

Abstract
Paramyxoviruses have been shown to produce proteins that inhibit interferon production and signaling. For human respiratory syncytial virus (RSV), the nonstructural NS1 and NS2 proteins have been shown to have interferon antagonist activity through an unknown mechanism. To understand further the functions of NS1 and NS2, we generated recombinant RSV in which both NS1 and NS2 were replaced by the PIV5 V protein, which has well-characterized IFN antagonist activities (ΔNS1/2-V). Expression of V was able to partially inhibit IFN responses in ΔNS1/2-V-infected cells. In addition, the replication kinetics of ΔNS1/2-V were intermediate between ΔNS1/2 and wild-type (rA2) in A549 cells. However, expression of V did not affect the ability of ΔNS1/2-V to activate IRF3 nuclear translocation and IFNβ transcription. These data indicate that V was able to replace some of the IFN inhibitory functions of the RSV NS1 and NS2 proteins, but also that NS1 and NS2 have functions in viral replication beyond IFN antagonism.
Keywords: paramyxovirus, interferon antagonism, nonstructural
INTRODUCTION
Respiratory syncytial virus (RSV) is the most important etiologic agent of pediatric viral respiratory infection and remains a major cause of morbidity and mortality among infants as well as immunocompromised subjects and the elderly (Collins et al., 2001). RSV is classified in the family Paramyxoviridae in the order Mononegavirales, and is the prototype member of the Pneumovirus genus. The nonsegmented, negative-sense (NNS) RNA genome of RSV is 15,222 nt long and contains 10 transcription units from which 11 proteins are translated. A number of these proteins are homologous to proteins from other paramyxoviruses, including nucleocapsid (N nucleocapsid, P phosphoprotein, L polymerase), assembly (M matrix), and attachment and fusion (G and F) proteins. In addition, RSV encodes two proteins in the M2 gene (M2-1 and M2-2) that are thought to play a role in regulating RNA synthesis by the RSV polymerase complex. Finally, the NS1 and NS2 proteins are nonstructural proteins which are dispensable for viral replication in vitro; however, deletion of either NS gene attenuates recombinant RSV (rRSV) significantly in vitro and markedly in vivo (Buchholz et al., 1999; Jin et al., 2000; Schlender et al., 2000; Teng and Collins, 1999; Whitehead et al., 1999). RSV enters cells by direct fusion of its envelope with the plasma membrane and replicates solely in the cytoplasm. Transcription of the genome by the viral RNA-dependent RNA polymerase is polar, such that the genes located proximal to the genomic promoter at the 3′ end of the viral RNA are transcribed earlier and to a greater extent than the promoter distal genes. Production of viral proteins leads to assembly of replicated genomes into encapsidated ribonucleoprotein (RNP) complexes that traffic to the plasma membrane where virion morphogenesis occurs. The primary site of infection is the respiratory epithelium, with infection and release occurring at the apical surface. A distinctive characteristic of RSV is that it does not induce long-lived immunity upon exposure, resulting in recurrent infection throughout life.
That viruses have developed mechanisms for inhibiting antiviral activities induced by IFNs has been well documented. A well-studied example of viral interferon antagonism is the parainfluenza virus 5 (PIV5), formerly SV5, V protein. PIV5 is a member of the Rubulavirinae genus of the family Paramyxoviridae (Lamb and Kolakofsky, 2001). The V protein of PIV5 is encoded by the V/P gene whose mRNA is “edited” during transcription, resulting in the insertion of 2 G residues which alters the reading frame and allows the production of the P protein, an essential cofactor for the viral polymerase (Lamb and Kolakofsky, 2001). PIV5 V shares its N-terminus with P, but encodes a cysteine-rich C-terminus, which is well conserved among paramyxoviruses, and is produced in relatively equimolar amounts to P (Lamb and Kolakofsky, 2001). V has been shown to assemble ubiquitin ligase complexes targeted to STAT1, resulting in the proteasome-mediated degradation of STAT1 (Ulane and Horvath, 2002). The polyubiquitylation of STAT1 by these V-dependent ubiquitin ligase machines requires species-specific recognition of STAT2 (Parisien et al., 2002). A number of other paramyxoviruses have also been shown to encode accessory proteins that target the JAK-STAT pathway to inhibit IFN signaling (Andrejeva et al., 2002b; Didcock et al., 1999; Horvath, 2004; Rodriguez et al., 2004; Young et al., 2000). In addition to inhibiting IFN signaling, PIV5 V can inhibit IFN production (He et al., 2002; Poole et al., 2002). Recently, Andrejeva et al. have shown that this inhibition results from the interaction of V with the DExD/H-box helicase MDA-5 and blocks the activation of the transcription factors IRF3 and NFκB (Poole et al., 2002). Other viruses have been shown to inhibit IFN production, though the mechanisms by which this effect occurs is not well described. One notable exception is hepatitis C virus NS3/4A, which has been found to disrupt IFN production by cleaving signal transduction proteins in both the RIG-I and TLR3 pathways (Foy et al., 2005; Li et al., 2005).
The nonstructural proteins of RSV also appear to have IFN inhibitory functions. NS1 and NS2 are encoded by the two promoter-proximal transcription units, making them the earliest and most abundantly transcribed genes. These small proteins (NS1, 139 a.a.; NS2, 124 a.a.) have no significant sequence homology with each other or with any cellular protein in the database. NS1 and NS2 appear to antagonize both the cellular antiviral response as well as the induction of IFN production (Bossert and Conzelmann, 2002; Bossert et al., 2003; Schlender et al., 2000; Spann et al., 2004). This antagonism likely requires the accumulation of NS1 and NS2 in the cell since RSV induces STAT1 phosphorylation at early time points postinfection (Kong et al., 2003); however, the mechanism for this antagonism is unclear. Later in infection, RSV appears to cause the degradation of STAT2, though this effect may be cell-type dependent (Ramaswamy et al., 2004). Recent evidence suggests that both NS1 and NS2 are important for STAT2 degradation by RSV (Lo et al., 2005; Ramaswamy et al., 2005). Thus, the attenuation of rRSV lacking NS1 and/or NS2 may be due in part to the lack of interferon antagonism by these viruses. However, even in interferon-deficient cells, the growth of rRSV lacking NS1 and/or NS2 is attenuated (Jin et al., 2000; Spann et al., 2004). Thus, the NS genes likely have functions required for optimal RSV replication in addition to IFN antagonism. There is some evidence that NS1 may interact with viral proteins M and P (Evans et al., 1996; Hengst and Kiefer, 2000) and expression of NS1 in a minireplicon system strongly inhibits RNA replication and transcription by the RSV polymerase, though the mechanism is unknown (Atreya et al., 1998). Therefore, these small nonstructural proteins encode multiple functions that are essential for optimal RSV replication.
RESULTS
Recovery of rRSV containing the V gene of PIV5 in place of the NS1 and NS2 genes of RSV
Recent studies have implicated NS1 and NS2 in antagonizing the host interferon response in RSV-infected cells (Bossert et al., 2003; Spann et al., 2004; Spann et al., 2005). As an initial step to determining the mechanism of this interferon antagonism, we replaced the NS1 and NS2 with the open reading frame of the PIV5 V gene (Figure 1a). Since the V mRNA can be “edited” during PIV5 infection, resulting in the insertion of 2 G residues, we cloned in two versions of the V ORF. The first contained the wild-type V ORF (Vwt) and the second contained a V ORF in which the editing site was mutated to prevent insertion of G residues (Vmut). Both rRSVs encoding V in place of NS1 and NS2 (ΔNS1/2-Vwt or ΔNS1/2-Vmut) were recovered essentially as described (Buchholz et al., 1999).
Figure 1.

Generation of V mutant rRSV. (a) Genomic structure of V mutant rRSV. (b) Expression of viral proteins in rRSV-infected cells. Vero cells were infected by the indicated viruses at a MOI of 3. Cell lysates were harvested at 16 h p.i. and separated by SDS-PAGE. Viral proteins were detected by Western blot analysis using antibodies directed against RSV structural proteins (top), V (middle), or NS1 and NS2 (bottom). (c) Electropherograms of sequences of V editing sites. Total RNA was extracted from cells infected by PIV5, ΔNS1/2-Vwt, and ΔNS1/2-Vmut at a MOI of 3 for 16 hours and subjected to RT-PCR using the V cloning primers. PCR fragments were isolated and sequenced.
We next examined if the mutant rRSVs expressed the proper complement of viral proteins. Therefore, we performed Western blot analysis of whole cell lysates derived from Vero cells infected by rA2, ΔNS1/2, ΔNS1/2-Vwt, or ΔNS1/2-Vmut (Figure 1b). As expected, these viruses all expressed the RSV structural proteins and the ΔNS1/2 derivatives did not express either nonstructural protein. In addition, the V mutant rRSVs expressed immunoreactive V protein that migrates at the same rate with PIV5 V in SDS-PAGE (Figure 1b, middle panel).
We constructed two versions of the V mutant rRSVs since it was formally possible that the RSV polymerase could recognize the PIV5 editing site and insert two Gs during transcription, altering the coding frame, and thus the protein sequence, of the C-terminus of V. The antibody used in the Western blot only recognizes the common N-terminus of the PIV5 P and V proteins, so V protein derived from edited or unedited mRNAs would be recognized similarly. V from edited mRNA in the ΔNS1/2-Vwt infected cells would be predicted to be 13 residues longer, though the amino acid composition of the C-terminus would be completely different. We were unable to detect a difference in electrophoretic mobility between V expressed from PIV5 compared with that from either rRSV by Western blot (Figure 1b). However, to ensure that no editing had occurred, we isolated total RNA from PIV5, ΔNS1/2-Vwt, and ΔNS1/2-Vmut infected cells, amplified the V mRNA by PCR using specific primers, and sequenced the resulting PCR fragment. As expected, the sequence of V mRNA derived from PIV5 infected cells becomes heterogeneous after the editing site, indicative of the insertion of G residues. However, PCR fragments derived from the V mutant rRSV showed homogeneous sequences throughout this region regardless of the presence of a functional editing site (Figure 1c). Thus, the RSV polymerase does not recognize the V editing site.
PIV5 V has previously been shown to be responsible for both inhibition of MDA-5 and STAT1 degradation, leading to antagonism of both IFNβ production and signaling, respectively (Andrejeva et al., 2004; Andrejeva et al., 2002a; Didcock et al., 1999; He et al., 2002). Therefore, we sought to show that the V expressed by the mutant rRSVs was functional by assessing the status of STAT1 in infected cells by Western blot. A549 cells were infected by rA2, ΔNS1/2, ΔNS1/2-Vwt, or ΔNS1/2-Vmut at a MOI of 3 and samples were harvested at 4-hour intervals postinfection (p.i.). The accumulation of V, STAT1, and N (infection control) protein was detected by Western blot (Figure 2). The expression of STAT1 decreased slightly over time in rA2-infected cells, but was detectable even 36 h p.i. (Figure 2a). In contrast, STAT1 levels increased markedly during the course of infection by ΔNS1/2 (Figure 2b). In both ΔNS1/2-Vmut and ΔNS1/2-Vwt infected cells, STAT1 levels decreased to undetectable levels by 20 h p.i. (Figure 2c and d). Interestingly, accumulation of detectable V did not occur until 20–24 h p.i. in V mutant rRSV-infected cells. Thus, depression of STAT1 occurred in infected cells with undetectable levels of V. Regardless, the expression of V by the mutant rRSVs was sufficient to completely knock out the presence of STAT1 in infected cells, indicating that the expressed V is functional.
Figure 2.
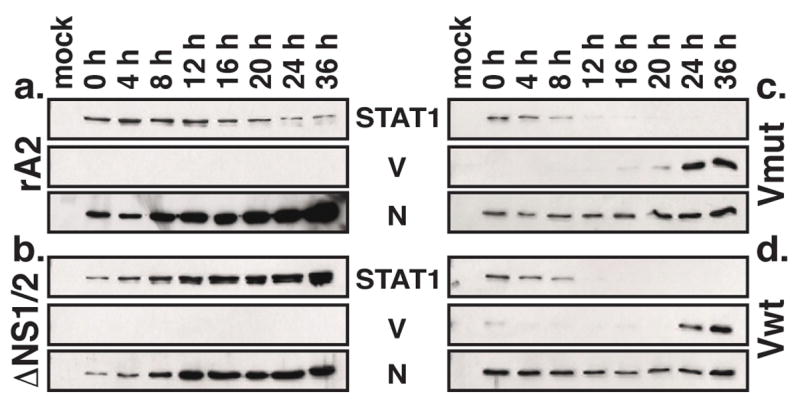
V expressed by rRSV causes STAT1 degradation. A549 cells were infected by rA2 (a), ΔNS1/2 (b), ΔNS1/2-Vwt (c), or ΔNS1/2-Vmut (d) at a MOI of 3. Total cell extracts were harvested at 4 hour intervals as indicated and subjected to Western blot analysis using antibodies to V and STAT1. N expression is shown as a control for infection.
PIV5 V partially rescues the interferon antagonism of RSV NS1 and NS2
Since NS1 and NS2 have been shown to have interferon antagonist activity, we next examined whether PIV5 V could inhibit IFN production and signaling in the context of RSV infection. Therefore, we examined the level of IFNβ mRNA in cells infected by the V mutant rRSVs. Total cellular RNA was isolated from infected A549 cells at 16 h p.i. and was subjected to Northern blot analysis using a radiolabeled probe for IFNβ (Figure 3). Infection by rA2 results in barely detectable accumulation of IFNβ mRNA after 16 h p.i., while ΔNS1/2-infected cells contain elevated amounts of IFNβ mRNA (Figure 3). Interestingly, infection by the V mutant rRSVs resulted in an intermediate level of IFNβ mRNA accumulation. Densitometric analysis indicated that IFNβ mRNA levels were approximately 2-fold greater in V mutant rRSV versus rA2-infected cells, while ΔNS1/2-infected cells contained > 6-fold higher levels. Thus, V expression was partially effective in replacing the inhibition of IFNβ production by NS1 and NS2.
Figure 3.
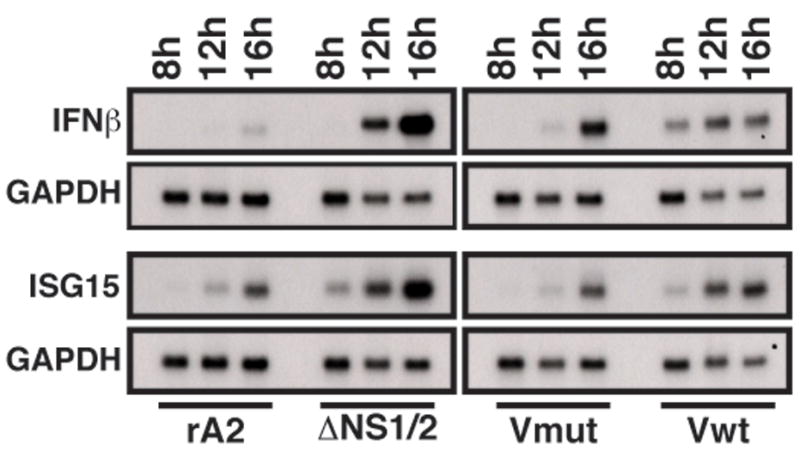
Accumulation of IFNβ and ISG15 mRNA in rRSV-infected cells. A549 cells were infected with the indicated viruses at a MOI of 3. At 8, 12, and 16 h p.i., total RNA was isolated from the infected cells and subjected to Northern blot analysis using radiolabled DNA probes to IFNβ (top) or ISG15 (bottom). Bands were visualized by autoradiography. After exposure, the blots were stripped and reprobed with a radiolabeled probe for GAPDH as a loading control (lower panels).
We next examined the transcription of the ISG15 gene, which can be activated both by interferon signaling through the JAK/STAT pathway as well as by IRF3. Infection by rA2 resulted in a low level of ISG15 mRNA accumulation, consistent with the low IFNβ levels, while ΔNS1/2 infection induced higher levels (Figure 3). As expected, ISG15 mRNA in V mutant rRSV-infected cells was expressed at similarly low levels as those in rA2-infected cells. These levels were approximately 2-fold lower than those in ΔNS1/2-infected cells by densitometry. These data suggest that V, like NS1/2, is able to inhibit JAK/STAT signaling in the context of RSV infection, albeit using a different mechanism.
Transcription from the IFNβ promoter requires the activation and nuclear translocation of IRF3. Previous studies have shown that infection of cells with RSV lacking NS1 and/or NS2 induces activation of IRF3 (Bossert et al., 2003; Spann et al., 2005). Therefore, we examined the subcellular localization of IRF3 in RSV-infected cells. A549 cells were infected by rA2, ΔNS1/2, or the V-mutant rRSVs and subjected to immunofluorescence assays using antibodies to RSV F and IRF3. The percentages of infected cells with nuclear IRF3 were determined at 9, 12, and 16 h p.i. (Figure 4a–c). At early time points after infection, IRF3 nuclear translocation was not significantly different among the infected cell populations (Figure 4a and b); however, by 16 h p.i., ΔNS1/2-infected cells had approximately twice as many cells with nuclear IRF3 compared with rA2 (Figure 4c). A549 cells infected by ΔNS1/2-Vmut or ΔNS1/2-Vwt behaved similarly to those infected by ΔNS1/2, showing ~60% of the cells having nuclear IRF3 at 16 h p.i. (Figure 4c). To confirm the nuclear localization of IRF3 was indicative of its activation, we performed a Western blot analysis for phosphorylated IRF3 (Figure 4d). Consistent with the immunofluorescence analysis, cells infected by ΔNS1/2 or the V mutant rRSVs showed similar levels of Ser396 phosphorylation. In addition, we performed an electrophoretic mobility shift assay (EMSA) using oligonucleotide probes for the interferon-stimulated response element (ISRE) from the ISG15 gene (Figure 4e). We found that nuclear extracts from rRSV-infected A549 cells formed complexes that specifically retarded the mobility of the probe and that this binding could be ablated by incubation with excess cold competitor. Thus, in contrast to the Northern blot results with IFNβ, expression of V did not alter the ability of ΔNS1/2 to induce IRF3 activation.
Figure 4.
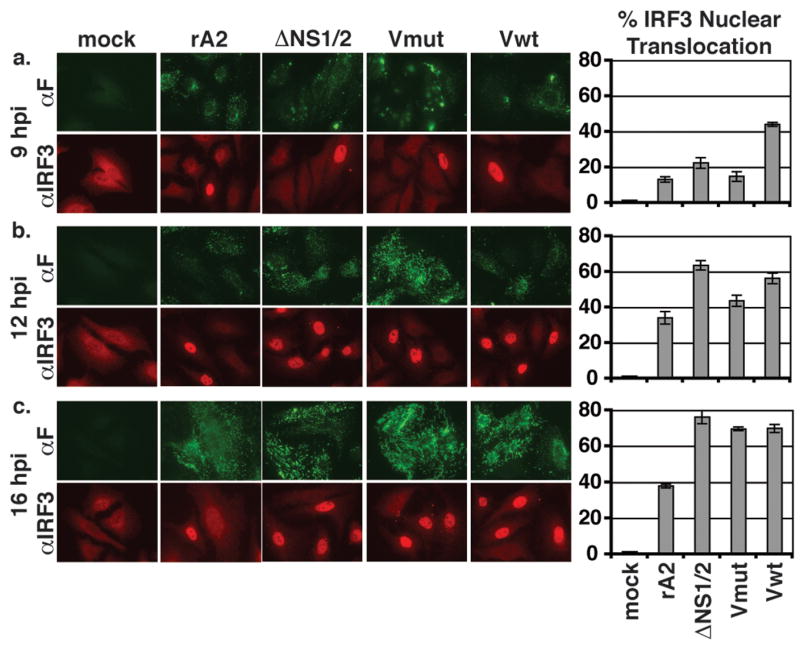
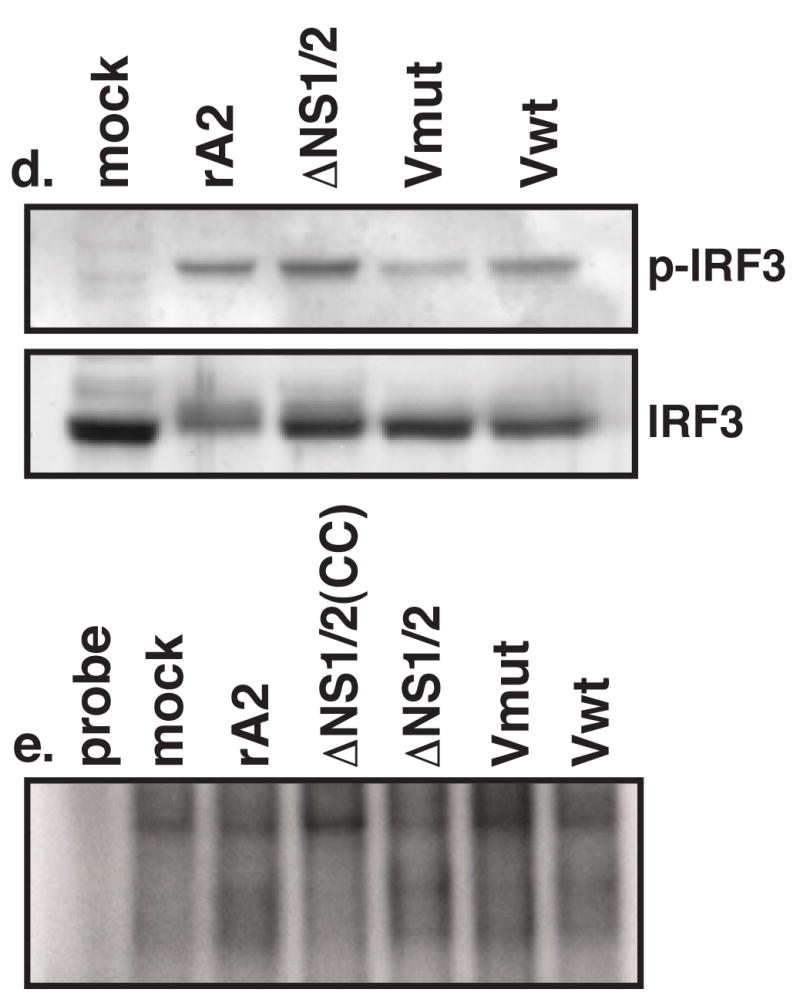
Activation of IRF-3 in rRSV-infected cells. A549 cells on coverslips were infected at a MOI of 3 by the indicated viruses. At 9 (a), 12 (b), or 16 (c) h p.i., coverslips were fixed in formalin, permeabilized, and subjected to indirect immunofluorescence using antibodies to F (green) and IRF-3 (red). The percentages of infected cell displaying nuclear IRF-3 (graphs) were determined by counting approximately 300 cells per coverslip. Each sample was assayed in triplicate. Error bars represent SEM. (d) A549 cells were infected at a MOI of 3 by the indicated viruses. Total cell lysates were harvested 16 h p.i. and subjected to Western blot analysis for phosphorylated (Ser396) and total IRF3. (e) EMSA for IRF3 binding. A549 cells were infected at a MOI of 3 by the indicated viruses. Nuclear extracts were harvested at 16 h p.i. and used in an EMSA with a radiolabeled ISRE probe. CC, 100X cold competitor.
PIV5 V does not rescue the viral replication of ΔNS1/2
Previous studies with rRSV lacking NS1 and NS2 indicated that these viruses display decreased plaque size and growth kinetics in cell culture (Jin et al., 2000; Spann et al., 2004). Therefore, we examined multiple-step replication of the V mutant rRSVs in a number of cell lines. Growth of ΔNS1/2 was significantly decreased in human (A549, HEp-2, HeLa) and monkey (Vero) cell lines compared with rA2 (Figure 5). Replication of the V mutant rRSVs was intermediate between rA2 and ΔNS1/2 in A549 cells (Figure 5a), mirroring the partial inhibition of IFN seen above. However, replication of the V mutant rRSVs was similar to that of ΔNS1/2 in IFN-deficient Vero cells (Figure 5b). Interestingly, ΔNS1/2 and the ΔNS1/2-V mutants displayed similar growth kinetics in HEp-2 and HeLa cell (Figures 5c and d). Also, V mutant rRSVs also formed plaques with similar decreased size and morphology as ΔNS1/2 in both HEp-2 and Vero cells, reflecting the multiple step replication results (data not shown). Plaque formation by any of the rRSV was not detectable in A549 or HeLa cells (data not shown). Therefore, expression of V cannot replace the functions of NS1 and NS2 in viral replication beyond IFN antagonism.
Figure 5.
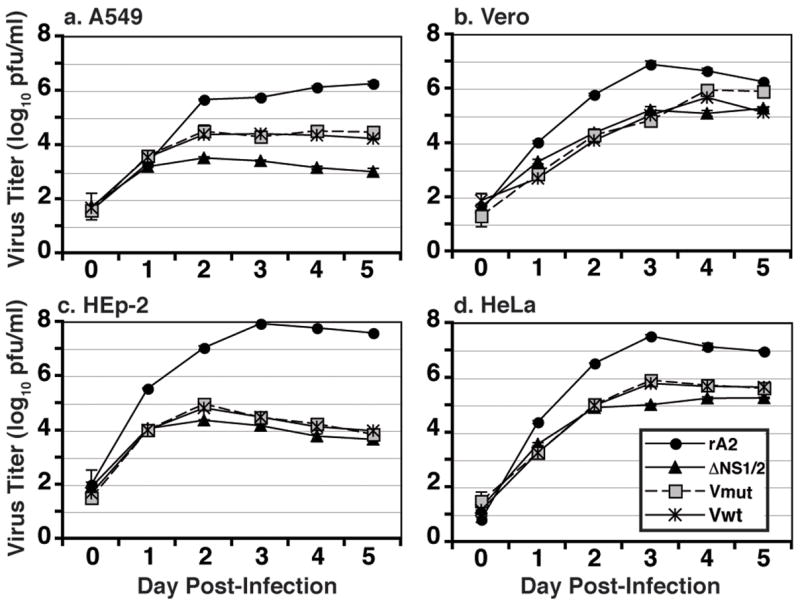
Multiple step growth analysis of V mutant rRSV. A549 (a), Vero (b), HEp-2 (c), or HeLa (d) cells were infected with the V mutant rRSV at a MOI of 0.01 and supernatant samples were harvested daily for 5 d p.i. Supernatants were clarified and viral titers were determined. The analysis was performed in triplicate. Shown are the mean titers; error bars represent the SEM.
PIV5 V has recently been shown to regulate RNA synthesis by the PIV5 polymerase (Lin et al., 2005). To determine whether expression of V altered the function of the RSV polymerase, we performed Northern blot analysis examining the production of viral RNAs in rRSV-infected cells. N mRNA accumulated over time to a similar extent in HEp-2 cells infected by rA2, ΔNS1/2, ΔNS1/2-Vwt, or ΔNS1/2-Vmut (Figure 6). In addition, the expected patterns of readthrough mRNAs were present, including the novel V-N dicistronic mRNA in the ΔNS1/2-V-infected cells. As expected, genome length RNA (Figure 6, G/AG) accumulated dramatically over time in rA2-infected cells but poorly in ΔNS1/2-infected cells. Accumulation of these RNA species in ΔNS1/2-V-infected cells was similar to that seen with ΔNS1/2 (Figure 6c and d). Thus, V does not appear to affect the function of the RSV polymerase.
Figure 6.
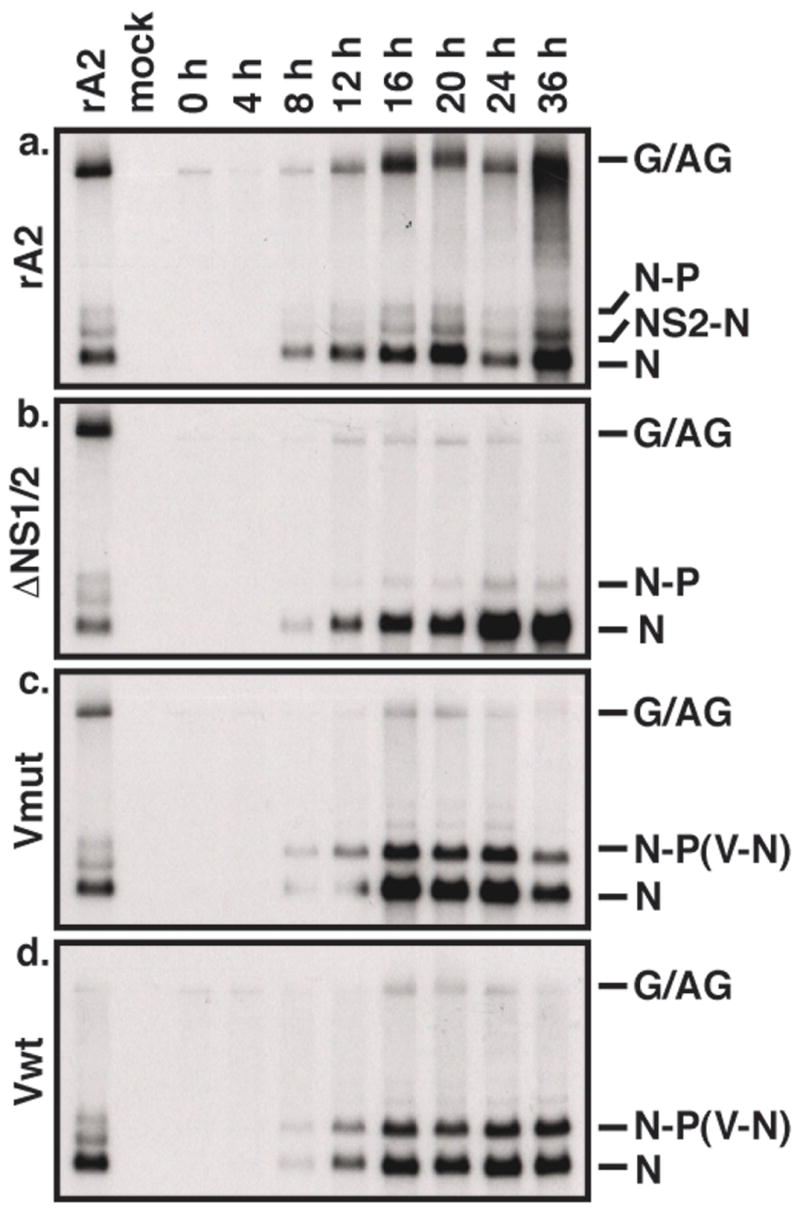
Viral RNA production in rRSV-infected cells. HEp-2 cells were infected by rA2 (a), ΔNS1/2 (b), ΔNS1/2-Vwt (c), or ΔNS1/2-Vmut (d) at a MOI of 3. Total cellular RNA was harvested at 4 hour intervals as indicated and subjected to Northern blot analysis using a radiolabeled DNA probe to RSV N. Viral RNA species are indicated on the right. G/AG, genome/antigenome.
DISCUSSION
We have shown that RSV encoding the PIV5 V protein in place of NS1 and NS2 is viable and express functional V. Our V mutant rRSVs encode the V gene at the promoter proximal position in the gene order. Therefore, V should be highly expressed in ΔNS1/2-V infected cells. However, PIV5 appears to produce significantly more V protein during infection than does ΔNS1/2-Vwt or ΔNS1/2-Vmut. Whether this differential regulation occurs at the level of transcription or translation is currently unknown. In PIV5 infected cells, the viral polymerase inserts 2 G residues at a specific site in the V mRNA during transcription. This process (mRNA “editing”) results in the production of the P mRNA whose product is essential for viral replication. Since paramyxovirus polymerases are thought to operate by similar mechanisms, we replaced NS1 and NS2 in rRSV with either the wild-type V gene or a mutant V that cannot be edited, to ensure that V would be properly expressed. However, we found that the RSV polymerase does not insert G residues into the wild-type V mRNA containing the editing site. Since editing sequences among the paramyxoviruses are highly homologous, our results suggest that the lack of editing of RSV mRNAs is likely due to differences in polymerase function rather than the absence of editing signals.
Recently, a great deal of attention has been focused on the interferon antagonist properties of paramyxovirus accessory proteins, of which PIV5 V is the best-studied example. Inhibition of IRF-3 activation and STAT1 degradation by V both require the unique C-terminus of the protein, though the exact residues responsible for these effects are unknown (Andrejeva et al., 2002a; He et al., 2002). RSV NS1 and NS2 have also been shown to inhibit IFN production and signaling (Bossert et al., 2003; Spann et al., 2004; Spann et al., 2005). Therefore, we investigated whether the IFN antagonism of V could functionally replace that of NS1 and NS2. The V expressed by rRSV was functional in terms of STAT1 degradation, indicating the presence of an intact V protein. Expression of V in place of NS1 and NS2 appeared to partially inhibit IFNβ mRNA accumulation in A549 cells. However, there was no apparent difference in the percentage of infected cells with nuclear IRF3 in A549 cells infected by ΔNS1/2 or ΔNS1/2-V, nor was there a significant difference in the Ser396-phosphorylation and ISRE binding of IRF3 in these cells. The reasons for this discrepancy are not clear. It is known that activation of IFNβ transcription is dependent on both IRF3 and NFκB. Thus, V may inhibit RSV-induced NFκB expression, leading to decreased accumulation of IFNβ mRNA. Alternatively, IFNβ production is thought to activate a positive feedback loop involving IRF7, which has been shown to be a master regulator for type I interferon responses (Honda et al., 2005). This feedback loop would be interrupted in cells in which the JAK/STAT pathway has been inhibited (e.g., NS1/2- or V-expressing cells), resulting in reduced expression of IFNβ. ISG15 transcription, which is induced by IFN, was relatively equal in rA2 and ΔNS1/2-V infected cells, suggesting that JAK/STAT signal transduction abrogated in these cells, in contrast to the increased levels seen with ΔNS1/2 infection. Thus, V was able to partially, but not fully, complement the IFN inhibitory effects of NS1 and NS2.
It is possible that V does not encode the full spectrum of IFN antagonist activities of NS1 and NS2. NS1 and NS2 have been shown to target STAT2 for degradation (Lo et al., 2005; Ramaswamy et al., 2006), but other activities for NS1 and NS2 have not been described. Also, it is possible that NS1 and NS2 act in tandem to subvert multiple IFN activation pathways. Thus, the IFN antagonism of either NS1 or NS2 alone may be able to be replaced by V. In this light, it is interesting to note that rRSV lacking NS1 (ΔNS1) or NS2 (ΔNS2) replicate more efficiently in cells that constitutively express V compared to non-expressing control cells (Young et al., 2003). However, wild-type rA2 as well as rRSV lacking SH or G also replicate to higher levels in V-expressing HEp-2 and 2fTGH cells compared to control, suggesting that the effect of V is not strictly related to its IFN antagonism.
Since expression of V by PIV5 inhibits IFN induction, our data suggest that PIV5 and RSV may activate IFN transcription by overlapping but distinct pathways. PIV5 V binds and inhibits the DEXD/H box helicase MDA-5 (Andrejeva et al., 2004). However, accumulation of detectable levels of V protein in V mutant rRSV-infected cells does not occur until later in infection. Thus, our results do not exclude the possibility that MDA-5 is involved in IFN induction by RSV. Given the sustained increase in IRF3 activation in V mutant rRSV-infected cells, it is likely that other molecules play more important roles in IFN induction. Other pathways that are activated by RNA virus infection include a related DEXD/H box helicase, RIG-I, and TLR3 (Conzelmann, 2005). Recent studies by us and others indicate that RIG-I and TLR3 are essential for IFN induction by RSV (Liu et al., 2007). We are currently determining the relative contribution of each of these pathways to RSV-induced IFN activation.
In addition to their IFN antagonist activities, NS1 and NS2 appear to play a role in viral replication. Accordingly, the plaque morphology and multiple step growth characteristics of ΔNS1/2-Vwt and ΔNS1/2-Vmut are similar to that of ΔNS1/2, markedly decreased compared to wild-type rA2. This growth deficiency appears to be partially relieved by V in A549 cells, but not HEp-2 or HeLa cells. The reason for this cell line specific effect is not known. It is possible that RSV replication is more sensitive to IFN in A549 cells compared to the other cell lines and the partial IFN inhibition due to V is sufficient to allow enhanced viral growth. In addition, A549 cells are derived from human lung carcinoma and may more closely resemble lung epithelial cells than HEp-2 (larynx carcinoma) or HeLa (cervical carcinoma) cells.
Even in IFN-deficient Vero cells, the ΔNS1/2-V mutants replicated similarly to ΔNS1/2, indicating that NS1 and/or NS2 perform functions important for viral replication unrelated to their IFN antagonist activities. NS1 has been previously shown to regulate RNA synthesis by the RSV polymerase in a minigenome system (Atreya et al., 1998). In addition, we have observed that accumulation of RSV N mRNA is slower in cells infected by ΔNS1 or ΔNS1/2 compared to rA2 over the course of a single round of replication (K. C. T. and M. N. T., unpublished data). Thus, NS1 may regulate RSV replication at the level of RNA synthesis. NS2 does not appear to affect RSV macromolecule synthesis. However, deletion of NS2 from recombinant RSV results in pinpoint plaque formation (Teng and Collins, 1999), indicating that NS2 may regulate cell-cell fusion, either by directly affecting F or altering cellular functions.
Deletion of either NS1 or NS2 results in viruses that are extremely attenuated in chimpanzees while inducing protective immunity against subsequent RSV challenge, indicating that these mutant RSVs may be good vaccine candidates (Jin et al., 2000; Teng et al., 2000; Whitehead et al., 1999). Indeed, initial studies in human volunteers with NS2-deleted viruses show promising safety and immunogenicity (Wright et al., 2006). Therefore, understanding the mechanisms by which NS1 and NS2 affect viral replication may enhance our ability to engineer effective vaccines and design antiviral therapies for RSV.
MATERIALS AND METHODS
Plasmid construction
To replace, the NS1 and NS2 ORFs of RSV with PIV5 V, unique restriction sites were engineered into pGEM-NS (Teng and Collins, 1999) by inverse PCR mutagenesis using DeepVent DNA polymerase (NEB). A NheI site was generated downstream of the translation initiation codon of NS1 and a BsiWI site was inserted upstream of the NS2 translation termination codon using the phosphorylated primers NS1 ATG NheI R and NS2 TAA BsiWI F (Table 1). PIV5 V containing a functional editing site (Vwt) or a mutant editing site (Vmut) was amplified from pBH360 and pBH361, respectively, using primers PIV5 V F and PIV5 V R, which encode NheI and BsiWI restriction sites, respectively. The V ORFs were cloned into pGEM-NS using the engineered sites and sequenced. The fragments for each mutant were introduced back into the antigenome cDNA for RSV, resulting in D53ΔNS1/2-Vwt and D53ΔNS1/2-Vmut, which were then used to recover recombinant RSV.
Table I.
Oligonucleotides used for cloning
| Primer name | Sequence |
|---|---|
| NS1 ATG NheI R | CAATGAATTGCTAGCATCTCTAACC |
| NS2 TAA BsiWI F | TAATCCACGTACGTAAATTTCAACACAATA |
| PIV5 V F | TTTGCTAGCGATCCCACTGATCTGAGCTTCTCCCC |
| PIV5 V R | CGGCGTACGTTAAGTATCTCGTTCACATTCAGAGC |
Recovery of recombinant RSV
Recovery of recombinant RSV from cloned DNA was performed as described previously (Collins et al., 1995). Briefly, monolayers of BSR-T7 cells in six-well plates were transfected using GeneJuice (Novagen) with a mixture of plasmids encoding the RSV N, P, L, and M2-1 proteins and either wild type or mutant antigenome cDNA (1, 1, 0.5, 0.5, and 1 μg each/well). The transfection mixture was removed after 20 h of incubation at 37°C and replaced with fresh medium (DMEM supplemented with 5% fetal bovine serum, Life Technologies). After an additional 48 h, the supernatants were harvested, clarified by centrifugation, and passaged onto fresh Vero cells. The recovered viruses were harvested 4 days later. Viral titers were determined by plaque assay on Vero cells. Plaques were visualized by immunostaining using a cocktail of three murine anti-F monoclonal antibodies followed by horseradish peroxidase-coupled anti-mouse IgG antibodies and 4CN substrate (Kirkegaard and Perry Laboratories) as described (Murphy et al., 1990). The presence of the expected mutations was confirmed in recombinant virus by RT-PCR of viral genomic RNA and nucleotide sequencing.
Northern blot and Western blot analysis
A549, Vero and HEp-2 cells were infected in three independent experiments with wild-type or V mutant viruses at a multiplicity of infection (MOI) of 3 PFU per cell. In each experiment, an aliquot of each inoculum was analyzed by plaque assay to confirm the titer. Cells were collected at zero, four, eight, 12, 16, 20, 24 and 36 h post-infection and divided into two aliquots: one was harvested to isolate total intracellular RNA using TRIzol reagent (Invitrogen), and the other was processed for protein analysis by the addition of 2X gel sample buffer (100 mM Tris-Cl pH 6.8, 4% SDS, 20% glycerol, 0.2% bromophenol blue, 200 mM DTT) and centrifugation through Qiashredders (Qiagen). RNA samples were subjected to electrophoresis in 1.0% or 1.2% agarose gels containing formaldehyde, transferred to nitrocellulose (Optitran, 0.2μm; Schleicher & Schuell), and hybridized with 32P-labeled double stranded DNA probes prepared by random hexamer labeling using the Megaprime DNA Labeling Systems (Amersham Life Science). For Western blot analysis, cell extracts were electrophoresed through 10% (for STAT1 and RSV structural proteins) or 12% (for V proteins) polyacrylamide Tris-glycine gels. Proteins were transferred to nitrocellulose membrane (BioTrace NT, Pall) then incubated with rabbit antibodies raised against purified RSV virions (Bukreyev et al., 1997) or with monoclonal antibodies specific to PIV5 V/P (Pk) or STAT1 (Santa Cruz). Bound antibodies were visualized by secondary incubation with horseradish peroxidase-coupled goat anti-rabbit IgG or goat anti-mouse IgG antibodies (Kirkegaard & Perry) followed by chemiluminescence (Pierce). To detect IRF3, proteins were transferred to Immobilon FL (Millipore) and incubated with anti-IRF3 (Santa Cruz) or anti-phospho-Ser396 IRF3 (Cell Signaling) per the manufacturer’s directions. Bound IRF3 antibodies were detected by alkaline phosphatase-coupled secondary antibodies and chemifluorescence (ECF, Amersham) followed by quantitation using a Typhoon phosphorimager (Molecular Dynamics).
Growth of rRSV in vitro
Triplicate cell culture monolayers of A549, Vero, HEp-2, and HeLa cells were infected with each of the V recombinant viruses, ΔNS1/2 or rA2 at an MOI of 0.01 PFU per cell. In each experiment, an aliquot of each inoculum was analyzed by plaque assay to confirm the titer. Cell culture media from infected cells was collected daily for 6 days and virus titers were determined by plaque assay in the Vero cell. Plaques were visualized by immunostaining as described above.
Immunofluorescence assay
A549 cells were plated on coverslips, incubated overnight at 37°C, then infected by the different rRSV at a MOI of 3 in triplicate. At 9, 12, and 16 hours p.i., the cells were fixed in formalin and permeabilized by the addition of 0.1% Triton X-100. Subcellular localization of IRF3 was detected by staining with rabbit anti-human IRF3 (Santa Cruz) followed by Cy3-coupled goat anti-rabbit Ig (Jackson Immunologicals). Virus-infected cells were detected by staining with anti-F monoclonal antibody (MAb1269, Beeler and van Wyke Coelingh, 1989) followed by Alexa488-coupled goat anti-mouse Ig (Jackson Immunologicals). Percentages of infected cells showing nuclear localization of IRF3 were determined by counting approximately 300 infected cells in 10 fields per coverslip. Each experimental time point was performed in triplicate so that each percentage represents between 900 – 1000 cells counted. Micrographs were taken using a Zeiss Axioplan epifluorescence microscope with a 63X Apochromat oil-immersion objective.
Electrophoretic Mobility Shift Assay (EMSA)
A549 cells were infected by rA2, ΔNS1/2, ΔNS1/2-Vmut or ΔNS1/2-Vwt at MOI of 3. Sixteen h (post-infection) p.i., cells were harvested, washed with ice-cold 1X PBS, then resuspended in 10 mM HEPES pH 7.9, 10 mM KCl, 0.1 mM EDTA, 0.1 mM EGTA, 1 mM dithiothreitol (DTT), and 0.5 mM phenylmethylsulfonyl fluoride (PMSF). The suspension was left on ice for 30 min and brought to a final concentration of 0.5% NP-40. Samples were vortexed for 10 s then spun for 30 s at 16,000× g. Supernatants were removed and the pellets were resuspended in 20 mM HEPES pH 7.9, 400 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1 mM DTT, and 0.5 mM PMSF. Samples were mixed on a rotator at 4°C for 15 min and spun at 16000× g for 5 min at 4°C. Nuclear extracts were assayed for IRF3 binding in gel shift analysis using a 32P-labeled double-stranded oligonucleotide corresponding to the ISRE of the ISG15 gene (5′-GATCGGAAAGGGAAACCGAAACTGAAGCC-3′). Complexes were formed by incubating the probe with 10 μg of nuclear extract for 30 min at room temperature in 20 mM Tris-Cl (pH 7.5), 100 mM NaCl, 5% glycerol, 2 mM MgCl2 and poly(dI-dC) (3 μg). Extracts were run on a 6% polyacrylamide gel prepared in 1X Tris/Borate/EDTA (TBE) at 20 mA for 3 h. Subsequently, the gel was dried and exposed to Kodak BioMax film at −80°C overnight.
Acknowledgments
The authors would like to thank Todd Borland for technical assistance and Dr. Lorraine Santy for the use of the fluorescence microscope. This work was funded in part by a grant from the NIAID to B.H. (R01 AI051372).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Andrejeva J, Childs KS, Young DF, Carlos TS, Stock N, Goodbourn S, Randall RE. The V proteins of paramyxoviruses bind the IFN-inducible RNA helicase, mda-5, and inhibit its activation of the IFN-beta promoter. Proc Natl Acad Sci U S A. 2004;101(49):17264–9. doi: 10.1073/pnas.0407639101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrejeva J, Poole E, Young DF, Goodbourn S, Randall RE. The p127 subunit (DDB1) of the UV-DNA damage repair binding protein is essential for the targeted degradation of STAT1 by the V protein of the paramyxovirus simian virus 5. J Virol. 2002a;76(22):11379–86. doi: 10.1128/JVI.76.22.11379-11386.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrejeva J, Young DF, Goodbourn S, Randall RE. Degradation of STAT1 and STAT2 by the V proteins of simian virus 5 and human parainfluenza virus type 2, respectively: consequences for virus replication in the presence of alpha/beta and gamma interferons. J Virol. 2002b;76(5):2159–67. doi: 10.1128/jvi.76.5.2159-2167.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atreya PL, Peeples ME, Collins PL. The NS1 protein of human respiratory syncytial virus is a potent inhibitor of minigenome transcription and RNA replication. J Virol. 1998;72(2):1452–1461. doi: 10.1128/jvi.72.2.1452-1461.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beeler JA, van Wyke Coelingh K. Neutralization epitopes of the F glycoprotein of respiratory syncytial virus: effect of mutation upon fusion function. Journal of Virology. 1989;63(7):2941–2950. doi: 10.1128/jvi.63.7.2941-2950.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bossert B, Conzelmann KK. Respiratory syncytial virus (RSV) nonstructural (NS) proteins as host range determinants: a chimeric bovine RSV with NS genes from human RSV is attenuated in interferon-competent bovine cells. J Virol. 2002;76(9):4287–93. doi: 10.1128/JVI.76.9.4287-4293.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bossert B, Marozin S, Conzelmann KK. Nonstructural proteins NS1 and NS2 of bovine respiratory syncytial virus block activation of interferon regulatory factor 3. J Virol. 2003;77(16):8661–8. doi: 10.1128/JVI.77.16.8661-8668.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchholz UJ, Finke S, Conzelmann KK. Generation of bovine respiratory syncytial virus (BRSV) from cDNA: BRSV NS2 is not essential for virus replication in tissue culture, and the human RSV leader region acts as a functional BRSV genome promoter. J Virol. 1999;73(1):251–9. doi: 10.1128/jvi.73.1.251-259.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bukreyev A, Whitehead SS, Murphy BR, Collins PL. Recombinant respiratory syncytial virus from which the entire SH gene has been deleted grows efficiently in cell culture and exhibits site- specific attenuation in the respiratory tract of the mouse. J Virol. 1997;71(12):8973–82. doi: 10.1128/jvi.71.12.8973-8982.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins PL, Chanock RM, Murphy BR. Respiratory syncytial virus. In: Knipe DM, Howley PM, editors. Fields Virology. 4. Lippincott, Williams and Wilkins; Philadelphia: 2001. pp. 1443–1485. [Google Scholar]
- Collins PL, Hill MG, Camargo E, Grosfeld H, Chanock RM, Murphy BR. Production of infectious human respiratory syncytial virus from cloned cDNA confirms an essential role for the transcription elongation factor from the 5′ proximal open reading frame of the M2 mRNA in gene expression and provides a capability for vaccine development. Proc Natl Acad Sci USA. 1995;92:11563–11567. doi: 10.1073/pnas.92.25.11563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conzelmann KK. Transcriptional activation of alpha/beta interferon genes: interference by nonsegmented negative-strand RNA viruses. J Virol. 2005;79(9):5241–8. doi: 10.1128/JVI.79.9.5241-5248.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Didcock L, Young DF, Goodbourn S, Randall RE. The V protein of simian virus 5 inhibits interferon signalling by targeting STAT1 for proteasome-mediated degradation. J Virol. 1999;73(12):9928–33. doi: 10.1128/jvi.73.12.9928-9933.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans JE, Cane PA, Pringle CR. Expression and characterisation of the NS1 and NS2 proteins of respiratory syncytial virus. Virus Res. 1996;43(2):155–61. doi: 10.1016/0168-1702(96)01327-5. [DOI] [PubMed] [Google Scholar]
- Foy E, Li K, Sumpter R, Jr, Loo YM, Johnson CL, Wang C, Fish PM, Yoneyama M, Fujita T, Lemon SM, Gale M., Jr Control of antiviral defenses through hepatitis C virus disruption of retinoic acid-inducible gene-I signaling. Proc Natl Acad Sci U S A. 2005;102(8):2986–91. doi: 10.1073/pnas.0408707102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He B, Paterson RG, Stock N, Durbin JE, Durbin RK, Goodbourn S, Randall RE, Lamb RA. Recovery of paramyxovirus simian virus 5 with a V protein lacking the conserved cysteine-rich domain: the multifunctional V protein blocks both interferon-beta induction and interferon signaling. Virology. 2002;303(1):15–32. doi: 10.1006/viro.2002.1738. [DOI] [PubMed] [Google Scholar]
- Hengst U, Kiefer P. Domains of human respiratory syncytial virus P protein essential for homodimerization and for binding to N and NS1 protein. Virus Genes. 2000;20(3):221–5. doi: 10.1023/a:1008188527858. [DOI] [PubMed] [Google Scholar]
- Honda K, Yanai H, Negishi H, Asagiri M, Sato M, Mizutani T, Shimada N, Ohba Y, Takaoka A, Yoshida N, Taniguchi T. IRF-7 is the master regulator of type-I interferon-dependent immune responses. Nature. 2005;434(7034):772–7. doi: 10.1038/nature03464. [DOI] [PubMed] [Google Scholar]
- Horvath CM. Silencing STATs: lessons from paramyxovirus interferon evasion. Cytokine Growth Factor Rev. 2004;15(2–3):117–27. doi: 10.1016/j.cytogfr.2004.02.003. [DOI] [PubMed] [Google Scholar]
- Jin H, Zhou H, Cheng X, Tang R, Munoz M, Nguyen N. Recombinant respiratory syncytial viruses with deletions in the NS1, NS2, SH, and M2-2 genes are attenuated in vitro and in vivo. Virology. 2000;273(1):210–8. doi: 10.1006/viro.2000.0393. [DOI] [PubMed] [Google Scholar]
- Kong X, San Juan H, Kumar M, Behera AK, Mohapatra A, Hellermann GR, Mane S, Lockey RF, Mohapatra SS. Respiratory syncytial virus infection activates STAT signaling in human epithelial cells. Biochem Biophys Res Commun. 2003;306(2):616–22. doi: 10.1016/s0006-291x(03)01008-8. [DOI] [PubMed] [Google Scholar]
- Lamb RA, Kolakofsky D. Paramyxoviridae: the viruses and their replication. In: Knipe DM, Howley PM, editors. Fields Virology. 4. Lippincott, Williams and Wilkins; Philadelphia: 2001. pp. 1305–1340. [Google Scholar]
- Li K, Foy E, Ferreon JC, Nakamura M, Ferreon AC, Ikeda M, Ray SC, Gale M, Jr, Lemon SM. Immune evasion by hepatitis C virus NS3/4A protease-mediated cleavage of the Toll-like receptor 3 adaptor protein TRIF. Proc Natl Acad Sci U S A. 2005;102(8):2992–7. doi: 10.1073/pnas.0408824102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y, Horvath F, Aligo JA, Wilson R, He B. The role of simian virus 5 V protein on viral RNA synthesis. Virology. 2005;338(2):270–80. doi: 10.1016/j.virol.2005.05.014. [DOI] [PubMed] [Google Scholar]
- Liu P, Jamaluddin M, Li K, Garofalo RP, Casola A, Brasier AR. Retinoic acid-inducible gene I mediates early antiviral response and Toll-like receptor 3 expression in respiratory syncytial virus-infected airway epithelial cells. J Virol. 2007;81(3):1401–11. doi: 10.1128/JVI.01740-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo MS, Brazas RM, Holtzman MJ. Respiratory syncytial virus nonstructural proteins NS1 and NS2 mediate inhibition of Stat2 expression and alpha/beta interferon responsiveness. J Virol. 2005;79(14):9315–9. doi: 10.1128/JVI.79.14.9315-9319.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy BR, Sotnikov AV, Lawrence LA, Banks SM, Prince GA. Enhanced pulmonary histopathology is observed in cotton rats immunized with formalin-inactivated respiratory syncytial virus (RSV) or purified F glycoprotein and challenged with RSV 3–6 months after immunization. Vaccine. 1990;8(5):497–502. doi: 10.1016/0264-410x(90)90253-i. [DOI] [PubMed] [Google Scholar]
- Parisien JP, Lau JF, Horvath CM. STAT2 acts as a host range determinant for species-specific paramyxovirus interferon antagonism and simian virus 5 replication. J Virol. 2002;76(13):6435–41. doi: 10.1128/JVI.76.13.6435-6441.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poole E, He B, Lamb RA, Randall RE, Goodbourn S. The V proteins of simian virus 5 and other paramyxoviruses inhibit induction of interferon-beta. Virology. 2002;303(1):33–46. doi: 10.1006/viro.2002.1737. [DOI] [PubMed] [Google Scholar]
- Ramaswamy M, Shi L, Monick MM, Hunninghake GW, Look DC. Specific inhibition of type I interferon signal transduction by respiratory syncytial virus. Am J Respir Cell Mol Biol. 2004;30(6):893–900. doi: 10.1165/rcmb.2003-0410OC. [DOI] [PubMed] [Google Scholar]
- Ramaswamy M, Shi L, Varga SM, Barik S, Behlke MA, Look DC. Respiratory syncytial virus nonstructural protein 2 specifically inhibits type I interferon signal transduction. Virology. 2005 doi: 10.1016/j.virol.2005.09.009. [DOI] [PubMed] [Google Scholar]
- Ramaswamy M, Shi L, Varga SM, Barik S, Behlke MA, Look DC. Respiratory syncytial virus nonstructural protein 2 specifically inhibits type I interferon signal transduction. Virology. 2006;344(2):328–39. doi: 10.1016/j.virol.2005.09.009. [DOI] [PubMed] [Google Scholar]
- Rodriguez JJ, Cruz CD, Horvath CM. Identification of the nuclear export signal and STAT-binding domains of the Nipah virus V protein reveals mechanisms underlying interferon evasion. J Virol. 2004;78(10):5358–67. doi: 10.1128/JVI.78.10.5358-5367.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlender J, Bossert B, Buchholz U, Conzelmann KK. Bovine respiratory syncytial virus nonstructural proteins NS1 and NS2 cooperatively antagonize alpha/beta interferon-induced antiviral response. J Virol. 2000;74(18):8234–42. doi: 10.1128/jvi.74.18.8234-8242.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spann KM, Tran KC, Chi B, Rabin RL, Collins PL. Suppression of the induction of alpha, beta, and lambda interferons by the NS1 and NS2 proteins of human respiratory syncytial virus in human epithelial cells and macrophages [corrected] J Virol. 2004;78(8):4363–9. doi: 10.1128/JVI.78.8.4363-4369.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spann KM, Tran KC, Collins PL. Effects of nonstructural proteins NS1 and NS2 of human respiratory syncytial virus on interferon regulatory factor 3, NF-kappaB, and proinflammatory cytokines. J Virol. 2005;79(9):5353–62. doi: 10.1128/JVI.79.9.5353-5362.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teng MN, Collins PL. Altered growth characteristics of recombinant respiratory syncytial viruses which do not produce NS2 protein. J Virol. 1999;73(1):466–73. doi: 10.1128/jvi.73.1.466-473.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teng MN, Whitehead SS, Bermingham A, St Claire M, Elkins WR, Murphy BR, Collins PL. Recombinant respiratory syncytial virus that does not express the NS1 or M2-2 protein is highly attenuated and immunogenic in chimpanzees. J Virol. 2000;74(19):9317–21. doi: 10.1128/jvi.74.19.9317-9321.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulane CM, Horvath CM. Paramyxoviruses SV5 and HPIV2 assemble STAT protein ubiquitin ligase complexes from cellular components. Virology. 2002;304(2):160–6. doi: 10.1006/viro.2002.1773. [DOI] [PubMed] [Google Scholar]
- Whitehead SS, Bukreyev A, Teng MN, Firestone CY, St Claire M, Elkins WR, Collins PL, Murphy BR. Recombinant respiratory syncytial virus bearing a deletion of either the NS2 or SH gene is attenuated in chimpanzees. J Virol. 1999;73(4):3438–42. doi: 10.1128/jvi.73.4.3438-3442.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright PF, Karron RA, Madhi SA, Treanor JJ, King JC, O’Shea A, Ikizler MR, Zhu Y, Collins PL, Cutland C, Randolph VB, Deatly AM, Hackell JG, Gruber WC, Murphy BR. The interferon antagonist NS2 protein of respiratory syncytial virus is an important virulence determinant for humans. J Infect Dis. 2006;193(4):573–81. doi: 10.1086/499600. [DOI] [PubMed] [Google Scholar]
- Young DF, Andrejeva L, Livingstone A, Goodbourn S, Lamb RA, Collins PL, Elliott RM, Randall RE. Virus replication in engineered human cells that do not respond to interferons. J Virol. 2003;77(3):2174–81. doi: 10.1128/JVI.77.3.2174-2181.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young DF, Didcock L, Goodbourn S, Randall RE. Paramyxoviridae use distinct virus-specific mechanisms to circumvent the interferon response. Virology. 2000;269(2):383–90. doi: 10.1006/viro.2000.0240. [DOI] [PubMed] [Google Scholar]