal of Medicinal Chemistry. 1997;40:4113–35. doi: 10.1021/jm970104t. [DOI] [PubMed] [Google Scholar]
- 22.Bertrand JA, Oleksyszyn J, Kam CM, Boduszek B, Presnell S, Plaskon RR, Suddath FL, Powers JC, Williams LD. Inhibition of trypsin and thrombin by amino(4-amidinophenyl)methanephosphonate diphenyl ester derivatives: X-ray structures and molecular models. Biochemistry. 1996;35:3147–55. doi: 10.1021/bi9520996. [DOI] [PubMed] [Google Scholar]
- 23.Tong L, Qian C, Massariol MJ, Bonneau PR, Cordingley MG, Lagace L. A new serine-protease fold revealed by the crystal structure of human cytomegalovirus protease. Nature. 1996;383:272–5. doi: 10.1038/383272a0. [DOI] [PubMed] [Google Scholar]
- 24.Qiu X, Culp JS, DiLella AG, Hellmig B, Hoog SS, Janson CA, Smith WW, Abdel-Meguid SS. Unique fold and active site in cytomegalovirus protease. Nature. 1996;383:275–9. doi: 10.1038/383275a0. [DOI] [PubMed] [Google Scholar]
- 25.Hoog SS, Smith WW, Qiu X, Janson CA, Hellmig B, McQueney MS, O'Donnell K, O'Shannessy D, DiLella AG, Debouck C, Abdel-Meguid SS. Active site cavity of herpesvirus proteases revealed by the crystal structure of herpes simplex virus protease/inhibitor complex. Biochemistry. 1997;36:14023–9. doi: 10.1021/bi9712697. [DOI] [PubMed] [Google Scholar]
- 26.Buisson M, Hernandez JF, Lascoux D, Schoehn G, Forest E, Arlaud G, Seigneurin JM, Ruigrok RW, Burmeister WP. The crystal structure of the Epstein-Barr virus protease shows rearrangement of the processed C terminus. J Mol Biol. 2002;324:89–103. doi: 10.1016/s0022-2836(02)01040-9. [DOI] [PubMed] [Google Scholar]
- 27.Khayat R, Batra R, Massariol MJ, Lagace L, Tong L. Investigating the role of histidine 157 in the catalytic activity of human cytomegalovirus protease. Biochemistry. 2001;40:6344–51. doi: 10.1021/bi010158b. [DOI] [PubMed] [Google Scholar]
- 28.LaPlante SR, Aubry N, Bonneau PR, Cameron DR, Lagace L, Massariol MJ, Montpetit H, Plouffe C, Kawai SH, Fulton BD, Chen Z, Ni F. Human cytomegalovirus protease complexes its substrate recognition sequences in an extended peptide conformation. Biochemistry. 1998;37:9793–801. doi: 10.1021/bi980555v. [DOI] [PubMed] [Google Scholar]
- 29.Bonneau PR, Grand-Maitre C, Greenwood DJ, Lagace L, LaPlante SR, Massariol MJ, Ogilvie WW, O'Meara JA, Kawai SH. Evidence of a conformational change in the human cytomegalovirus protease upon binding of peptidyl-activated carbonyl inhibitors. Biochemistry. 1997;36:12644–52. doi: 10.1021/bi970366x. [DOI] [PubMed] [Google Scholar]
- 30.Koshland DE., Jr Protein and biological control. Sci Am. 1973;229:52–64. doi: 10.1038/scientificamerican1073-52. [DOI] [PubMed] [Google Scholar]
- 31.Liang PH, Brun KA, Feild JA, O'Donnell K, Doyle ML, Green SM, Baker AE, Blackburn MN, Abdel-Meguid SS. Site-directed mutagenesis probing the catalytic role of arginines 165 and 166 of human cytomegalovirus protease. Biochemistry. 1998;37:5923–9. doi: 10.1021/bi9726077. [DOI] [PubMed] [Google Scholar]
- 32.Hedstrom L. Serine protease mechanism and specificity. Chem Rev. 2002;102:4501–24. doi: 10.1021/cr000033x. [DOI] [PubMed] [Google Scholar]
- 33.Perona JJ, Craik CS. Evolutionary divergence of substrate specificity within the chymotrypsin-like serine protease fold. J Biol Chem. 1997;272:29987–90. doi: 10.1074/jbc.272.48.29987. [DOI] [PubMed] [Google Scholar]
- 34.Perona JJ, Craik CS. Structural basis of substrate specificity in the serine proteases. Protein Sci. 1995;4:337–60. doi: 10.1002/pro.5560040301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Buisson M, Valette E, Hernandez JF, Baudin F, Ebel C, Morand P, Seigneurin JM, Arlaud GJ, Ruigrok RW. Functional determinants of the Epstein-Barr virus protease. Journal of Molecular Biology. 2001;311:217–28. doi: 10.1006/jmbi.2001.4854. [DOI] [PubMed] [Google Scholar]
- 36.Lagunoff M, Bechtel J, Venetsanakos E, Roy AM, Abbey N, Herndier B, McMahon M, Ganem D. De novo infection and serial transmission of Kaposi's sarcoma-associated herpesvirus in cultured endothelial cells. J Virol. 2002;76:2440–8. doi: 10.1128/jvi.76.5.2440-2448.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jing H, Babu YS, Moore D, Kilpatrick JM, Liu XY, Volanakis JE, Narayana SV. Structures of native and complexed complement factor D: implications of the atypical His57 conformation and self-inhibitory loop in the regulation of specific serine protease activity. J Mol Biol. 1998;282:1061–81. doi: 10.1006/jmbi.1998.2089. [DOI] [PubMed] [Google Scholar]
- 38.Xu Y, Narayana SV, Volanakis JE. Structural biology of the alternative pathway convertase. Immunol Rev. 2001;180:123–35. doi: 10.1034/j.1600-065x.2001.1800111.x. [DOI] [PubMed] [Google Scholar]
- 39.Pray TR, Nomura AM, Pennington MW, Craik CS. Auto-inactivation by cleavage within the dimer interface of Kaposi's sarcoma-associated herpesvirus protease. J Mol Biol. 1999;289:197–203. doi: 10.1006/jmbi.1999.2791. [DOI] [PubMed] [Google Scholar]
- 40.Leslie AGW. Recent changes to the MOSFLM package for processing film and image plate data. Joint CCP4 + ESF-EAMCB Newsletter on Protein Crystallography; 1992. [Google Scholar]
- 41.Evans PR. Proceedings of CCP4 Study Weekend, 1993, on Data Collection & Processing.1993. [Google Scholar]
- 42.Vagin A, Teplyakov A. MOLREP: an automated program for molecular replacement. J Appl Cryst. 1997:1022–1025. [Google Scholar]
- 43.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–32. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 44.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr. 1997;53:240–55. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 45.Winn MD, Isupov MN, Murshudov GN. Use of TLS parameters to model anisotropic displacements in macromolecular refinement. Acta Crystallogr D Biol Crystallogr. 2001;57:122–33. doi: 10.1107/s0907444900014736. [DOI] [PubMed] [Google Scholar]
- 46.Krissinel E, Henrick K. Detection of Protein Assemblies in Crystals. In: Berthold MRea., editor. Computational Life Sciences. Vol. 3695. Springer Berlin; Heidelberg: 2005. pp. 163–174. [Google Scholar]
- 47.Eisenberg D, McLachlan AD. Solvation energy in protein folding and binding. Nature. 1986;319:199–203. doi: 10.1038/319199a0. [DOI] [PubMed] [Google Scholar]
- 48.Fersht AR. Chemical basis of biological specificity. Pure and Applied Chemistry. 1982;54:1819–1824. [Google Scholar]
- 49.Harris JL, Backes BJ, Leonetti F, Mahrus S, Ellman JA, Craik CS. Rapid and general profiling of protease specificity by using combinatorial fluorogenic substrate libraries. Proc Natl Acad Sci U S A. 2000;97:7754–9. doi: 10.1073/pnas.140132697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wallace AC, Laskowski RA, Thornton JM. LIGPLOT: a program to generate schematic diagrams of protein-ligand interactions. Protein Eng. 1995;8:127–34. doi: 10.1093/protein/8.2.127. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.