

Abstract
Neuroimaging studies indicate reduced volumes of certain gray matter regions in survivors of prematurity with periventricular leukomalacia (PVL). We hypothesized that subacute and/or chronic gray matter lesions are increased in incidence and severity in PVL cases compared to non-PVL cases at autopsy. Forty-one cases of premature infants were divided based on cerebral white matter histology: PVL (n = 17) with cerebral white matter gliosis and focal periventricular necrosis; diffuse white matter gliosis (DWMG) (n = 17) without necrosis; and “
Negative” group (n = 7) with no abnormalities. Neuronal loss was found almost exclusively in PVL, with significantly increased incidence and severity in the thalamus (38%), globus pallidus (33%), and cerebellar dentate nucleus (29%) compared to DWMG cases. The incidence of gliosis was significantly increased in PVL compared to DWMG cases in the deep gray nuclei (thalamus/basal ganglia; 50–60% of PVL cases), and basis pontis (100% of PVL cases). Thalamic and basal ganglionic lesions occur almost exclusively in infants with PVL. Gray matter lesions occur in a third or more of PVL cases suggesting that white matter injury generally does not occur in isolation, and that the term “perinatal panencephalopathy” may better describe the scope of the neuropathology.
Keywords: Basal ganglia, Brainstem, Perinatal panencephalopathy, Neurodevelopmental disability, Perinatal hypoxia–ischemia, Thalamus, White matter gliosis
Introduction
Periventricular leukomalacia (PVL), a major disorder of the immature cerebral white matter, has long been considered the underlying neuropathologic substrate of cerebral palsy in premature infants who survive into childhood [41]. The substrate of the cognitive impairments in these children, however, is less certain, given that cognition is typically attributed to gray matter (neuronal cell body), as opposed to white matter (oligodendrocyte), function, and the incidence of gray matter injury in PVL has historically been considered minimal [1, 4, 8]. Understanding the neuroanatomic basis of impaired cognition is of increasing importance in the care of premature infants: currently, in extremely premature infants (born at <1,500 g), the most common neurological disabilities involve cognition and learning, affecting 20–50% of such infants, compared to cerebral palsy which affects 10% [39, 42]. Attempts have been made to correlate neuropathologic findings in autopsied brains with neurologic sequelae in survivors of premature birth, however, much of this work has focused on the role of white matter injury and hemorrhages and not gray matter injury [13]. In the seminal paper of the neuropathology of PVL in 1969, Banker and Larroche reported only “mild neuronal injury” in the cerebral cortex, hippocampus, subiculum, basis pontis, and cerebellar dentate nucleus and Purkinje cells in the majority of PVL brains [4]. Subsequent neuropathologic studies reported only rare “anoxic neuronal injury” in association with PVL [1, 8], or downplayed the importance of observed gray matter lesions and did not emphasize their potential importance to neurologic sequelae in survivors. Thus, the conventional teaching has been that cerebral white matter is especially vulnerable to hypoxia-ischemia in the premature brain, with relative sparing of the gray matter, and that gray matter injury dominates only in older infants, children, and adults [23, 24], even though recent evidence shows that neuronal/axonal injury is common in the perinatal brain [5, 25]. This teaching is increasingly challenged by modern quantitative volumetric MRI studies of premature infants who exhibit reduced volumes of the cerebral cortex [35], thalamus [17, 18], basal ganglia [17, 18], and hippocampus [19, 34]. The anatomic substrate for the gray matter volumetric deficits associated with the apparent white matter disease remains unknown.
In the following study of the neuropathology of premature infants autopsied at this institution in the modern era of neonatal intensive care, we aimed to determine: (1) whether gray matter abnormalities in premature infants are more common in the presence of PVL than in the absence of this lesion; and (2) whether the constellation of any gray matter abnormalities provides insight into the basis of the cognitive impairments in living premature infants. In this study, we stratified the cases according to three patterns of cerebral white matter histology: (1) PVL (n = 17), defined as diffuse cerebral white matter gliosis combined with focal (macro- and/or microscopic) periventricular necrosis and; (2) diffuse white matter gliosis (DWMG) (n = 17), defined as diffuse gliosis without focal necrosis; and (3) “Negative” (n = 7), without diffuse gliosis and focal necrosis. While the pathogenesis of the white matter gliosis in the DWMG group is unknown, we considered it in a separate category from PVL (gliosis combined with focal necrosis) because the cerebral white matter pathology is distinct, without macro- and/or microscopic periventricular cysts. In the following study, we analyzed the three groups independently of each other. We hypothesized that PVL cases have a significantly greater incidence and degree of gray matter injury than non-PVL, i.e., DWMG and Negative cases; that this injury involves structures critical for cognition and learning, i.e., deep gray nuclei, cerebral cortex, and hippocampus; and that the pattern of gray matter injury mimics the pattern of volume reduction in the deep gray nuclei and cerebral cortex detected by neuroimaging studies in long-term survivors.
Materials and methods
Case selection criteria
The neuropathology of all premature infants (<37 gestational weeks at birth) autopsied between 1997–1999 at Children’s Hospital Boston was retrospectively reviewed. This time-frame was selected because it represents a modern era of intensive care management of premature infants in a Level 3 neonatal intensive care nursery. It also represents a period at our hospital when the brain and spinal cord were extensively sampled in a relatively standardized fashion, whether or not macroscopic lesions were apparent, thereby permitting a systematic neuropathologic survey in a large dataset. Parental authorization of the use of autopsy human tissue for research was given in each case.
Microscopic slide review
A median of 15 (range 9–20) hematoxylin–eosin (H&E) or H&E/Luxol-fast-blue stained sections was examined from each case. These sections included cerebral cortex from all lobes, thalamus (at the level of lateral geniculate nucleus and including the dorsomedial and lateral posterior nuclei), hypothalamus, caudate, putamen, globus pallidus, hippocampus (level of lateral geniculate nucleus), amygdala, cerebellar dentate nucleus, cerebellar cortex (including Purkinje cells and granule cell layer), midbrain, pons, and medulla.
We scored the density of neuronal necrosis, neuronal loss, and gliosis in the most severely affected high power fields of each of the gray matter sites. Two of three neuropathologists (CRP, RDF, HCK) reviewed the slides together at any one time, with a three-way consensus achieved on difficult cases. The brainstem tegmentum was graded separately from the basis pontis and inferior olive in recognition of their susceptibility to injury in the perinatal period [23]. We defined neuronal necrosis as hypereosinophilic neurons with pyknotic nuclei, or, in cases with neuronal immaturity and scant cytoplasm, karyorrhexis [2]. We used the following scale: 0, no necrosis; 1, a few scattered necrotic neurons in a high-powered field (hpf; 400×); 2, isolated clusters of necrotic neurons/hpf; and 3, larger, confluent areas of necrotic neurons/hpf. We scored neuronal loss as: 0, no neuronal loss/hpf; 1, mild, scattered neuronal dropout/hpf; 2, moderate, focal areas of neuronal dropout/hpf; and 3, severe, confluent areas of neuronal dropout/hpf. We interpreted neuronal necrosis as an acute or agonal change, occurring within 24–48 h of death [2], while neuronal loss and gliosis were interpreted as markers of subacute or chronic injury, indicative of insult occurring 3–5 days or more prior to death [2]. We combined cases for analysis that had grades 2/3 and 3/3 of neuronal loss because these grades are unequivocally recognized by standard microscopic examination, and therefore represent, in our opinion, a substantial degree of injury. We scored gliosis as: 0, no reactive astrocytes/hpf; 1, 1–10 reactive astrocytes/hpf; 2, 11–20 reactive astrocytes/hpf; and 3, >20 reactive astrocytes/hpf. Reactive astrocytes were defined as stellate configured cells with abundant (“hypertrophic”) eosinophilic cytoplasm and an enlarged, often eccentrically placed nucleus with delicate chromatin. The cytoplasm of these cells was immunopositive for glial fibrillary acidic protein (GFAP), the well-established astrocytic marker. In scoring neuronal necrosis, neuronal loss and gliosis, we evaluated the entire available region of each gray matter structure in the section, and assessed the most severely injured region, which was virtually always representative of all fields.
We assessed white matter in the cerebral lobes, corpus callosum, posterior limb of the internal capsule, and cerebellum for PVL and diffuse white matter gliosis (DWMG). PVL is defined by the combined presence of: (1) focal necrosis in the periventricular region; and (2) diffuse reactive gliosis in the surrounding white matter [24]. DWMG is defined by the presence of gliosis in the cerebral white matter unaccompanied by periventricular foci of necrosis [24]. Both PVL and DWMG are characterized by reactive astrocytes throughout the white matter, and are thus associated with a pattern of “diffuse” white matter injury. The density of white matter gliosis was scored according to the same scale used for gray matter gliosis (grades 0–3).
Glial fibrillary acidic protein (GFAP) immunohistochemistry
Four-micron thick formalin-fixed paraffin-embedded sections of frontal cortex could be cut from 27 of the 41 cases and were immunostained with mouse anti-GFAP antibody (1:500, #SM1-22R, Covance, Berkeley, CA). Negative controls were performed without primary antibody. Scoring of GFAP stained sections was performed by counting positive cells/hpf, in the most intensely immunopositive region of frontal cortex after a survey of all fields. Reactive astrocytes were defined as those cells with substantial cytoplasmic GFAP staining around a nucleus. Other non-reactive cortical astrocytes with limited cytoplasmic staining had GFAP positive processes that were generally perpendicular to the glial limitans were counted separately. The grading system was: 0, no staining; 1, 1–10 cells/hpf; 2, 11–20 cells/hpf; and 3 > 20 cells/hpf. Two observers (CRP, HCK) scored each case without knowledge of the white matter group.
O4 and GFAP double-labeling immunofluorescence
Myelination gliosis is commonly encountered in the newborn brain and must be discerned from reactive gliosis. So-called myelination glia are oligodendrocyte precursors that form during myelination; these cells have large nuclei with chromatin intermediate in density between oligodendrocytes and astrocytes and large slightly basophilic cell bodies so they can potentially be mistaken for reactive astrocytes [36]. To help discern reactive gliosis from myelination gliosis fresh tissue that was immediately fixed in 4% paraformaldehyde and sectioned at 40–50 μm was available from 4 PVL, 1 DWMG and 4 of the Negative cases. Double labeling was performed sequentially beginning with the mouse anti-O4 monoclonal antibody (1:750; gift from Dr. Steven Pfeiffer) to detect developing oligodendrocytes and followed with rabbit anti-GFAP antibodies (1:200, Z0334, Dako) to detect astrocytic differentiation. Relevant secondary antibodies conjugated with FITC or Texas Red were used and sections were visualized with Nikon Eclipse E800 microscope (Nikon, Melville, NY) outfitted with Spot image capture software (Diagnostics Instruments Incorporated, Sterling Heights, MI).
Statistical analysis
The 41 cases were divided into three groups according to cerebral white matter histology: (1) a PVL group; (2) a DWMG group; and (3) a “Negative” white matter group with no diffuse gliosis or focal periventricular necrosis in the cerebral white matter [23, 24]. Demographic characteristics were compared between the PVL, DWMG and Negative groups using Wilcoxon rank sum tests for continuous variables and χ2 tests for categorical variables (Table 1). The number of gray matter sites involved with an injury, i.e., acute neuronal necrosis, neuronal loss, and gliosis, was counted for each case in each group, and plotted relative to postconceptional age (i.e., gestational age plus postnatal age, PCA; in weeks) to graphically depict the total number of gray matter sites that were injured in each case. To test the hypothesis that the incidence and severity (grade 2–3/3) of neuronal necrosis, neuronal loss, and gliosis in all gray matter sites analyzed varies significantly among PVL, DWMG, and Negative cases, Fisher exact tests were used. To control for the potential impact of age on these analyses, analysis of covariance of diagnosis on lesion severity were performed, controlling for PCA. Nonsignificant interaction effects between diagnosis and age were subsequently eliminated from the models. In all analyses, P < 0.05 was considered significant.
Table 1.
Clinicopathologic variables of the three white matter study groups for comparison of gray matter injuries
| Mean ± SD; median, (range) or percent | P value | ||||
|---|---|---|---|---|---|
| PVL group n = 17 | DWMG group n = 17 | Negative group n = 7 | Three-way | PVL versus DWMG | |
| Demographics | |||||
| Gestational age (weeks) | 32.8 ± 3.1; 34, (26–36) | 31.6 ± 3.8; 33, (24–36) | 26.5 ± 2.3; 27, (23–30) | 0.003 | 0.282 |
| Postnatal age (weeks) | 3.7 ± 4.1; 2.3, (0.1–15) | 3.4 ± 4.0; 1.2, (0.1–12.0) | 0.8 ± 1.2; 0.1, (0.1–3.0) | 0.141 | |
| Postconceptional age (weeks) | 36.5 ± 5.4; 35.5, (26–52) | 34.8 ± 4.5; 35.0, (28.4–48.0) | 27.3 ± 2.8; 28.3, (23.1–30.3) | 0.001 | 0.293 |
| Length of ICU stay (days) | 13.8 ± 15.9; 9.5, (0–56) | 23.8 ± 34.6; 5.0, (1–96) | 5.0 ± 8.9; 1.5, (0.04–23.0) | 0.419 | |
| Race: Caucasian | 8/17; 47% | 9/17; 53% | 5/7; 71% | 0.801 | |
| African-American | 2/17; 12% | 2/17; 12% | 1/7; 14% | ||
| Unknown | 7/17; 41% | 6/17; 35% | 1/7; 14% | ||
| Percent male | 9/17, 53% | 7/17, 41% | 4/7, 57% | 0.702 | |
| Postmortem interval (h) | 16.0 ± 7.7; 18, (2–30) | 23.4 ± 19.6; 17, (2–72) | 36.3 ± 42.4; 22, (16–132) | 0.364 | |
| Birth weight (g) | 1576 ± 717; 1380, (780–2700) | 1832 ± 1048; 2100, (610–3800) | 925 ± 408; 930, (440–1400) | 0.232 | |
| Brain weight (g) | 245.0 ± 81.1; 257.5, (110–340) | 255.0 ± 72.1; 245, (124–380) | 127.6 ± 40.7; 140.0, (63.8–178.0) | 0.003 | 0.905 |
| Body weight (g) | 2423.1 ± 1165.0; 2225, (760–4875) | 3276.2 ± 4706.0; 2290, (1150–21300) | 947.9 ± 359.2; 915, (440–1400) | 0.002 | 0.815 |
| Body length (cm) | 42.0 ± 7.8; 41.5, (26–56) | 43.2 ± 4.4; 44.0, (36–51) | 34.7 ± 4.0; 32.5, (30–41) | 0.007 | 0.589 |
| Twin or other multiple gestation | 3/17, 18% | 4/17, 24% | 2/7, 29% | 0.824 | |
| Multiple congenital anomaliesa | 4/17, 24% | 2/17, 12% | 3/7, 43% | 0.242 | |
| Cesarean section | 10/17, 59% | 12/17, 71% | 3/7, 43% | 0.436 | |
| Cardiorespiratory factors | |||||
| Apgar score at 1 min | 4.1 ± 2.4; 4, (1–8) | 4.7 ± 2.9; 5, (0–9) | 3.5 ± 2.2; 3, (1–7) | 0.691 | |
| Apgar score at 5 min | 5.2 ± 2.9; 6, (1–9) | 6.6 ± 2.9; 7, (0–10) | 5.0 ± 2.8; 5.5, (1–8) | 0.405 | |
| Acute respiratory distress syndrome | 7/17, 41% | 10/17, 59% | 5/7, 71% | 0.344 | |
| Mechanical ventilation | 13/17, 77% | 13/17, 77% | 7/7, 100% | 0.359 | |
| Length of mechanical ventilation (days) | 8.4 ± 11.6; 3.0, (0–38) | 3.5 ± 6.3; 1.0, (1–19) | 4.4 ± 8.2; 1.0, (0.04–23.0) | 0.383 | |
| Extracorpeal membrane oxygenation | 1/17%, 6% | 0/17%, 0% | 0/7, 0% | 0.485 | |
| Continuous positive airway pressure | 0/17, 0% | 3/17, 18% | 1/7, 14% | 0.202 | |
| Cardiopulmonary resuscitation any time during hospitalization | 5/17, 29% | 7/17, 41% | 5/7, 71% | 0.165 | |
| Congenital heart diseasea | 2/17, 12% | 2/17, 12% | 1/7, 14% | 0.983 | |
| Infectious/inflammatory factors | |||||
| Pneumoniaa | 4/17, 24% | 2/17, 12% | 0/7, 0% | 0.303 | |
| Maternal fever at delivery | 0/15, 0% | 2/9, 22% | 0/4, 0% | 0.103 | |
| Maternal history of urinary tract infection | 0/15, 0% | 0/10, 0% | 1/4, 25% | 0.039 | NA |
| Chorioamnionitis | 1/15, 7% | 2/10, 20% | 1/5, 20% | 0.562 | |
| Necrotizing enterocolitisa | 1/17, 6% | 4/17, 24% | 2/7, 29% | 0.415 | |
| Clinical diagnosis of sepsis | 8/17, 47% | 8/17, 47% | 3/7, 43% | 0.980 | |
If a three-way P value was significant (P < 0.05), a two-way P value between the PVL and DWMG groups was performed. NA, not applicable since the incidence was 0 in the PVL and DWMG groups
NS, not significant
a diagnosed at autopsy
Results
Clinical and autopsy data
Seventeen cases (41%) fulfilled the criteria for PVL, while 17 cases (41%) had DWMG, and there were 7 so-called Negative cases (17%).
Pregnancy, labor and delivery
The Negative group was significantly younger in terms of gestational age (GA) than the PVL and DWMG groups, but there was no significant difference in GA between the PVL and DWMG groups (Table 1). In terms of PCA, the PVL and DWMG groups were significantly older than the Negative group (Table 1). The Negative group had significantly lower brain weight and body weight, and shorter body length, but not birth weight, than the PVL and DWMG groups. The low somatic and brain measurements in the Negative group compared to the PVL and DWMG groups reflect the early gestational age at birth and younger postnatal age at death (Table 1). The incidence of various clinical variables, e.g., chorioamnionitis, maternal fever at delivery, history of Cesarean section, and congenital anomalies were not significantly different among the three groups. The mean Apgar scores were less than seven at 1 and 5 min in all three groups (Table 1).
Syndromes
Twenty-four percent of cases had a constellation of findings classified as a genetic/developmental syndrome, e.g., Treacher-Collins syndrome, Potter’s sequence, and osteogenesis imperfecta in three PVL cases; Down’s syndrome in a DWMG case; and Fryns syndrome, Beckwith-Wiedemann syndrome, and VACTERL association in three Negative cases. Excluding these cases from the analysis had no significant effect on the results for the different acquired lesions analyzed semi-quantitatively, i.e neuronal loss and gliosis (data not shown), and thus, their data were combined with that of the non-syndromic cases in the complete analysis reported below.
Postnatal period
Although there were no statistically significant differences in postnatal age (PNA) among the three groups, the median PNA was only 0.1 week in the Negative group in contrast to 2.3 weeks in the PVL group and 1.2 weeks in the DWMG group (Table 1). The younger PNA and significantly different PCA and GA lead us to limit our gray matter comparisons to those between the PVL and DWMG groups, although the data from the Negative group is reported in all of the tables for completeness. Cardiorespiratory disorders were common in all three groups, with no significant differences (Table 1). Acute respiratory distress syndrome was noted in 41% of PVL cases, 59% of DWMG cases, and 71% of Negative cases. Seventy-seven percent of PVL and DWMG cases, and 100% of the Negative cases required ventilation (Table 1). The duration of ventilation and incidence of cardiopulmonary resuscitation was not significantly different among the three groups. Infectious and inflammatory disorders e.g., pneumonia, necrotizing enterocolitis, and sepsis, occurred in all groups, and did not differ significantly among the groups (Table 1).
White matter findings
Eighty-five percent of all cases studied had diffuse gliosis in the cerebellum, and 82% in the cerebral hemispheres. Forty-one percent of the cases had PVL, and 41% had DWMG. Macroscopically evident periventricular cysts (<5 mm in diameter) were noted in one PVL case, while chalky-white necrotic foci (2–3 mm) were visible in two cases. In all of the other PVL cases (82%), necrotic foci were only detected microscopically, and were <1 mm in diameter. Necrotic foci were typically found within a few millimeters from the ventricles. All cerebral lobes demonstrated a similar incidence of PVL (23–28%), except for the temporal lobe (12%). DWMG particularly involved the internal capsule (67%) and corpus callosum (64%) (Table 2). Necrotic foci were also identified in nonperiventricular regions, notably the internal capsule (19%) and corpus callosum (9%) (Table 2). The incidence of PVL increased with age, but only significantly so in the frontal lobe (Table 2). In contrast, the incidence of DWMG significantly increased in almost all sites with increasing gestational and postnatal ages (Table 2). By 37+ weeks, DWMG was present in 100% of the cases in the fronto-parieto-temporal lobes, 90% of the cases in the cerebellum, and 80–86% of the cases in the internal capsule and corpus callosum (Table 2). The degree of severity of DWMG significantly increased with both GA and PNA in all cases in the frontal, parietal, temporal, occipital, and cerebellar white matter (P < 0.04; data not shown). Moreover, the coefficients in the regression models for GA and PNA were similar, indicating that the degree of gliosis increased the same amount for each extra week of gestation and for each extra week of postnatal life (data not shown). In effect, the degree of gliosis increased constantly with PCA (gestational age plus postnatal age).
Table 2.
Distribution of white matter lesions in 41 autopsied premature infants by anatomic site and postconceptional age (weeks)
| White matter site | Overall incidence | Incidence by postconceptional age (weeks) | P value Logistic Regression of incidence and postconceptional weeks | ||
|---|---|---|---|---|---|
| 23–29 | 30–36 | 37+ | |||
| Frontal lobe | |||||
| PVL | 11/40 (28%) | 0/8 (0%) | 6/22 (27%) | 5/10 (50%) | 0.025 |
| DWMG | 31/40 (78%) | 2/8 (25%) | 19/22 (86%) | 10/10 (100%) | 0.004 |
| Temporal lobe | |||||
| PVL | 4/33 (12%) | 0/6 (0%) | 3/22 (14%) | 1/5 (20%) | NS |
| DWMG | 22/33 (67%) | 1/6 (17%) | 16/22 (73%) | 5/5 (100%) | 0.006 |
| Parietal lobe | |||||
| PVL | 7/28 (25%) | 0/6 (0%) | 6/16 (38%) | 1/6 (17%) | NS |
| DWMG | 20/28 (71%) | 1/6 (17%) | 13/16 (82%) | 6/6 (100%) | 0.010 |
| Occipital lobe | |||||
| PVL | 8/35 (23%) | 1/5 (20%) | 5/23 (22%) | 2/7 (29%) | NS |
| DWMG | 28/35 (80%) | 3/5 (60%) | 19/23 (83%) | 6/7 (86%) | NS |
| Corpus callosum | |||||
| PVL | 2/22 (9%) | 0/3 (0%) | 1/14 (7%) | 1/5 (20%) | NS |
| DWMG | 14/22 (64%) | 0/3 (0%) | 10/14 (71%) | 4/5 (80%) | 0.049 |
| Internal capsule | |||||
| PVL | 5/27 (19%) | 0/2 (0%) | 3/18 (17%) | 2/7 (29%) | NS |
| DWMG | 18/27 (67%) | 0/2 (0%) | 12/18 (67%) | 6/7 (86%) | NS |
| Cerebellum | |||||
| PVL | 3/40 (8%) | 0/8 (0%) | 2/22 (9%) | 1/10 (10%) | NS |
| DWMG | 34/40 (85%) | 4/8 (50%) | 21/22 (95%) | 9/10 (90%) | 0.012 |
P values denote significant differences in the incidence of DWMG with postconceptional age.
NS, not significant; PVL, periventricular leukomalacia; DWMG, diffuse white matter gliosis in the cerebral and cerebellar hemisphere
To exclude the possibility that myelination glia were scored along with reactive astrocytes in these infants immunofluoresence staining was performed to co-localize O4 a marker of the developing oligodendrocyte, and GFAP, an astrocytic marker. No co-localization of these proteins was identified in of the three groups of cases studied (Fig. 1).
Fig. 1.
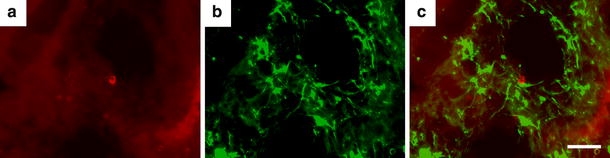
Immunoflourescence images of parieto-occipital white matter from a PVL case at 39 postconceptional weeks. O4 labeling (a) is shown in red and GFAP labeling (b) is shown in green. Merged images (c) show no co-localization of O4 and GFAP suggesting two distinct cell populations, i.e. GFAP-positive astrocytes and O4- positive oligodendrocyte precursors, are present. The scale bar represents 50 μm
Gray matter lesions associated with PVL in the premature infant
Acute neuronal necrosis
Acute neuronal necrosis, which is considered a marker of terminal/agonal injury, was common, and occurred diffusely across gray matter regions in all three groups. Sixty-six percent of PVL, 59% of DWMG, and 43% of Negative cases had two or more gray matter sites with acute neuronal necrosis (data not shown). Significant differences in the incidence of acute neuronal necrosis between the three groups were noted only in the cerebellar cortex (53%, PVL; 13%, DWMG; 0%, Negative; P = 0.008) and frontal cortex (56%, PVL; 41%, DWMG; 0%, Negative; P = 0.039). The incidence of neuronal necrosis was not significantly different at any gray matter site when adjusted for GA and PNA (data not shown).
Neuronal loss and gliosis
Neuronal loss and gliosis, considered markers of subacute and chronic injury, were more prevalent and of greater severity in PVL cases compared to non-PVL cases (DWMG and Negative groups) (Figs. 2 and 3, Tables 3 and 4). PVL cases showed more damage to the deep nuclear structures than was encountered in non-PVL cases. In PVL cases, the thalamus and globus pallidus had significantly higher incidences of neuronal loss (38 and 33%, respectively) and more severe neuronal loss (38 and 33%, respectively) than did the DWMG and Negative groups (both, 0%) (Fig. 2, Table 3). The incidence of gliosis was also significantly higher in the thalamus (56%), caudate (60%), putamen (50%) and globus pallidus (60%) in PVL than in DWMG (12–47%) and Negative cases (0–14%). The cerebellar dentate nucleus showed a significantly higher incidence of neuronal loss in PVL (29%) compared to the DWMG (6%) and Negative (14%) groups. PVL cases (29%) had significantly more severe neuronal loss in the cerebellar dentate compared to the DWMG and Negative groups (both, 0%) (Table 3). Gliosis of the basis pontis was seen in 100% of PVL cases and only 79% of DWMG and 29% of Negative cases (P = 0.001; Table 4). The hippocampus also had substantial neuronal loss and gliosis (Tables 2 and 3). PVL cases showed relatively mild cerebral cortical neuronal loss, compared to other neuroanatomic sites, while the incidence of gliosis ranged from 20% (temporal cortex) to 31% (frontal cortex) (Tables 3 and 4). By contrast, the cerebral cortex in all lobes from DWMG and Negative cases was totally free of neuronal loss and was infrequently gliotic (all <10%) (Tables 3 and 4).
Fig. 2.
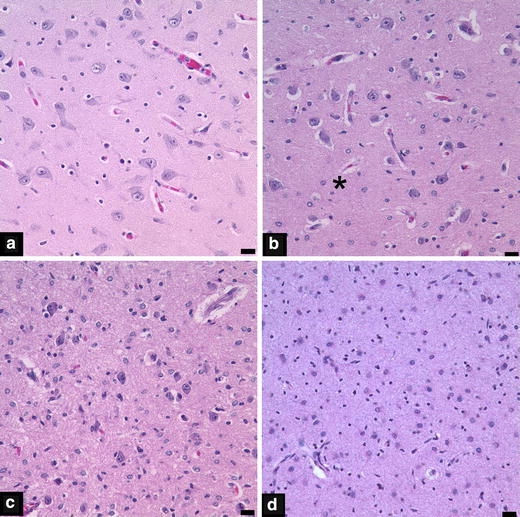
Photomicrographs from thalami illustrating neuronal loss scores of 0 (a), 1 (b), 2 (c) and 3 (d). The asterisk in panel b denotes a focal area of neuronal loss. The scale bar represents 20 μm
Fig. 3.
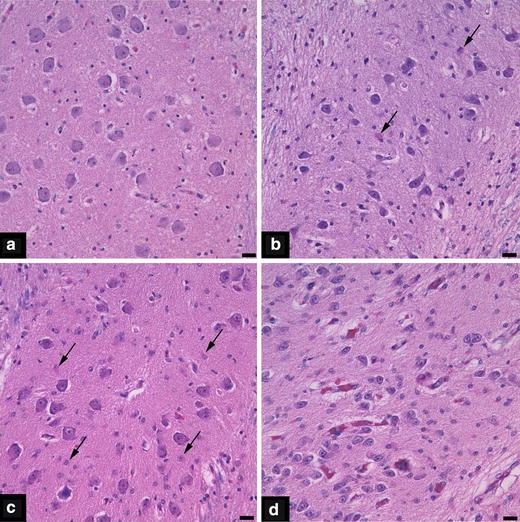
Photomicrographs from the inferior olivary nuclei depicting gliosis scores of 0 (a), 1 (b), 2 (c) and 3 (d). Arrows in panels b and c indicate some of the reactive astrocytes that are present. The scale bar represents 20 μm
Table 3.
Incidence and severity of neuronal loss in PVL, DWMG and Negative cases
| Overall incidence | Incidence of severity 2–3 | |||||||
|---|---|---|---|---|---|---|---|---|
| PVL | DWMG | Negative | P value | PVL | DWMG | Negative | P value | |
| Neuronal loss | ||||||||
| Cerebral cortex | ||||||||
| Frontal cortex | 13% (2/16) | 0% (0/17) | 0% (0/7) | 0.477 | 6% (1/16) | 0% (0/17) | 0% (0/7) | 0.575 |
| Temporal cortex | 0% (0/15) | 0% (0/13) | 0% (0/6) | 1.000 | 0% (0/15) | 0% (0/13) | 0% (0/6) | 1.000 |
| Parietal cortex | 8% (1/13) | 0% (0/11) | 0% (0/6) | 1.000 | 8% (1/13) | 0% (0/11) | 0% (0/6) | 1.000 |
| Occipital Cortex | 0% (0/15) | 0% (0/16) | 0% (0/4) | 1.000 | 0% (0/15) | 0% (0/16) | 0% (0/4) | 1.000 |
| Deep gray nuclei | ||||||||
| Thalamus | 38% (6/16) | 0% (0/17) | 0% (0/7) | 0.005 | 38% (6/16) | 0% (0/17) | 0% (0/7) | 0.005 |
| Hypothalamus | 20% (2/10) | 0% (0/10) | 0% (0/2) | 0.567 | 10% (1/10) | 0% (0/10) | 0% (0/2) | 1.000 |
| Caudate | 13% (2/15) | 0% (0/16) | 0% (0/7) | 0.329 | 13% (2/15) | 0% (0/16) | 0% (0/7) | 0.329 |
| Putamen | 13% (2/16) | 0% (0/17) | 0% (0/7) | 0.477 | 13% (2/16) | 0% (0/17) | 0% (0/7) | 0.477 |
| Globus pallidus | 33% (5/15) | 0% (0/15) | 0% (0/6) | 0.028 | 33% (5/15) | 0% (0/15) | 0% (0/6) | 0.028 |
| Cerebellum and relay nuclei | ||||||||
| Basis pontis | 21% (3/14) | 0% (0/14) | 0% (0/7) | 0.206 | 14% (2/14) | 0% (0/14) | 0% (0/7) | 0.341 |
| Inferior olive | 15% (2/13) | 8% (1/13) | 20% (1/5) | 0.807 | 8% (1/13) | 8% (1/13) | 20% (1/5) | 0.549 |
| Cerebellar cortex | 24% (4/17) | 6% (1/16) | 14% (1/7) | 0.449 | 24% (4/17) | 6% (1/16) | 14% (1/7) | 0.449 |
| Dentate | 29% (4/14) | 0% (0/15) | 0% (0/6) | 0.031 | 29% (4/14) | 0% (0/15) | 0% (0/6) | 0.031 |
| Limbic structures | ||||||||
| Hippocampus | 33% (5/13) | 0% (0/14) | 14% (1/7) | 0.055 | 33% (5/15) | 0% (0/14) | 14% (1/7) | 0.055 |
| Amygdala | 0% (0/6) | 0% (0/3) | 0% (0/2) | 1.000 | 0% (0/6) | 0% (0/3) | 0% (0/2) | 1.000 |
| Substantia inominata | 29% (2/7) | 0% (0/3) | 0% (0/1) | 1.000 | 29% (2/7) | 0% (0/3) | 0% (0/1) | 1.000 |
| Brainstem | 14% (2/14) | 0% (0/14) | 0% (0/5) | 0.629 | 14% (2/14) | 0% (0/14) | 0% (0/5) | 0.629 |
PVL, periventricular leukomalacia; DWMG, diffuse white matter gliosis in the cerebral and cerebellar hemisphere. P values denote differences in the incidence or severity of neuronal loss at these neuroanatomic sites between PVL and DWMG groups with postconceptional age
Table 4.
Incidence and severity of gliosis in PVL, DWMG and Negative cases
| Overall incidence | Incidence of severity 2–3 | |||||||
|---|---|---|---|---|---|---|---|---|
| PVL | DWMG | Negative | P value | PVL | DWMG | Negative | P value | |
| Gliosis | ||||||||
| Cerebral cortex | ||||||||
| Frontal cortex | 31% (5/16) | 6% (1/17) | 0% (0/7) | 0.102 | 13% (2/16) | 0% (0/17) | 0% (0/7) | 0.477 |
| Temporal cortex | 20% (3/15) | 8% (1/13) | 0% (0/6) | 0.495 | 0% (0/15) | 0% (0/13) | 0% (0/6) | 1.000 |
| Parietal cortex | 23% (3/13) | 9% (1/11) | 0% (0/6) | 0.499 | 8% (1/13) | 9% (1/11) | 0% (0/6) | 1.000 |
| Occipital cortex | 27% (4/15) | 0% (0/16) | 0% (0/4) | 0.054 | 13% (2/15) | 0% (0/16) | 0% (0/4) | 0.395 |
| Deep gray nuclei | ||||||||
| Thalamus | 56% (9/16) | 18% (3/17) | 14% (1/7) | 0.031 | 19% (3/16) | 0% (0/17) | 14% (1/7) | 0.161 |
| Hypothalamus | 40% (4/10) | 10% (1/10) | 50% (1/2) | 0.264 | 20% (2/10) | 0% (0/10) | 50% (1/2) | 0.130 |
| Caudate | 60% (9/15) | 19% (3/16) | 14% (1/7) | 0.028 | 13% (2/15) | 6% (1/16) | 14% (1/7) | 0.659 |
| Putamen | 50% (8/16) | 12% (2/17) | 14% (1/7) | 0.044 | 19% (3/16) | 0% (0/17) | 0% (0/7) | 0.130 |
| Globus Pallidus | 60% (9/15) | 47% (7/15) | 0% (0/6) | 0.040 | 20% (3/15) | 7% (1/15) | 0% (0/6) | 0.492 |
| Cerebellum and relay nuclei | ||||||||
| Basis pontis | 100% (14/14) | 79% (11/14) | 29% (2/7) | 0.001 | 36% (5/14) | 21% (3/14) | 14% (1/7) | 0.684 |
| Inferior olive | 92% (12/13) | 92% (12/13) | 80% (4/5) | 0.549 | 62% (8/13) | 54% (7/13) | 20% (1/5) | 0.400 |
| Cerebellar cortex | 29% (5/17) | 6% (1/16) | 14% (1/7) | 0.259 | 12% (2/17) | 6% (1/16) | 0% (0/7) | 1.000 |
| Dentate | 43% (6/14) | 13% (2/15) | 17% (1/6) | 0.177 | 21% (3/14) | 0% (0/15) | 0% (0/6) | 0.125 |
| Limbic structures | ||||||||
| Hippocampus | 47% (7/15) | 7% (1/14) | 29% (2/7) | 0.056 | 20% (3/15) | 0% (0/14) | 29% (2/7) | 0.143 |
| Amygdala | 50% (3/6) | 0% (0/3) | 0% (0/2) | 0.3273 | 0% (0/6) | 0% (0/3) | 0% (0/2) | 1.000 |
| Substantia inominata | 29% (2/7) | 0% (0/3) | 0% (0/1) | 1.000 | 0% (0/7) | 0% (0/3) | 0% (0/1) | 1.000 |
| Brainstem | 43% (6/14) | 20% (3/14) | 20% (1/20) | 0.518 | 7% (1/14) | 0% (0/14) | 0% (0/5) | 1.000 |
PVL, periventricular leukomalacia; DWMG, diffuse white matter gliosis in the cerebral and cerebellar hemisphere. P values denote differences in the incidence or severity of gliosis at these neuroanatomic sites between PVL and DWMG groups with postconceptional age
Glial fibrillary acidic protein immunohistochemistry performed on sections of frontal cortex from PVL (n = 10), DWMG (n = 13) and Negative cases (n = 4) showed astrocytes of two different general morphologies (Fig. 4). Greater numbers of reactive astrocytes with abundant GFAP positive cytoplasm tended to occur in the frontal cortex of PVL cases (mean score 0.75 ± 0.22), compared to DWMG cases (0.48 ± 0.19) or (0.59 ± 0.38); however, when controlled for PCA this trend was not statistically significant. Non-reactive astrocytes were found in similar frequency among PVL, DWMG and Negative cases, and no such trend, as that noted for reactive astrocytes appeared (data not shown).
Fig. 4.
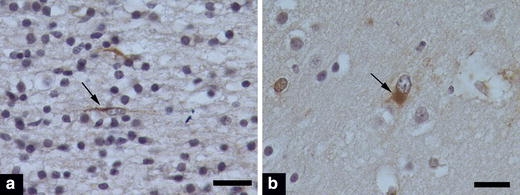
GFAP immunohistochemical staining of frontal cortex illustrating non-reactive astrocytes (a) with a linear GFAP-positive process that is perpendicular to glial limitans, which is at the right of this image (not depicted) and a reactive astrocyte (b), with abundant GFAP-positive cytoplasm, and an eccentrically placed, enlarged nucleus. The scale bar represents 20 μm
Discussion
This neuropathologic analysis shows clearly that gray matter abnormalities are more common in the presence of PVL than in its absence (summarized in Fig. 5). Moreover, and remarkably, neuronal loss and gliosis in the cerebral cortex and deep nuclear structures are essentially confined to those infants with PVL. Thus, not a single infant with DWMG exhibited neuronal loss in the cerebral cortex, hippocampus, and deep gray nuclei. Similarly, gliosis was very unusual in these areas in the infants with DWMG. Although the incidence and severity of lesions in this autopsy series may not be completely representative of the brain pathology in premature infants who survive beyond the perinatal period, the findings are nevertheless important to understand the neuroanatomic substrate and pathogenesis of the neurological sequelae in long-term survivors.
Fig. 5.
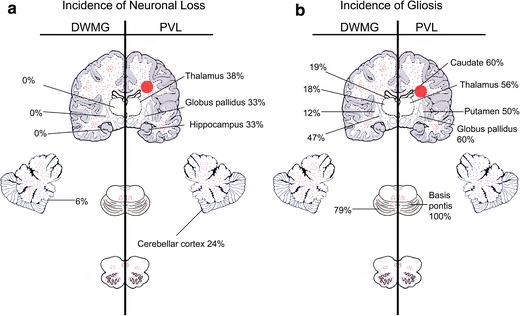
Summary diagram comparing gray matter sites with a significantly higher incidence (percentages) of neuronal loss (a) and gliosis (b) in PVL (right of panel) and DWMG (left of panel) cases. Gliosis of the cerebral and cerebellar white matter, basis pontis, brainstem tegmentum and inferior olives is depicted by small red dots, and focal, periventricular necrosis in the cerebral white matter (PVL) is denoted by a large red periventricular circle
The pathogenesis of the gray matter lesions in PVL is likely due to the same phenomena implicated in the white matter lesion. The pathogenesis of PVL likely involves cerebral ischemia-reperfusion in the respiratory compromised preterm infant, in combination with one or more infectious/inflammatory and other, yet to be defined, derangements [24]. Thus, the topographic patterns of gray and white matter damage in the premature brain likely reflect a complex interplay of the differential vulnerabilities of the regions to glutamate, free radical, and cytokine toxicity. These differential vulnerabilities appear to be based upon the maturational stage of neurons and oligodendrocytes [3, 23, 40], i.e., the targeted cell types in gray and white matter injury, respectively, and upon the developmental profiles of glutamate and cytokine receptors [9, 23] and antioxidant systems [9], as well as multiple related factors [23, 40]. The term “perinatal panencephalopathy” best describes, in our opinion, the combined gray and white matter injury delineated in this study that is typical of perinatal neuropathology of prematurity. We regard PVL as a major part of this disorder that should now be considered, we believe, in the context of total brain injury. Since the majority of patients in the PVL, DWMG, and Negative groups required mechanical ventilation of comparable durations, and showed substantial involvement by inflammatory/infectious processes, it is difficult to decipher the factors responsible for the substantial brain injury in the PVL group. It is very likely that there are specific clinical factors at work that we do not yet know, or were not analyzed in this study, e.g., lowest oxygen levels, alterations in acid-base status, and dysfunction in cerebral autoregulation, which are difficult to analyze in a meaningful way from complicated neonatal records. Thus, this study is hypothesis-generating for a prospective analysis of the key clinical factors involved in the pathogenesis of perinatal panencephalopathy.
The presence of isolated hypertrophic astrocytes in the cerebral white matter of premature infants, as reported in this series in the DWMG group, has been recognized for decades, but its significance remains unknown. Focal necrosis and diffuse hypertrophic astrocytes that are associated with “globules” and “acutely damaged glia” have been considered histological manifestations of the same disorder of immature cerebral white matter for which the term acquired perinatal telencephalopathy (PTL) has been coined [27]. Yet, hypoxic–ischemic white matter injury may follow a continuum of damage, from mild (gliosis [hypertrophic astrocytes] alone) to severe (periventricular necrosis combined with gliosis) [27]. Astrocytes, however, may also normally undergo hypertrophy in the late fetal and perinatal white matter as an obligatory developmental change, potentially due to the “physiological oxidative stress” of active myelin sheath synthesis, and thus may not be a marker of pathology at all [15]. These so-called myelination glia are immature oligodendrocytes that express markers such as O4, and are morphologically similar to GFAP positive reactive astrocytes. Coimmunofluoresence studies show no overlap in the expression of O4 and GFAP. The significant differences in age and survival encountered among the PVL, DWMG and Negative groups in this study precluded using the Negative group as a control representing “no white matter injury”. The Negative group consisted of infants who were born after significantly shorter gestational periods and who survived for significantly shorter time-periods postnatally than those in the PVL or DWMG groups. Thus, it is possible that the infants in the Negative group showed no white matter gliosis because the white matter is not vulnerable to injury at this early age, immature astrocytes are not capable of mounting a hypertrophic reaction to injury at this early time-point, and/or the patients did not survive long enough for astrocytic hypertrophy to develop. Further studies are needed to examine the significance of astrocytic hypertrophy in developmental pathology. The challenge is heightened by the unavoidable fact that live-born infants dying during the last half of gestation are not “normal”, but rather, typically die in intensive care units with multiple complications of prematurity that are known to adversely affect the brain.
This study suggests that neuronal loss and/or gliosis in the perinatal period in gray matter sites critical to cognition, memory, and learning, i.e., thalamus [7, 38], basal ganglia [33], hippocampus [7, 26], and cerebellum [21, 37], play a role in cognitive defects in long-term survivors of prematurity. The neuroanatomic structures involved with neuronal loss and/or gliosis correlates well with the neuroimaging data, which has shown volumetric deficits in the thalamus and basal ganglia [17, 18], and to a lesser degree, the hippocampus [19, 34], and cerebral cortex [35] in survivors of prematurity. This thalamic damage could be important in the pathogenesis of subsequent cognitive impairments. Of note, afferent thalamocortical axons fail to reach the cortex when the subplate neurons are ablated and abnormal cortical lamination results [10–12, 14, 22, 30]. Selective subplate neuronal loss occurs in hypoxic-ischemic injury in neonatal rats [31], underscoring the possibility of homologous injury in human premature infants [16] and the need for in depth studies of the subplate-thalamic-cortical unit in humans. Premature infants are also at high risk for cerebellar injury [6, 20, 28, 29, 32], given the mounting evidence that the cerebellum plays a role in cognition [21, 37], our finding of substantial damage in this structure and its brainstem relay nuclei suggests that it could contribute to cognitive defects in survivors. In addition, injury to the cerebellum, as well as the thalamus and basal ganglia (globus pallidus), may contribute to the motor deficits of prematurity. Traditionally, the spastic motor deficits, i.e., cerebral palsy, in preterm infants has been attributed to damage to axons in the necrotic foci in PVL that are coursing through the periventricular white matter from the motor cortex to the spinal cord [41]. Our data suggest that at least some of the common, less severe motor deficits are due to gray, as well as white, matter damage.
In conclusion, this study draws attention to the combination of white and gray matter injury in the brains of preterm infants dying in the perinatal period by the term “perinatal panencephalopathy”. Our findings suggest that future treatment strategies should target both white and gray matter damage to prevent the neurologic deficits in survivors of prematurity.
Acknowledgment
The authors would like to thank Ms. Lena Liu for her help with neurohistology and Mr. Richard A. Belliveau for his computer expertise and help with the figures.
Footnotes
Statement of financial support: CRP is supported by KO8 NS049090 from NINDS. This study was supported by grants from NINDS (PO1-NS38475) and NICHD (Children’s Hospital Mental Retardation Research Center) (P30-HD18655).
References
- 1.Armstrong D, Norman MG (1974) Periventricular leucomalacia in neonates. Complications and sequelae. Arch Dis Child 49:367–375 [DOI] [PMC free article] [PubMed]
- 2.Auer RN, Sutherland GR (2002) Hypoxia and related conditions. In: Graham DI, Lantos PL (eds) Greenfield’s neuropathology, 7th edn. Arnold, London, pp 234–280
- 3.Back SA, Luo NL, Borenstein NS, Volpe JJ, Kinney HC (2002) Arrested oligodendrocyte lineage progression during human cerebral white matter development: dissociation between the timing of progenitor differentiation and myelinogenesis. J Neuropathol Exp Neurol 61:197–211 [DOI] [PubMed]
- 4.Banker BQ, Larroche JC (1962) Periventricular leukomalacia of infancy. A form of neonatal anoxic encephalopathy. Arch Neurol 7:386–410 [DOI] [PubMed]
- 5.Bell JE, Becher JC, Wyatt B, Keeling JW, McIntosh N (2005) Brain damage and axonal injury in a Scottish cohort of neonatal deaths. Brain 128:1070–1081 [DOI] [PubMed]
- 6.Bodensteiner JB, Johnsen SD (2005) Cerebellar injury in the extremely premature infant: newly recognized but relatively common outcome. J Child Neurol 20:139–142 [DOI] [PubMed]
- 7.Constantinidis C, Procyk E (2004) The primate working memory networks. Cogn Affect Behav Neurosci 4:444–465 [DOI] [PMC free article] [PubMed]
- 8.DeReuck J, Chattha AS, Richardson Jr EP (1972) Pathogenesis and evolution of periventricular leukomalacia in infancy. Arch Neurol 27:229–236 [DOI] [PubMed]
- 9.Folkerth RD, Keefe RJ, Haynes RL, Trachtenberg FL, Volpe JJ, Kinney HC (2004) Interferon-gamma expression in periventricular leukomalacia in the human brain. Brain Pathol 14:265–274 [DOI] [PMC free article] [PubMed]
- 10.Ghosh A (1995) Subplate neurons and the patterning of thalamocortial connections. Ciba Found Symp 193:150–172; discussion 192–159 [DOI] [PubMed]
- 11.Ghosh A, Antonini A, McConnell SK, Shatz CJ (1990) Requirement for subplate neurons in the formation of thalamocortical connections. Nature 347:179–181 [DOI] [PubMed]
- 12.Ghosh A, Shatz CJ (1993) A role for subplate neurons in the patterning of connections from thalamus to neocortex. Development 117:1031–1047 [DOI] [PubMed]
- 13.Golden JA, Gilles FH, Rudelli R, Leviton A (1997) Frequency of neuropathological abnormalities in very low birth weight infants. J Neuropathol Exp Neurol 56:472–478 [DOI] [PubMed]
- 14.Hanganu IL, Kilb W, Luhmann HJ (2001) Spontaneous synaptic activity of subplate neurons in neonatal rat somatosensory cortex. Cereb Cortex 11:400–410 [DOI] [PubMed]
- 15.Haynes RL, Folkerth RF, Szweda LI, Volpe JJ, Kinney HC (2006) Lipid peroxidation during human cerebral myelination. J Neuropathol Exp Neurol 65:894–904 [DOI] [PubMed]
- 16.Iai M, Takashima S (1999) Thalamocortical development of parvalbumin neurons in normal and periventricular leukomalacia brains. Neuropediatrics 30:14–18 [DOI] [PubMed]
- 17.Inder TE, Warfield SK, Wang H, Huppi PS, Volpe JJ (2005) Abnormal cerebral structure is present at term in premature infants. Pediatrics 115:286–294 [DOI] [PubMed]
- 18.Inder TE, Wells SJ, Mogridge NB, Spencer C, Volpe JJ (2003) Defining the nature of the cerebral abnormalities in the premature infant: a qualitative magnetic resonance imaging study. J Pediatr 143:171–179 [DOI] [PubMed]
- 19.Isaacs EB, Lucas A, Chong WK, Wood SJ, Johnson CL, Marshall C, Vargha-Khadem F, Gadian DG (2000) Hippocampal volume and everyday memory in children of very low birth weight. Pediatr Res 47:713–720 [DOI] [PubMed]
- 20.Johnsen SD, Tarby TJ, Lewis KS, Bird R, Prenger E (2002) Cerebellar infarction: an unrecognized complication of very low birthweight. J Child Neurol 17:320–324 [DOI] [PubMed]
- 21.Kalashnikova LA, Zueva YV, Pugacheva OV, Korsakova NK (2005) Cognitive impairments in cerebellar infarcts. Neurosci Behav Physiol 35:773–779 [DOI] [PubMed]
- 22.Kanold PO, Kara P, Reid RC, Shatz CJ (2003) Role of subplate neurons in functional maturation of visual cortical columns. Science 301:521–525 [DOI] [PubMed]
- 23.Kinney HC, Armstrong D (2002) Perinatal neuropathology. In: Graham DI, Lantos PL (eds) Greenfield’s neuropathology, 7th edn. Arnold, London, pp 543–551
- 24.Kinney HC, Haynes RL, Folkerth RD (2004) White matter lesions in the perinatal period. In: Golden JA (ed) Developmental neuropathology, ISN Neuropathology, Basel, p 386
- 25.Kinney HC, Panigrahy A, Newburger JW, Jonas RA, Sleeper LA (2005) Hypoxic-ischemic brain injury in infants with congenital heart disease dying after cardiac surgery. Acta Neuropathol (Berl) 110:563–578 [DOI] [PubMed]
- 26.Leutgeb S, Leutgeb JK, Moser MB, Moser EI (2005) Place cells, spatial maps and the population code for memory. Curr Opin Neurobiol 15:738–746 [DOI] [PubMed]
- 27.Leviton A, Gilles FH (1984) Acquired perinatal leukoencephalopathy. Ann Neurol 16:1–8 [DOI] [PubMed]
- 28.Limperopoulos C, Soul JS, Gauvreau K, Huppi PS, Warfield SK, Bassan H, Robertson RL, Volpe JJ, du Plessis AJ (2005) Late gestation cerebellar growth is rapid and impeded by premature birth. Pediatrics 115:688–695 [DOI] [PubMed]
- 29.Limperopoulos C, Soul JS, Haidar H, Huppi PS, Bassan H, Warfield SK, Robertson RL, Moore M, Akins P, Volpe JJ, du Plessis AJ (2005) Impaired trophic interactions between the cerebellum and the cerebrum among preterm infants. Pediatrics 116:844–850 [DOI] [PubMed]
- 30.McQuillen PS, Ferriero DM (2005) Perinatal subplate neuron injury: implications for cortical development and plasticity. Brain Pathol 15:250–260 [DOI] [PMC free article] [PubMed]
- 31.McQuillen PS, Sheldon RA, Shatz CJ, Ferriero DM (2003) Selective vulnerability of subplate neurons after early neonatal hypoxia-ischemia. J Neurosci 23:3308–3315 [DOI] [PMC free article] [PubMed]
- 32.Messerschmidt A, Brugger PC, Boltshauser E, Zoder G, Sterniste W, Birnbacher R, Prayer D (2005) Disruption of cerebellar development: potential complication of extreme prematurity. AJNR 26:1659–1667 [PMC free article] [PubMed]
- 33.Monchi O, Petrides M, Strafella AP, Worsley KJ, Doyon J (2006) Functional role of the basal ganglia in the planning and execution of actions. Ann Neurol 59:257–264 [DOI] [PubMed]
- 34.Nosarti C, Al-Asady MH, Frangou S, Stewart AL, Rifkin L, Murray RM (2002) Adolescents who were born very preterm have decreased brain volumes. Brain 125:1616–1623 [DOI] [PubMed]
- 35.Peterson BS, Vohr B, Staib LH, Cannistraci CJ, Dolberg A, Schneider KC, Katz KH, Westerveld M, Sparrow S, Anderson AW, Duncan CC, Makuch RW, Gore JC, Ment LR (2000) Regional brain volume abnormalities and long-term cognitive outcome in preterm infants. JAMA 284:1939–1947 [DOI] [PubMed]
- 36.Friede RL (1989) Developmental neuropathology, 2nd edn. Springer, Berlin, p 22
- 37.Schmahmann JD, Caplan D (2006) Cognition, emotion and the cerebellum. Brain 129:290–292 [DOI] [PubMed]
- 38.Sur M, Rubenstein JL (2005) Patterning and plasticity of the cerebral cortex. Science 310:805–810 [DOI] [PubMed]
- 39.Volpe JJ (2003) Cerebral white matter injury of the premature infant-more common than you think. Pediatrics 112:176–180 [DOI] [PubMed]
- 40.Volpe JJ (2001) Neurobiology of periventricular leukomalacia in the premature infant. Pediatr Res 50:553–562 [DOI] [PubMed]
- 41.Volpe JJ (2001) Neurology of the newborn. Saunders, Philadelphia, pp 362–363
- 42.Woodward LJ, Edgin JO, Thompson D, Inder TE (2005) Object working memory deficits predicted by early brain injury and development in the preterm infant. Brain 128:2578–2587 [DOI] [PubMed]