

Abstract
Certain hexavalent chromium [Cr(VI)] compounds are known genotoxic respiratory carcinogens, which induce apoptosis as a predominant mode of cell death. Selection of cells that are resistant to apoptosis may be a factor in tumour progression. We developed sub-populations of telomerase-transfected human fibroblasts (BJ-hTERT) that survived a 99% clonogenically lethal exposure to Cr(VI) (B-5Cr). B-5Cr cells were markedly resistant to apoptosis induced by several agents and exhibited increased clonogenic survival, especially at apoptogenic doses. B-5Cr cells did not exhibit altered cellular uptake of Cr(VI) and retained a normal p53 response to Cr(VI) exposure. We conducted large-scale gene expression analysis at different time-points after a secondary genotoxic Cr(VI) insult in B-5Cr and BJ-hTERT cells using Affymetrix Genechip® human genome arrays. Cr(VI) exposure led to differential regulation of many genes, which affect a diverse set of cellular activities such as transcription, signal transduction, stress response, cell adhesion, DNA repair, apoptosis and cell cycle modulation. We compared Cr(VI)-induced altered gene expression in the B-5Cr cells to that in the parental cells and identified 223, 147 and 204 genes with at least a two-fold difference in expression at 4, 8 and 18 h after exposure, respectively. Cluster analysis by gene function revealed altered expression of genes involved in apoptosis, cell cycle regulation and DNA repair. Our data suggest an alteration in gene expression that may favor cell survival and/or incomplete DNA repair after genotoxic exposure. Selection of cells with altered expression of these genes may constitute the early stages of tumour progression.
Introduction
The cellular response to exposure to DNA damaging agents usually involves cell cycle arrest followed by a cellular fate that is determined by the extent of DNA damage [1]. When damage is irreparable, cells are removed from the proliferating population by undergoing either apoptosis or terminal growth arrest, presumably to avoid the propagation of damaged DNA. Thus, DNA damaging agents are typically apoptosis-inducing agents at levels that overwhelm intracellular DNA repair mechanisms [2]. Hexavalent chromium [Cr(VI)] is a potent DNA damaging agent that has been shown to cause apoptosis and prolonged growth arrest as the predominant cellular fates in response to exposure levels that induce some cell death [3-7]. However, at high exposure levels, a very small fraction of cells survive and regain replicative potential. These cells may have sustained extensive DNA damage yet somehow are able to escape death. It is unknown what factors allow this very small survivor population to maintain replicative potential. The phenotypic and potential genotypic differences in the surviving fraction of cells may convey a selective survival advantage, which may disrupt cell death/growth homeostasis and predispose these cells to neoplastic progression.
Certain inhaled forms of Cr(VI)-containing compounds are strongly associated with lung toxicity and an increased incidence of lung cancer [8, 9]. These compounds are recognised occupational human carcinogens and the generation of chromium waste by chromate industries comprises a potential environmental health risk [10-13]. Cr(VI) ions can enter cells through an anion transporter and there they are subject to metabolic reduction to reactive genotoxic products [14-16]. These products, as well as the oxidative stress generated by the reduction process may contribute to diverse genotoxic and cytotoxic effects. The structural DNA damage that results from Cr exposure is well documented and includes Cr-DNA ternary adducts, strand breaks, oxidised bases [17, 18], DNA-protein crosslinks, abasic sites and Cr-intrastrand crosslinks [19-25] (for review, see [26]). Structural damage may lead to functional DNA damage (reviewed in [6]), such as DNA and RNA polymerase arrest [27, 28], mutagenesis [29-31], and/or altered gene expression [32-34].
The main cellular targets for Cr(VI) toxicity are lung epithelial cells and fibroblasts exposed to moderate to high concentrations of Cr(VI) in the immediate microenvironment of inhaled particles [35, 36]. To study Cr(VI) genotoxicity over time, a population of relevant cells that maintain a consistent and normal response to genotoxic stress over subsequent population doublings is required. Normal diploid fibroblasts rapidly change their responses to genotoxicity as the population grows in culture and approaches senescence [37, 38], and transformation of fibroblasts with viral oncoproteins extend cell lifespan through mechanisms that alter cell cycle control and increase genomic instability [39, 40]. Therefore, we used human fibroblasts (BJ) immortalised by telomerase transfection (BJ-hTERT; Geron Corp., Menlo Park, CA) for these experiments. BJ-hTERT cells provide an excellent model for studying Cr(VI) toxicity because they are immortal but maintain a normal response to genotoxic stress [41, 42]. Moreover, we have found that the responses of these cells to Cr(VI) are indistinguishable from those of human lung fibroblasts.
We have previously shown that cell populations exposed to Cr(VI) exhibit a different spectrum of cellular fates, which include apoptosis, terminal growth arrest (TGA) or regaining replicative potential, depending on the extent of DNA damage [7]. As the dose of Cr(VI) increases, the extent of DNA damage increases and we observe a shift in cellular fate from clonogenic survival to terminal growth arrest and then to apoptosis. For genotoxic carcinogens, in order to induce neoplastic transformation, doses that also induce apoptosis are usually required. In the case of Cr(VI), exposure to levels of Cr compounds that are associated with lung cancer usually cause respiratory conditions associated with large amounts of cell death, such as perforation of the nasal septum or respiratory tract ulcerations (reviewed in [6]). At these concentrations, the vast majority of exposed cells are removed from the proliferating population, but a small number may survive. Regaining replicative potential appears to require either escape from or resistance to apoptosis or TGA.
Genotoxicity is often thought to be a prerequisite for neoplastic transformation. Mistakes made in the repair of genetic lesions, or cell cycling in the absence of DNA repair, may cause either an increase in the DNA mutational spectra or a decrease in genomic stability, which may lead to uncontrolled cellular proliferation. The cells that survive after exposure to apoptogenic levels of a DNA damaging agent are the precursor pool from which neoplastic variants will emerge. Survivors may have developed an extrinsic alteration in gene expression due to the Cr(VI)-induced DNA damage, or they may have been selected for because of a different intrinsic gene expression pattern that conveys a survival advantage. Furthermore, by way of this survival advantage, such a sub-population of cells may, in fact, be selected for by Cr(VI) exposure, thereby increasing the amount of cells with the resistant phenotype. If early tumour growth requires a net accumulation of cells due to alterations in the growth/death ratio, then selection of cells with resistance to apoptosis may facilitate the early steps of neoplastic progression. In this study we developed a sub-population of cells that survived a relatively high dose of Cr(VI) (less than 0.5% clonogenic survival). It is unclear whether these cells survived by chance or if they have a pre-existing selective advantage over the rest of the population. Therefore we set out to examine the survivor phenotype and any potential genotypic changes in the survivor sub-population. Phenotypic differences that would convey resistance to Cr(VI)-induced death may involve molecular components of the apoptotic or terminal growth arrest signalling pathways. Other phenotypic differences that may provide cells with an intrinsic resistance to apoptosis may involve sensing DNA damage, generation of and/or response to oxidative stress and the ability to maintain mitochondrial stability. These phenotypic differences may be the result of altered gene expression. Therefore we examined gene expression in survivor and control cells using a large-scale Affymetrix gene array. We were able to screen 12,625 genes and ESTs for expression changes after Cr(VI) exposure and between survivor and control cell populations. Gene expression differences in survivor cells may shed light on the roles that different genes play in genotoxic selective survival.
Materials and methods
Cell culture
BJ-hTERT fibroblasts (Geron Corp., Menlo Park, CA), and the BJ Cr(VI)-survivor sub-populations were maintained in Dulbecco’s minimal essential media (D-MEM)/Medium 199 (M199) (4:1) containing 10% fetal bovine serum (Hyclone Laboratories, Inc., Logan, UT), and 5 μg/ml gentamicin (Life Technologies, Gaithersburg, MD). Additionally, BJ-hTERT medium contained 10 μg/ml hygromycin B (Life Technologies) as a selection agent for the hTERT transgene. Cells were incubated in a 95% air/5% CO2 humidified atmosphere at 37 °C and the medium was replaced every 48 h.
Treatment of cells with chromium or hydrogen peroxide
Sodium chromate (Na2CrO4·4H2O) (J.T. Baker Chemical Company, Phillipsburg, NJ) was dissolved in deionised H2O and sterilised through a 0.2 μm filter before use. Cells were treated with a final concentration of 0-6, or 9 μM sodium chromate for 4, 8, 18 or 24 h in complete medium. H2O2 was diluted with deionised H2O and sterilised through a 0.2 μm filter before use. Cells were treated with 0.5 or 1 mM H2O2 for 24 h in complete medium prior to phosphatidylserine translocation analysis.
Phosphatidylserine translocation assay
After 24 h, the cells were rinsed with phosphate buffered saline (PBS), the medium was replaced and the cells were incubated for an additional 24 h before biochemical analysis. This assay was conducted as previously described [7, 43]. Briefly, cells were seeded at 105 cells/60 mm2 dish and incubated for 24 h prior to sodium chromate exposure. Following sodium chromate treatment, cells were gently harvested by trypsinization and combined with non-adherent cells from the culture medium. The cells were centrifuged at 600 × g for 5 min. Cell pellets were washed once in PBS and resuspended in 100 μl binding buffer (10 mM HEPES, pH 7.4, 150 mM NaCl, 5 mM KCl, 1 mM MgCl2, 1.8 mM CaCl2) containing 2 μl annexin(V)-FLUOS™ (Roche, Indianapolis, IN). Samples were incubated in the dark at room temperature for 15 min. An amount of 30 μl was loaded on a microscope slide and the percentage of annexin(V)-FLUOS stained cells was determined using a Olympus AX70 microscope (Olympus, Lake Success, NY) with a fluorescent filter set suitable for FLUOS analysis (excitation 460-490, emission 515).
Clonogenicity analysis
BJ-hTERT cells were seeded at 105 cells/60 mm dish and incubated for 24 h prior to Cr(VI) exposure. After the 24 h exposure, cells were collected by trypsinization, counted, and reseeded at 7 × 103 cells/60 mm2 dish in triplicate. The plates were incubated for 12-13 days, then rinsed with PBS and incubated with crystal violet stain (80% methanol, 2% formaldehyde, 2.5 g/l crystal violet) for 15-30 min at room temperature. The plates were thoroughly rinsed with dH2O and allowed to dry. Colonies containing greater than 20 cells were counted, the triplicates were averaged, and clonogenicity was determined as percent of control.
Cell growth curves
BJ-hTERT cells were seeded at 105 cells/60 mm dish and incubated for 24 h prior to Cr(VI) exposure. Replicate dishes were seeded for each dose tested and all of the replicates within the group received the same treatment. One replicate was taken every other day over the 17-day time-course and counted to determine total cell number at each dose and time. Cells were gently harvested with trypsin, centrifuged at 600 × g for 5 min, and the cell pellets were resuspended in 1 ml PBS. Total cell number was determined using a Coulter Mutisizer II cell counter (Coulter, Louton, UK).
Cr(VI) uptake analysis
An amount of 5 × 105 cells/sample were exposed to 0-90 μM Na2CrO4 spiked with Na2Cr51O4 for 3 h at 37 °C. The cells were centrifuged at 300 × g for 5 min at 4 °C. Cell pellets were washed twice in PBS and lysed in 500 μl lysis buffer (100 mM Tris-Cl (pH 8.0), 200 mM NaCl, 10 mM EDTA, 4% SDS). An amount of 100 μl of each sample was transferred to a 5 ml scintillation vial and combined with 1 ml Ecolite scintillation cocktail (ICN, Irvine, CA). DPM/105 cells was determined on a Beckman LS6500 scintillation counter (Beckman Instruments, Fullerton, CA).
Preparation of cell lysates for p53 analysis
Cells were seeded at 106 cells/75 cm flask and incubated for 24 h prior to 9 μM Na2CrO4 exposure. Two hours after treatment, cells were harvested by gentle cell scraping and combined with non-adherant cells from the culture medium. The cells were centrifuged at 300 × g for 5 min at 4 °C and were resuspended in 200 μl of ice-cold RIPA buffer (150 mM NaCl, 50 mM Tris-HCl (pH 7.4), 5 mM EDTA, 1% Igepal™ (Sigma), 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulfate) containing 0.1 mg/ml PMSF, 30 μl/ml aprotinin and 1 mM sodium orthovanidate. Cell suspensions were sheared through a 21 gauge hypodermic needle and the resulting lysates were incubated in RIPA plus the inhibitors for 20 min on ice, then centrifuged at 10,000 × g for 20 min at 4 °C. The resulting supernatants were stored at -20 °C until used for PAGE and Western blot analysis.
Protein electrophoresis and p53 Western blotting
Cell lysates were separated on a 10% SDS-polyacrylamide gel and transferred to a polyvinylidene difluoride (PVDF) membrane by electroblotting as previously described [43]. Blots were probed with a p53 Ab6 primary antibody (Calbiochem, Cambridge, MA), followed by a horseradish peroxidase-linked secondary antibody (Amersham Pharmacia Biotech, Piscataway, NJ). The secondary antibody was visualised by ECL Western blotting detection reagents (NEN Life Science Products Inc., Boston, MA), after exposure to X-ray film (Amersham Pharmacia).
Preparation of total RNA for Affymetrix Genechip® micro arrays
Total RNA preparation was done using the RNA-Bee™ RNA Isolation Kit (Tel-Test Inc., Friendswood, TX) as directed by the manufacturer.
Affymetrix Genechip® micro array analysis
Expression profiling
Six micrograms of total RNA was converted to cDNA using the SuperScript Choice System (Invitrogen) and a T7-(dT24) primer (GENESET Corp.). cDNA was purified by phenol-chloroform-isoamyl alcohol extraction. Clean cDNA was used for the in vitro synthesis of biotin-labelled cRNA using the BioArray RNA transcript labelling kit (ENZO). cRNA was cleaned using RNeasy mini kits (QIAGEN) and fragmented using a magnesium acetate buffer (200 mM tris-acetate, pH 8.2, 500 mM potassium acetate, 150 mM magnesium acetate). Ten micrograms of labelled cRNA was hybridised to Affymetrix Human_HG U95Av2 arrays (http://www.affymetrix.com/products/arrays/specific/hgu95.affx) for 16 h. The GeneChips® were washed and stained according to the manufacturer’s recommendations (Affymetrix). Each chip was scanned using a confocal laser scanner after an initial staining with streptavidin phycoerythrin, and after second antibody staining used to amplify the signal.
Data analysis
Data analysis was performed using Affymetrix Microarray Suite 5.0 software to generate an absolute analysis for each chip. Each chip was scaled globally to a target intensity value of 800 to allow for inter-array comparisons.
For the baseline study comparing treated versus untreated BJ-hTERT cells we ran a comparison analysis between both conditions in duplicate. A list of upregulated and downregulated genes with a greater than two-fold change was created using the “iterative comparison” analysis [44]. This is a very stringent method of analysis, as genes that survive the filtering are up or downregulated two-fold in all comparisons.
Absolute intensity values for each chip from the time course study were screened for probe saturation using the “Array Data Manipulation” program created by Tanya Teslovich at Children’s National Medical Center, Washington, DC. The saturation program identifies probes that have become saturated in the second scan, after probe signals are amplified using goat IgG. Probe saturation was common when the PMTs for Affymetrix scanners were calibrated to a higher value (10 times higher than the current settings). Please see http://www.affymetrix.com/support/technical/product_updates/scanner_gain_update.pdf for more information on scanner PMT settings. The program considers a probe saturated if its signal in the first scan (before antibody amplification) is greater than 1500 and the (scan 2/scan 1) ratio is less than a user-specified minimum value of 0.8. If the probe is saturated on any array in the experiment, then the ‘scan 1’ signal intensity is used for that gene across all chips. After the files have been de-saturated, they are loaded into GeneSpring (http://www.silicongenetics.com) for temporal clustering analysis and graphical views of gene ontology groups. Time-points 4, 8 and 18 h were normalised to the control, time-point 0, for each of the different cell types. Genes that were absent across all chips were eliminated through filtering. GeneSpring was also used to cluster genes by their ontology group.
Results
Chromium-resistant fibroblasts, derived from human foreskin fibroblasts immortalised by human telomerase gene transfection (BJ-hTERT), were used in comparative studies with parental cells. The chromium survivor sub-population (B-5Cr) was developed by collecting the fraction of cells that emerged in culture after being exposed to a 5 μM dose of Na2CrO4. The B-5Cr sub-population was compared to BJ-hTERT for differences in susceptibility to apoptosis induced by different toxic agents using the phosphatidylserine translocation assay (Fig. 1). Susceptibility to apoptosis in cells exposed to another Cr(VI) insult (0, 6 or 9 μMNa2CrO4 for 24 h) is shown in Fig. 1A. The B-5Cr sub-population was significantly resistant to Cr(VI)-induced apoptosis at both 6 and 9 μM Na2CrO4. Susceptibility to apoptosis in cells exposed to hydrogen peroxide (H2O2) (0, 0.5 or 1 mM H2O2 for 24 h) is shown in Fig. 1B. Again, the B-5Cr sub-population was significantly resistant to H2O2-induced apoptosis at both doses tested.
Fig. 1.
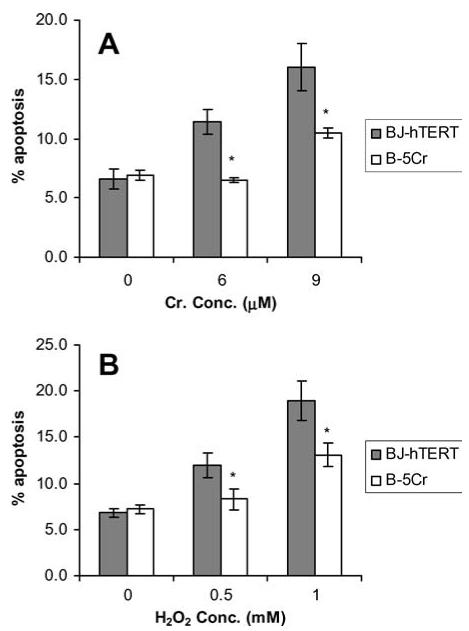
Apoptosis resistance in the B-5Cr survivor sub-population. B-5Cr cells were compared to parental BJ-hTERT cells for susceptibility to apoptosis. Cells received either (A) chronic (6-9 μM; 24 h) treatments of Na2CrO4, or (B) chronic (0.5-1 mM; 24 h) treatments of H2O2, and were analysed for phosphatidylserine translocation 24 h after treatment. The percentages of apoptotic cells represent the mean ± S.E. of at least three independent experiments. Comparisons were made between individual correlated samples compared to control, and p values were determined with a Student’s t-test and a normal distribution was verified with ANOVA and F-test. Asterisk (*) indicates a statistically significant decrease from control at p < 0.05.
The differential cumulative effects of Cr(VI) exposure on these cell populations was determined by comparing clonogenic survival and long-term (17 days) cell growth curves after Cr(VI) treatment. Clonogenicity is an indicator of long-term cell survival and replicative potential after toxic exposure. Clonogenicity was evaluated in B-5Cr compared to BJ-hTERT cells exposed to 0-6 μM Na2CrO4 for 24 h (Fig. 2A). A dose-dependent decrease in clonogenicity was shown in all of the cell populations as the Na2CrO4 concentration increased from 0 to 6 μM. The survivor B-5Cr population exhibited resistance to Cr-induced clonogenic lethality compared to the parental control, especially for concentrations of Cr(VI) that cause apoptosis (2-6 μM). This difference increased with Cr(VI) concentration and was significant at the highest dose tested.
Fig. 2.

Altered clonogenicity and cell growth potential in the B-5Cr survivor sub-population. (A) The cloning efficiency of survivor sub-population (B-5Cr) cells exposed to increasing concentrations of Na2CrO4 (1-6 μM, 24 h) was analysed and compared to BJ-hTERT. The number of colonies counted at the indicated concentrations of Na2CrO4 are expressed as a percentage of the 0 μM control for each group. Results are plotted on a semi-log graph and are the mean of at least three independent experiments. Comparisons were made between individual correlated samples compared to control, and p values were determined with a Student’s t-test and a normal distribution was verified with ANOVA and F-test. Asterisk (*) indicates a statistically significant increase from parental control at p < 0.05. (B) Growth curves were plotted for B-5Cr and BJ-hTERT cells after exposure to 5 μM Na2CrO4 for 24 h. Na2CrO4 was added to the dishes at day 0 and removed after 24 h. Total cell number was plotted over a 17-day period and indicates sample population cell loss, and sample population growth recovery throughout the time course for each cell population. Results are one representative experiment.
To determine differential cell growth potential, we examined cell growth over a 17-day period in BJ-hTERT and B-5Cr cells exposed to 5 μM Na2CrO4 for 24 h (Fig. 2B). In both cell populations, growth was suppressed over a 7-day period, but a fraction of surviving cells emerged after day 7 in the B-5Cr samples, while surviving cells did not emerge in the parental control sample until day 15.
We sought to determine if the difference in Cr(VI) susceptibility between these populations was caused by altered cellular capacity to take up Na2CrO4. Cr(VI) uptake was similar in both cell populations exposed to a wide range of Cr(VI) doses (Fig. 3).
Fig. 3.
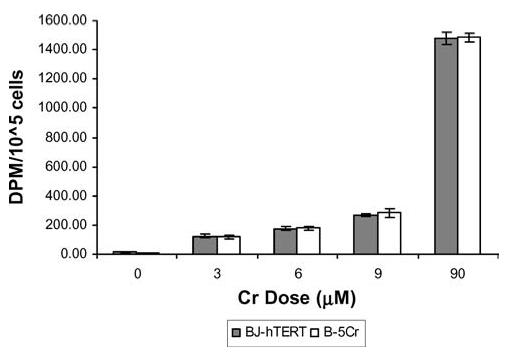
Resistance to Cr(VI)-induced apoptosis in survivor sub-populations is not due to altered Cr(VI) cellular uptake. BJ-hTERT and B-5Cr survivor sub-population cells were exposed to increasing concentrations of Na2CrO4 spiked with Na251CrO4 for 3 h. The samples were then lysed and analysed for intracellular 51Cr using a scintillation counter. Results are the mean ± S.E. of three independent experiments. Comparisons were made between individual correlated samples compared to control, and p-values were determined with a Student’s t-test and a normal distribution was verified with ANOVA and F-test.
We and others have shown that Cr-induced apoptosis proceeds in a p53-directed fashion [4, 5, 45]. The dysregulation/mutation of the p53 protein has been shown to be a key factor in cellular death resistance [46]. Therefore, we studied the ability of Cr(VI) to stabilise and thus, upregulate the expression of p53 in the survivor cell population. As shown in Fig. 4, there was no difference in p53 response to Cr(VI) exposure between the BJ-hTERT and B-5Cr cells.
Fig. 4.
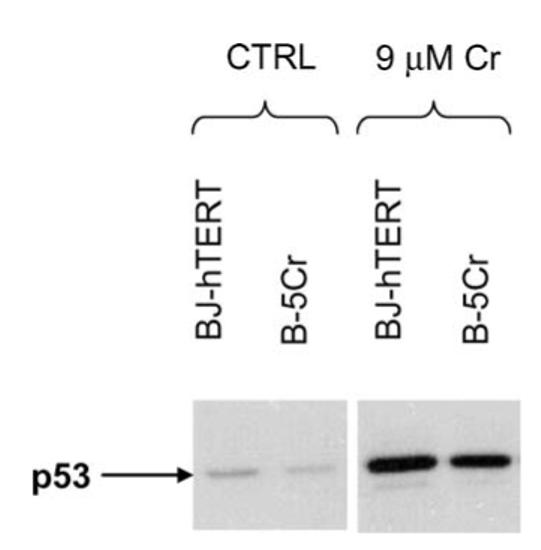
No change in p53 protein expression in the B-5Cr survivor subpopulation. p53 protein expression was determined in untreated (CTRL) cells or after a 24 h exposure to 9 μM Na2CrO4 (9 μM Cr) in BJ-hTERT and B-5Cr cells. Protein samples were isolated 2 h after treatment and analysed by Western blotting with a p53-specific monoclonal antibody. Blots are representative of three independent experiments.
Gene expression profiling after chromium exposure in BJ-hTERT
We used the Affymetrix human genome U95Av2 chip to profile the changes in gene expression in BJ-hTERT fibroblasts after exposure to Cr(VI). Total RNA was collected and purified from BJ-hTERT cells prior to and after exposure to 5 μM Na2CrO4 for 18 h. Pairwise comparative analysis between Cr(VI)-treated samples and the untreated controls was performed in duplicate using the Affymetrix Microarray Suite Software. Genes were identified that were differentially expressed in the treated cells (≥2.0-fold change in expression at p < 0.05) compared to the untreated control. A total of 1,001 of the 12,625 (8.7%) genes and ESTs met these criteria for differential expression. Of the 1,001 genes, 599 (4.7%) were upregulated, and 502 (4.0%) were downregulated.
To identify the coordinate regulation of functionally related gene groups we loaded the Affymetrix gene expression data into GeneSpring (http://www.silicongenetics.com) to sort differentially expressed genes into determined gene ontology groups. GeneSpring categorises genes based on known function and/or cellular location. Table 1 shows the number and percentages of genes that have at least a two-fold change in expression after Cr(VI) exposure within 31 different select ontology groups. Apoptosis related function genes showed a higher percentage of differentially expressed genes then the combined data set (total genes on chip). Of these, 37.5% of the genes categorised in the apoptosis inhibitor ontology subset were differentially expressed. Cell cycle related function genes also had a higher percentage of differential expression (33.8%) especially the cyclin-dependent kinase inhibitor subset (83.3%), but three important signal transduction ontology groups, the MAPKK, JAK and STAT pathways, did not show dramatic differences in differential expression. Interestingly, a higher amount of transcription factor genes were differentially expressed after Cr(VI) exposure (21.3%), and of these, the transcription initiation (50%) and transcription termination (100%) subsets had the highest percentage of genes affected.
Table 1.
Baseline gene expression after Cr(VI) exposure in BJ-hTERT
| Genes ≥ two-fold change |
||||||
|---|---|---|---|---|---|---|
| Upregulated |
Downregulated |
|||||
| Ontology group | Genes in group (#) | # | % | # | % | Total (%) |
| Total genes on chip | 12,625 | 599 | 4.7 | 502 | 4.0 | 8.7 |
| Apoptosis related function | 38 | 6 | 15.8 | 2 | 5.3 | 21.1 |
| Apoptosis inhibitor | 8 | 2 | 25.0 | 1 | 12.5 | 37.5 |
| Oncogene | 140 | 6 | 4.3 | 12 | 8.6 | 12.9 |
| Tumour suppressor | 20 | 2 | 10.0 | 2 | 10.0 | 20.0 |
| Cell cycle related function | 68 | 14 | 20.6 | 9 | 13.2 | 33.8 |
| Cyclin | 39 | 3 | 7.7 | 9 | 23.1 | 30.8 |
| Cell cycle regulator | 5 | 0 | 0.0 | 1 | 20.0 | 20.0 |
| Cyclin-dependent kinase inhibitors | 6 | 3 | 50.0 | 2 | 33.3 | 83.3 |
| Extra cellular matrix | 43 | 3 | 7.0 | 2 | 4.7 | 11.6 |
| Collagen | 53 | 3 | 5.7 | 7 | 13.2 | 18.9 |
| Microfibrils | 5 | 0 | 0.0 | 0 | 0.0 | 0.0 |
| Mitochondrial | 103 | 6 | 5.8 | 6 | 5.8 | 11.7 |
| Mitochondrial membrane | 9 | 0 | 0.0 | 0 | 0.0 | 0.0 |
| Cytochrome | 46 | 8 | 17.4 | 2 | 4.3 | 21.7 |
| DNA repair | 14 | 0 | 0.0 | 2 | 14.3 | 14.3 |
| RNA pol II transcription factor | 21 | 0 | 0.0 | 2 | 9.5 | 9.5 |
| Telomeric binding factor | 12 | 1 | 8.3 | 2 | 16.7 | 25.0 |
| RNA pol III tanscription factor | 4 | 0 | 0.0 | 0 | 0.0 | 0.0 |
| Transcription factor | 216 | 31 | 14.4 | 15 | 6.9 | 21.3 |
| Transcription initiation | 4 | 2 | 50.0 | 0 | 0.0 | 50.0 |
| Transcription termination | 3 | 1 | 33.3 | 2 | 66.7 | 100.0 |
| Transcriptional activator | 10 | 2 | 20.0 | 1 | 10.0 | 30.0 |
| Transcriptional repressor | 7 | 1 | 14.3 | 0 | 0.0 | 14.3 |
| JAK cascade | 5 | 0 | 0.0 | 0 | 0.0 | 0.0 |
| MAPKK cascade | 11 | 0 | 0.0 | 1 | 9.1 | 9.1 |
| STAT cascade | 65 | 3 | 4.6 | 2 | 3.1 | 7.7 |
| Translation elongation factor | 13 | 0 | 0.0 | 0 | 0.0 | 0.0 |
| Translation initiation factor | 34 | 0 | 0.0 | 2 | 5.9 | 5.9 |
| Antioxidants | 3 | 0 | 0.0 | 0 | 0.0 | 0.0 |
| Ubiquitin | 81 | 5 | 6.2 | 9 | 11.1 | 17.3 |
| Cell adhesion molecule | 29 | 4 | 13.8 | 5 | 17.2 | 31.0 |
The table shows the number of total genes on the chip, and in different gene ontology groups that showed at least a two-fold change in expression. Results are the mean of duplicate experiments.
Differential temporal gene expression profiling between cell populations after Cr(VI) exposure
We examined the differences in gene expression in the B-5Cr survivor sub-population compared to the parental BJ-hTERT control after exposure to 5 μM Na2CrO4 in one experiment. The gene expression profiles for each cell population were examined using GeneSpring before and at various time-points after exposure (4, 8 and 18 h) that were selected to show an early and intermediate temporal response. Genes were normalised to time-point 0 for each respective cell type. At least a 1.5-fold change in expression from the parental control was used for any gene to be considered having altered expression in this part of the study. B-5Cr had a large number of altered genes at the later (18 h) time-point. Analysis of the genes with altered expression revealed a wide variety of functional groups and suggests a gene expression pattern that may favor cell survival after genotoxic exposure.
To examine the different temporal changes in expression in response to Cr(VI) treatment between the survivor subpopulation and the parental controls, the genes modulated by Cr(VI) at one or more time-points were subjected to K means clustering analysis using the GeneSpring software. Clustering analysis involves the grouping of genes according to similarities in their expression profiles across multiple time-points. Clusters are then compared to the same genes for each cell type, and genes that showed distinct differences in temporal expression patterns between cell types were examined.
We identified specific genes of interest within the cluster groups, and their differential expression patterns are shown in Fig. 5. An example of a gene that is upregulated after Cr(VI) exposure in both cell populations is p21 (WAF1, CYP1) (Fig. 5A). Some genes that are upregulated in the BJ-hTERT parental cells but not in B-5Cr include: GADD45, Caspase 3, Map Kinase Phosphatase 5 (MKP5), Myc, the c-rel oncogene (Fig. 5A), and the voltage-dependent anion channel (VDAC) gene (Fig. 5B). Some genes that are upregulated in only the survivor B-5Cr cells are the DNA repair endonuclease subunit (XPF), collagenase type 4, Bcl-xL and the ligand mediated apoptosis signalling receptor DR6 (Fig. 5B). Finally, the UV radiation resistance associated gene (UV-RAG) is downregulated in the BJ-hTERT cells, but not in B-5Cr (Fig. 5B).
Fig. 5.


Altered gene expression patterns over time in specific genes of interest: (A) p21(Waf), GADD45, Caspase 3 (CPP-32), MKP5, Myc, c-rel; (B) VDAC, XPF, CLG4, BCL-xL, DR6, UVRAG. Results are one representative experiment.
Discussion
An initial consequence of genetic injury is cell cycle checkpoint arrest but genotoxins may also activate cell death pathways of apoptosis or “terminal” growth arrest. Cellular survival responses to genotoxic insult may produce intrinsic death resistance; such a selective growth advantage may allow for the emergence of a transformed phenotype. The net cell expansion associated with pre-malignant progression is likely to be comprised of both increased proliferative potential and resistance to cell death. In order to investigate potential differences in the survivor population of cells exposed to Cr(VI), BJ-hTERT cells were exposed to 5 μM Na2CrO4 for 24 h. The surviving fraction of cells was allowed to re-emerge over several weeks until a polyclonal cell strain (B-5Cr) was developed. Certain cell culture characteristics were notably altered as the B-5Cr cells recovered replicative competency after initial exposure (data not shown). For example, the cellular growth rate was higher in B-5Cr compared to BJ-hTERT cells.
The death resistance in the B-5Cr cells suggests that the progenitor cells of this line survived because of an intrinsic selective advantage over the rest of the population. A number of molecular mechanisms that convey death resistance may be different or altered in cells surviving genotoxic stress [47]. One possibility is that the survivor cells may have a reduced capacity to activate or increase levels of p53 in response to genotoxic damage. The p53 protein is important in relaying DNA damage-associated signals into apoptosis or growth-arresting responses, through transactivation of numerous apoptosis- and cell cycle-regulating genes [48], for review see [49]. While Cr(VI)-induced apoptosis in human lung fibroblasts (HLF) is dependent on the presence of p53, which is activated in response to Cr(VI) treatment [4, 5], upregulation of p53 protein levels in response to Cr(VI) was not altered in the B-5Cr cells. Accordingly, the transcription of p21waf1/cyp1, a p53-responsive gene, was virtually identical in both BJ-hTERT and B-5Cr cells, further indicating that the enhanced apoptotic resistance was not due to selective alterations in p53.
Another explanation for Cr(VI)-induced resistance to subsequent Cr(VI)-induced death may be related to alterations in Cr(VI) uptake or efflux mechanisms resulting in a decreased intracellular concentration of Cr(VI). Such a mechanism has been shown in human epithelial cell lines resistant to killing by CrO3 [50], and has been observed with other toxic metals such as arsenic, platinum, and antimony [51, 52]. The resistance of the B-5Cr cells to both Cr (VI)- and H2O2-induced cell death would suggest that the mechanism was not a function of alteration of cellular Cr(VI) accumulation. Indeed, this was subsequently confirmed in uptake studies of Na251CrO4 (Fig. 3).
Only a limited amount of information is available examining gene expression in normal human diploid cells following Cr(VI) exposure. In a study of A549 cells exposed to a single acute dose of Cr(VI), the expression patterns of several genes involved in the oxidative stress response (GPx, Cu/Zn SOD), calcium mobilization (calcineurin A2), signalling (NEN-1, MAPKAP kinase), cell cycle regulation (INK4p19, p34 CDC2) and metabolism (PDK2, Na,K-ATPase) were altered in response to Cr(VI) exposure [53]. In a study examining the expression of 216 genes in the lungs and livers of Cr(VI) exposed (intratracheally) rats, significant Cr(VI)-related alterations in gene expression were only observed in the lungs of rats [54] which mirrored the selective toxicity of Cr(VI) in this tissue [55]. Of the genes affected by Cr(VI), drug metabolizing enzymes (NADPH-cytochrome P450 reductase, cytochrome b5 reductase), oxidative stress (GPx), drug resistance (MDR protein), signal transduction (MAP kinase), cell cycle (cyclins D1-3) and apoptosis (Bcl-x) were induced at least two-fold after treatment. Recently, a comparative analysis of gene expression was performed on adenovirus 12-SV40 hybrid immortalised human bronchoepithelial cells exposed to Cr(VI) (10 μM, 4 h) and several other toxic metals [56]. Of the 1,200 genes examined, 44 were differentially expressed after Cr(VI) exposure relative to the other metals and mitomycin C. Cr(VI) moderately induced the expression of genes involved in protein metabolism (ubiquitin), histone acetylation (HATB2) and glucose transport (GLUT1) and downregulated the expression of genes involved in bone metabolism (BMP4), transcription (hEGR1, CREB2), survival signalling (PKB/AKT), protein trafficking (HSP-90A), growth factor signalling (FGFR1), xenobiotic metabolism (GSHPX1, CYP1B1) and cell growth control (c-myc, cyclin K). Our own recent work using normal diploid human lung fibroblasts found that Cr(VI) treatment transiently activated p53-responsive genes (GADD45, p21), upregulated the expression of pro-apoptotic genes (bcl-Xs), and downregulated the expression of anti-apoptotic genes (bcl-w and bcl-Xl) [57]. Moreover, Cr(VI)-induced upregulation of the key cell cycle inhibitors p21 and p15, was accompanied by decreased cyclin A expression.
To begin to understand the networks of genes that are affected by Cr(VI) exposure and also govern death resistance in the B-5Cr cells, we performed a microarray gene expression analysis using the Affymetrix human genome U95Av2 array. Out of a total of 12,625 genes and ESTs we identified approximately 1,200 genes displaying altered expression after Cr(VI) exposure. A large percentage of genes in many of the ontology groups shown in Table 1 were differentially expressed, but there was no significant trend in either upregulated or downregulated expression in any particular group in the BJ-hTERT parental cell line. Many of the groups had an apparent alteration in the regulation of many genes, both up and downregulated, while other groups were less affected, which is similar to what has been previously reported in other studies [56].
We further fractionated these gene changes as a function of time after exposure and compared death resistant and “wild type” cells. Cluster analysis by gene function revealed altered differential expression of genes primarily involved in apoptosis regulation, cell cycle regulation and DNA repair. Previous data indicate that cell death/growth arrest pathways, such as those regulated by GADD45, caspase and bclX, are dramatically affected by Cr(VI) exposure in normal human diploid fibroblasts [43, 53, 57]. The lack of Cr(VI) induction of GADD45 and caspase 3 in the B-5Cr cells, compared to that in BJ-hTERT cells, along with the upregulation of the anti-apoptotic Bcl-xL is consistent with their death-resistant phenotype. In contrast, the apparent transcriptional upregulation of the DR6 member of the TNF receptor superfamily should be consistent with an apoptogenic phenotype, but perhaps is indicative of a compensatory response to death resistance in the face of genotoxic insult.
Signalling through MAPK pathways results in the activation of a number of transcription factors, resulting in either cell proliferation or cell death/growth arrest, which is dependent upon the activation state and crosstalk among MAPK family members [58-61]. A growing family of MAPK phosphatases (MKPs) exerts negative regulation of MAPK activities [62]. MKP5 has been shown to be able to deactivate all MAPK members, with some selectivity for the JNK/SAPK members [63, 64]. Cr-induced activation of ERK, JNK and p38 pathways has been shown in a number of different cell lines, while the activation pattern is dependent on the dose/duration of the exposure [57, 65-67]. It has been suggested that Cr(VI) can potentially activate inhibitory MKPs [66]. The present data supports a role for Cr(VI) in MKP5 transcriptional upregulation, which may play a role in death resistance, as its upregulation was abrogated in the B-5Cr cells.
Enhanced expression of the c-myc oncogene has been shown in Cr-transformed C3H/10T1/2 Cl8 mouse embryo cell lines [68], while the NF-(B subunit c-Rel has been shown to malignantly transform cells in culture [69, 70]. Cr(VI) has been shown to both inhibit and activate the transcriptional activity of NF-(B [71, 72], which may be related to differences in Cr concentration and cell type [73]. In the present study, Cr-induced upregulation of these oncogenes was observed in the normal BJ-hTERT cells, but abrogated in the B-5Cr cells. It is unclear how to interpret these findings at this time, but it may be related to an upregulated survival response in the death-resistant cells, as inappropriate activation of oncogenes has been shown to induce a premature senescent phenotype, consistent with clonogenic death [74, 75].
The upregulation/lack of downregulation of the respective DNA damage response genes, XPF and UV-RAG after Cr exposure in B-5Cr cells is intriguing, particularly in light of their association with nucleotide excision repair (NER) and recombination pathways [76-79]. We and others have shown that both NER and homologous recombination repair mechanisms are associated with the repair of Cr-induced DNA lesions [80, 81] (for review, see [26]).
It has previously been shown that chemically transformed (Hut-11A) human skin fibroblasts secreted an active collagenase that induced extensive degradation of type IV collagen [82]. It is tempting to speculate that the specific Cr(VI)-induced upregulation of collagenase type IV in the death-resistant B-5Cr cells represents a predisposition of these cells to remodeling of the extracellular matrix, which is a hallmark of both fibrosis and tumour cell invasion.
Transport across the outer mitochondrial membrane is tightly regulated by a multi-protein pore complex, whose central component is the mitochondrial voltage-dependent anion channel (VDAC). Alterations in VDAC function lead to the mitochondrial membrane permeability transition, which results in the release of the inter-membrane space components into the cytosol (reviewed in [83-86]). Our studies have verified that release of cytochrome c from the mitochondria is the point of no return for Cr(VI)-induced apoptosis, which can be inhibited by cyclosporin A [87]. Data from the present study have potentially uncovered a novel toxic stress response pathway involving differential transcriptional upregulation of the mitochondrial VDAC protein in normal BJ-hTERT fibroblasts, and its abrogation in death-resistant B-5Cr cells. This discovery leads to the intriguing hypothesis that the repair/recovery process after toxic insult may involve the selection of cells with intrinsic or induced mitochondrial dysregulation leading to death resistance.
It is unclear whether resistance to apoptosis in the B-5Cr cells was due to selection of cells with intrinsic or induced phenotypic differences. We know that the progenitor cells of this sub-population received a dose of Cr(VI) that potentially caused DNA damage, but there may have been other selective pressures that could induce phenotypic changes that translate as increased apoptosis resistance. For example, it is possible that the survivor sub-population somehow gained a selective advantage during the process of continued cycling in culture with very few neighbouring cells. Whatever the case, the initial Cr(VI) exposure-induced death in the majority of the population and drove the selection of these cells regardless of what may be the ultimate apoptotic resistance-inducing condition.
An intriguing observation is that the B-5Cr sub-population has subsequently diminished its resistant phenotype, which was apparently a function of long-term storage, as it was not dependent on cell passage number. We speculate that this could represent the further clonal selection of cells with a more adherent, but less resistant phenotype. To better understand the molecular mechanisms of Cr-induced death resistance, we derived clones from the early B-5Cr populations derived in the present study. Indeed, we initially found two obvious phenotypes: death resistance and altered morphology (increased cytoplasm content and increased cell volume). Moreover, the death-resistant clones have maintained their phenotype after long-term passage and storage. Current work is aimed at elucidating the signalling pathways responsible for resistance in these stable subclones.
In conclusion, our data suggests an alteration in gene expression that may favor cell survival and/or incomplete DNA repair after genotoxic exposure in sub-populations of BJ-hTERT human fibroblasts, which have acquired resistance to Cr-induced clonogenic lethality that is not related to altered uptake. Selection of cells with altered expression of these genes may constitute the early stages of tumour progression.
Acknowledgments
This work was supported by NIH Grants, ES05304 and ES0996, to S.R.P.
References
- 1.Hartwell LH, Kastan MB. Cell cycle control and cancer. Science. 1994;266:1821–1828. doi: 10.1126/science.7997877. [DOI] [PubMed] [Google Scholar]
- 2.Evan GI, Vousden KH. Proliferation, cell cycle and apoptosis in cancer. Nature. 2001;411:342–348. doi: 10.1038/35077213. [DOI] [PubMed] [Google Scholar]
- 3.Blankenship LJ, Carlisle DL, Wise JP, Orenstein JM, Dye LE, III, Patierno SR. Induction of apoptotic cell death by particulate lead chromate: differential effects of vitamins C and E on genotoxicity and survival. Toxicol Appl Pharmacol. 1997;146:270–280. doi: 10.1006/taap.1997.8237. [DOI] [PubMed] [Google Scholar]
- 4.Carlisle DL, Pritchard DE, Singh J, Patierno SR. Chromium(VI) induces p53-dependent apoptosis in diploid human lung and mouse dermal fibroblasts. Mol Carcinog. 2000;28:111–118. doi: 10.1002/1098-2744(200006)28:2<111::aid-mc7>3.0.co;2-y. [DOI] [PubMed] [Google Scholar]
- 5.Carlisle DL, Pritchard DE, Singh J, Owens BM, Blankenship LJ, Orenstein JM, Patierno SR. Apoptosis and P53 induction in human lung fibroblasts exposed to chromium (VI): effect of ascorbate and tocopherol. Toxicol Sci. 2000;55:60–68. doi: 10.1093/toxsci/55.1.60. [DOI] [PubMed] [Google Scholar]
- 6.Singh J, Carlisle DL, Pritchard DE, Patierno SR. Chromium-induced genotoxicity and apoptosis: Relationship to chromium carcinogenesis (review) Oncol Rep. 1998;5:1307–1318. doi: 10.3892/or.5.6.1307. [DOI] [PubMed] [Google Scholar]
- 7.Pritchard DE, Ceryak S, Ha L, Fornsaglio JL, Hartman SK, O’Brien TJ, Patierno SR. Mechanism of apoptosis and determination of cellular fate in chromium(VI)-exposed populations of telomerase-immortalized human fibroblasts. Cell Growth Differ. 2001;12:487–496. [PubMed] [Google Scholar]
- 8.Chromium, nickel and welding [erratum appears in IARC Monogr Eval Carcinog Risks Hum 1991; 51: 483] IARC Monogr Eval Carcinog Risks Hum. 1990;49:1–648. [PMC free article] [PubMed] [Google Scholar]
- 9.Hayes RB. Review of occupational epidemiology of chromium chemicals and respiratory cancer. Sci Total Environ. 1988;71:331–339. doi: 10.1016/0048-9697(88)90205-7. [DOI] [PubMed] [Google Scholar]
- 10.Fishbein L. Sources, transport and alterations of metal compounds: an overview. I. Arsenic, beryllium, cadmium, chromium, and nickel. Environ Health Perspect. 1981;40:43–64. doi: 10.1289/ehp.814043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.The chromium problem: Research needs and risk assessment. Environ Health Perspect. 1991;92 doi: 10.1289/ehp.91923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.IARC . Monographs on the evaluation of the carcinogenic risk of chemicals to humans. 1982. pp. 167–170. [PubMed] [Google Scholar]
- 13.Burke DJ. Complexity in the spindle checkpoint. Curr Opin Genet Dev. 2000;10:26–31. doi: 10.1016/s0959-437x(99)00040-4. [DOI] [PubMed] [Google Scholar]
- 14.Wetterhahn KE, Hamilton JW, Aiyar J, Borges KM, Floyd R. Mechanism of chromium(VI) carcinogenesis. Reactive intermediates and effect on gene expression. Biol Trace Element Res. 1989;21:405–411. doi: 10.1007/BF02917282. [DOI] [PubMed] [Google Scholar]
- 15.Rossi SC, Wetterhahn KE. Chromium(V) is produced upon reduction of chromate by mitochondrial electron transport chain complexes. Carcinogenesis. 1989;10:913–920. doi: 10.1093/carcin/10.5.913. [DOI] [PubMed] [Google Scholar]
- 16.Stearns DM, Wetterhahn KE. Reaction of chromium(VI) with ascorbate produces chromium(V), chromium(IV), and carbon-based radicals. Chem Res Toxicol. 1994;7:219–230. doi: 10.1021/tx00038a016. [DOI] [PubMed] [Google Scholar]
- 17.Sugden KD, Martin BD. Guanine and 7,8-dihydro-8-oxo-guanine-specific oxidation in DNA by chromium(V) Environ Health Perspect. 2002;110(Suppl 5):725–728. doi: 10.1289/ehp.02110s5725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sugden KD, Campo CK, Martin BD. Direct oxidation of guanine and 7,8-dihydro-8-oxoguanine in DNA by a high-valent chromium complex: A possible mechanism for chromate genotoxicity. Chem Res Toxicol. 2001;14:1315–1322. doi: 10.1021/tx010088+. [DOI] [PubMed] [Google Scholar]
- 19.Miller CA, III, Costa M. Characterization of DNA-protein complexes induced in intact cells by the carcinogen chromate. Mol Carcinog. 1988;1:125–133. doi: 10.1002/mc.2940010208. [DOI] [PubMed] [Google Scholar]
- 20.Standeven AM, Wetterhahn KE. Chromium (VI) toxicity: Uptake, reduction, and DNA damage. J Am Cell Toxicol. 1989;8:1275–1283. [Google Scholar]
- 21.Singh J, Mclean JA, Pritchard DE, Montaser A, Patierno SR. Sensitive quantitation of chromium-DNA adducts by inductively coupled plasma mass spectrometry with a direct injection high-efficiency nebulizer. Toxicol Sci. 1998;46:260–265. doi: 10.1006/toxs.1998.2512. [DOI] [PubMed] [Google Scholar]
- 22.Xu J, Manning FC, Patierno SR. Preferential formation and repair of chromium-induced DNA adducts and DNA-protein crosslinks in nuclear matrix DNA. Carcinogenesis. 1994;15:1443–1450. doi: 10.1093/carcin/15.7.1443. [DOI] [PubMed] [Google Scholar]
- 23.Xu J, Bubley GJ, Detrick B, Blankenship LJ, Patierno SR. Chromium(VI) treatment of normal human lung cells results in guanine-specific DNA polymerase arrest, DNA-DNA cross-links and S-phase blockade of cell cycle. Carcinogenesis. 1996;17:1511–1517. doi: 10.1093/carcin/17.7.1511. [DOI] [PubMed] [Google Scholar]
- 24.Singh J, Bridgewater LC, Patierno SR. Differential sensitivity of chromium-mediated DNA interstrand crosslinks and DNA-protein crosslinks to disruption by alkali and EDTA. Toxicol Sci. 1998;45:72–76. doi: 10.1006/toxs.1998.2489. [DOI] [PubMed] [Google Scholar]
- 25.Bridgewater LC, Manning FC, Patierno SR. Base-specific arrest of in vitro DNA replication by carcinogenic chromium: Relationship to DNA interstrand crosslinking. Carcinogenesis. 1994;15:2421–2427. doi: 10.1093/carcin/15.11.2421. [DOI] [PubMed] [Google Scholar]
- 26.O’Brien TJ, Ceryak S, Patierno SR. Complexities of chromium carcinogenesis: role of cellular response, repair and recovery mechanisms. Mutat Res. 2003;533:3–36. doi: 10.1016/j.mrfmmm.2003.09.006. [DOI] [PubMed] [Google Scholar]
- 27.Bridgewater LC, Manning FC, Woo ES, Patierno SR. DNA polymerase arrest by adducted trivalent chromium. Mol Carcinog. 1994;9:122–133. doi: 10.1002/mc.2940090304. [DOI] [PubMed] [Google Scholar]
- 28.Manning FC, Xu J, Patierno SR. Transcriptional inhibition by carcinogenic chromate: relationship to DNA damage. Mol Carcinog. 1992;6:270–279. doi: 10.1002/mc.2940060409. [DOI] [PubMed] [Google Scholar]
- 29.Patierno SR, Banh D, Landolph JR. Transformation of C3H/10T1/2 mouse embryo cells to focus formation and anchorage independence by insoluble lead chromate but not soluble calcium chromate: Relationship to mutagenesis and internalization of lead chromate particles. Cancer Res. 1988;48:5280–5288. [PubMed] [Google Scholar]
- 30.Patierno SR, Landolph JR. Soluble vs insoluble hexavalent chromate. Relationship of mutation to in vitro transformation and particle uptake. Biol Trace Element Res. 1989;21:469–474. doi: 10.1007/BF02917290. [DOI] [PubMed] [Google Scholar]
- 31.Zhitkovich A, Song Y, Quievryn G, Voitkun V. Non-oxidative mechanisms are responsible for the induction of mutagenesis by reduction of Cr(VI) with cysteine: role of ternary DNA adducts in Cr(III)-dependent mutagenesis. Biochemistry. 2001;40:549–560. doi: 10.1021/bi0015459. [DOI] [PubMed] [Google Scholar]
- 32.Tully DB, Collins BJ, Overstreet JD, Smith CS, Dinse GE, Mumtaz MM, Chapin RE. Effects of arsenic, cadmium, chromium, and lead on gene expression regulated by a battery of 13 different promoters in recombinant HepG2 cells. Toxicol Appl Pharmacol. 2000;168:79–90. doi: 10.1006/taap.2000.9014. [DOI] [PubMed] [Google Scholar]
- 33.Wetterhahn KE, Hamilton JW. Molecular basis of hexavalent chromium carcinogenicity: Effect on gene expression. Sci Total Environ. 1989;86:113–129. doi: 10.1016/0048-9697(89)90199-x. [DOI] [PubMed] [Google Scholar]
- 34.Hamilton JW, Kaltreider RC, Bajenova OV, Ihnat MA, McCaffrey J, Turpie BW, Rowell EE, Oh J, Nemeth MJ, Pesce CA, Lariviere JP. Molecular basis for effects of carcinogenic heavy metals on inducible gene expression. Environ Health Perspect. 1998;106(Suppl 4):1005–1015. doi: 10.1289/ehp.98106s41005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wise JP, Orenstein JM, Patierno SR. Inhibition of lead chromate clastogenesis by ascorbate: Relationship to particle dissolution and uptake. Carcinogenesis. 1993;14:429–434. doi: 10.1093/carcin/14.3.429. [DOI] [PubMed] [Google Scholar]
- 36.Wise JP, Sr, Stearns DM, Wetterhahn KE, Patierno SR. Cell-enhanced dissolution of carcinogenic lead chromate particles: The role of individual dissolution products in clastogenesis. Carcinogenesis. 1994;15:2249–2254. doi: 10.1093/carcin/15.10.2249. [DOI] [PubMed] [Google Scholar]
- 37.Wright WE, Pereira-Smith OM, Shay JW. Reversible cellular senescence: implications for immortalization of normal human diploid fibroblasts. Mol Cell Biol. 1989;9:3088–3092. doi: 10.1128/mcb.9.7.3088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Campisi J. Replicative senescence: an old lives’ tale? Cell. 1996;84:497–500. doi: 10.1016/s0092-8674(00)81023-5. [DOI] [PubMed] [Google Scholar]
- 39.Shay JW, Pereira-Smith OM, Wright WE. A role for both RB and p53 in the regulation of human cellular senescence. Exp Cell Res. 1991;196:33–39. doi: 10.1016/0014-4827(91)90453-2. [DOI] [PubMed] [Google Scholar]
- 40.Counter CM, Avilion AA, LeFeuvre CE, Stewart NG, Greider CW, Harley CB, Bacchetti S. Telomere shortening associated with chromosome instability is arrested in immortal cells which express telomerase activity. EMBO J. 1992;11:1921–1929. doi: 10.1002/j.1460-2075.1992.tb05245.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jiang XR, Jimenez G, Chang E, Frolkis M, Kusler B, Sage M, Beeche M, Bodnar AG, Wahl GM, Tlsty TD, Chiu CP. Telomerase expression in human somatic cells does not induce changes associated with a transformed phenotype. Nat Genet. 1999;21:111–114. doi: 10.1038/5056. [DOI] [PubMed] [Google Scholar]
- 42.Vaziri H, Squire JA, Pandita TK, Bradley G, Kuba RM, Zhang H, Gulyas S, Hill RP, Nolan GP, Benchimol S. Analysis of genomic integrity and p53-dependent G1 checkpoint in telomerase-induced extended-lifespan human fibroblasts. Mol Cell Biol. 1999;19:2373–2379. doi: 10.1128/mcb.19.3.2373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ha L, Ceryak S, Patierno SR. Chromium (VI) activates ataxia telangiectasia mutated (ATM) protein: Requirement of ATM for both apoptosis and recovery from terminal growth arrest. J Biol Chem. 2003;278:17885–17894. doi: 10.1074/jbc.M210560200. [DOI] [PubMed] [Google Scholar]
- 44.Chen YW, Zhao P, Borup R, Hoffman EP. Expression profiling in the muscular dystrophies: Identification of novel aspects of molecular pathophysiology. J Cell Biol. 2000;151:1321–1336. doi: 10.1083/jcb.151.6.1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ye J, Wang S, Leonard SS, Sun Y, Butterworth L, Antonini J, Ding M, Rojanasakul Y, Vallyathan V, Castranova V, Shi X. Role of reactive oxygen species and p53 in chromium(VI)-induced apoptosis. J Biol Chem. 1999;274:34974–34980. doi: 10.1074/jbc.274.49.34974. [DOI] [PubMed] [Google Scholar]
- 46.Igney FH, Krammer PH. Death and anti-death: Tumour resistance to apoptosis. Nat Rev Cancer. 2002;2:277–288. doi: 10.1038/nrc776. [DOI] [PubMed] [Google Scholar]
- 47.Hofseth LJ. The adaptive imbalance to genotoxic stress: genome guardians rear their ugly heads. Carcinogenesis. 2004;25:1787–1793. doi: 10.1093/carcin/bgh196. [DOI] [PubMed] [Google Scholar]
- 48.Flatt PM, Polyak K, Tang LJ, Scatena CD, Westfall MD, Rubinstein LA, Yu J, Kinzler KW, Vogelstein B, Hill DE, Pietenpol JA. p53-dependent expression of PIG3 during proliferation, genotoxic stress, and reversible growth arrest. Cancer Lett. 2000;156:63–72. doi: 10.1016/s0304-3835(00)00441-9. [DOI] [PubMed] [Google Scholar]
- 49.Levine AJ. p53, the cellular gatekeeper for growth and division: review [55 refs] Cell. 1997;88:323–331. doi: 10.1016/s0092-8674(00)81871-1. [DOI] [PubMed] [Google Scholar]
- 50.Son KH, Zhang M, Rucobo E, Nwaigwe D, Montgomery F, Leffert H. Derivation and study of human epithelial cell lines resistant to killing by chromium trioxide. J Toxicol Environ Health A. 2004;67:1027–1049. doi: 10.1080/15287390490447304. [DOI] [PubMed] [Google Scholar]
- 51.Tamas MJ, Wysocki R. Mechanisms involved in metalloid transport and tolerance acquisition. Curr Genet. 2001;40:2–12. doi: 10.1007/s002940100234. [DOI] [PubMed] [Google Scholar]
- 52.Wernyj RP, Morin PJ. Molecular mechanisms of platinum resistance: Still searching for the Achilles’ heel. Drug Resist Updat. 2004;7:227–232. doi: 10.1016/j.drup.2004.08.002. [DOI] [PubMed] [Google Scholar]
- 53.Ye J, Shi X. Gene expression profile in response to chromium-induced cell stress in A549 cells. Mol Cell Biochem. 2001;222:189–197. [PubMed] [Google Scholar]
- 54.Izzotti A, Cartiglia C, Balansky R, D’Agostini F, Longobardi M, De Flora S. Selective induction of gene expression in rat lung by hexavalent chromium. Mol Carcinog. 2002;35:75–84. doi: 10.1002/mc.10077. [DOI] [PubMed] [Google Scholar]
- 55.D’Agostini F, Izzotti A, Bennicelli C, Camoirano A, Tampa E, De Flora S. Induction of apoptosis in the lung but not in the liver of rats receiving intra-tracheal instillations of chromium(VI) Carcinogenesis. 2002;23:587–593. doi: 10.1093/carcin/23.4.587. [DOI] [PubMed] [Google Scholar]
- 56.Andrew AS, Warren AJ, Barchowsky A, Temple KA, Klei L, Soucy NV, O’Hara KA, Hamilton JW. Genomic and proteomic profiling of responses to toxic metals in human lung cells. Environ Health Perspect. 2003;111:825–835. doi: 10.1289/ehp.111-1241504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ceryak S, Zingariello C, O’Brien T, Patierno SR. Induction of pro-apoptotic and cell cycle-inhibiting genes in chromium(VI)-treated human lung fibroblasts: lack of effect of ERK. Mol Cell Biochem. 2004;255:139–149. doi: 10.1023/b:mcbi.0000007270.82431.3e. [DOI] [PubMed] [Google Scholar]
- 58.Thomas G. MAP kinase by any other name smells just as sweet. Cell. 1992;68:3–6. doi: 10.1016/0092-8674(92)90199-m. [DOI] [PubMed] [Google Scholar]
- 59.Ballif BA, Blenis J. Molecular mechanisms mediating mammalian mitogen-activated protein kinase (MAPK) kinase (MEK)-MAPK cell survival signals. Cell Growth Differ. 2001;12:397–408. [PubMed] [Google Scholar]
- 60.Moelling K, Schad K, Bosse M, Zimmermann S, Schweneker M. Regulation of Raf-Akt Cross-talk. J Biol Chem. 2002;277:31099–31106. doi: 10.1074/jbc.M111974200. [DOI] [PubMed] [Google Scholar]
- 61.Murphy LO, Smith S, Chen RH, Fingar DC, Blenis J. Molecular interpretation of ERK signal duration by immediate early gene products. Nat Cell Biol. 2002;4:556–564. doi: 10.1038/ncb822. [DOI] [PubMed] [Google Scholar]
- 62.Keyse SM. Protein phosphatases and the regulation of mitogen-activated protein kinase signalling: review [49 refs] Curr Opin Cell Biol. 2000;12:186–192. doi: 10.1016/s0955-0674(99)00075-7. [DOI] [PubMed] [Google Scholar]
- 63.Zhou B, Wang ZX, Zhao Y, Brautigan DL, Zhang ZY. The specificity of extracellular signal-regulated kinase 2 dephosphorylation by protein phosphatases. J Biol Chem. 2002;277:31818–31825. doi: 10.1074/jbc.M203969200. [DOI] [PubMed] [Google Scholar]
- 64.Theodosiou A, Smith A, Gillieron C, Arkinstall S, Ashworth A. MKP5, a new member of the MAP kinase phosphatase family, which selectively dephosphorylates stress-activated kinases. Oncogene. 1999;18:6981–6988. doi: 10.1038/sj.onc.1203185. [DOI] [PubMed] [Google Scholar]
- 65.Samet JM, Graves LM, Quay J, Dailey LA, Devlin RB, Ghio AJ, Wu W, Bromberg PA, Reed W. Activation of MAPKs in human bronchial epithelial cells exposed to metals. Am J Physiol. 1998;275:L551–L558. doi: 10.1152/ajplung.1998.275.3.L551. [DOI] [PubMed] [Google Scholar]
- 66.Chuang SM, Liou GY, Yang JL. Activation of JNK, p38 and ERK mitogen-activated protein kinases by chromium(VI) is mediated through oxidative stress but does not affect cytotoxicity. Carcinogenesis. 2000;21:1491–1500. [PubMed] [Google Scholar]
- 67.Chuang SM, Yang JL. Comparison of roles of three mitogen-activated protein kinases induced by chromium(VI) and cadmium in non-small-cell lung carcinoma cells. Mol Cell Biochem. 2001;222:85–95. [PubMed] [Google Scholar]
- 68.Landolph JR. Molecular mechanisms of transformation of C3H/10T1/2 C1 8 mouse embryo cells and diploid human fibroblasts by carcinogenic metal compounds: review [58 refs] Environ Health Perspect. 1994;102(Suppl 3):119–125. doi: 10.1289/ehp.94102s3119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gilmore TD, Kalaitzidis D, Liang MC, Starczynowski DT. The c-Rel transcription factor and B-cell proliferation: a deal with the devil. Oncogene. 2004;23:2275–2286. doi: 10.1038/sj.onc.1207410. [DOI] [PubMed] [Google Scholar]
- 70.Hanson JL, Hawke NA, Kashatus D, Baldwin AS. The nuclear factor kappaB subunits RelA/p65 and c-Rel potentiate but are not required for Ras-induced cellular transformation. Cancer Res. 2004;64:7248–7255. doi: 10.1158/0008-5472.CAN-03-3898. [DOI] [PubMed] [Google Scholar]
- 71.Shumilla JA, Wetterhahn KE, Barchowsky A. Inhibition of NF-kappa B binding to DNA by chromium, cadmium, mercury, zinc, and arsenite in vitro: Evidence of a thiol mechanism. Arch Biochem Biophys. 1998;349:356–362. doi: 10.1006/abbi.1997.0470. [DOI] [PubMed] [Google Scholar]
- 72.Chen F, Bower J, Leonard SS, Ding M, Lu Y, Rojanasakul Y, Kung HF, Vallyathan V, Castranova V, Shi X. Protective roles of NF-kappa B for chromium(VI)-induced cytotoxicity is revealed by expression of Ikappa B kinase-beta mutant. J Biol Chem. 2002;277:3342–3349. doi: 10.1074/jbc.M101089200. [DOI] [PubMed] [Google Scholar]
- 73.Kaltreider RC, Pesce CA, Ihnat MA, Lariviere JP, Hamilton JW. Differential effects of arsenic(III) and chromium(VI) on nuclear transcription factor binding. Mol Carcinog. 1999;25:219–229. [PubMed] [Google Scholar]
- 74.Hahn WC, Weinberg RA. Modelling the molecular circuitry of cancer. Nat Rev Cancer. 2002;2:331–341. doi: 10.1038/nrc795. [DOI] [PubMed] [Google Scholar]
- 75.Mathon NF, Lloyd AC. Cell senescence and cancer. Nat Rev Cancer. 2001;1:203–213. doi: 10.1038/35106045. [DOI] [PubMed] [Google Scholar]
- 76.Lehmann AR. DNA repair-deficient diseases, xeroderma pigmentosum, Cockayne syndrome and trichothiodystrophy. Biochimie. 2003;85:1101–1111. doi: 10.1016/j.biochi.2003.09.010. [DOI] [PubMed] [Google Scholar]
- 77.Rosselli F, Briot D, Pichierri P. The Fanconi anemia pathway and the DNA interstrand cross-links repair. Biochimie. 2003;85:1175–1184. doi: 10.1016/j.biochi.2003.11.002. [DOI] [PubMed] [Google Scholar]
- 78.Eastman QM, Villey IJ, Schatz DG. Detection of RAG protein-V(D)J recombination signal interactions near the site of DNA cleavage by UV cross-linking. Mol Cell Biol. 1999;19:3788–3797. doi: 10.1128/mcb.19.5.3788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Mo X, Bailin T, Sadofsky MJ. RAG1 and RAG2 cooperate in specific binding to the recombination signal sequence in vitro. J Biol Chem. 1999;274:7025–7031. doi: 10.1074/jbc.274.11.7025. [DOI] [PubMed] [Google Scholar]
- 80.Reynolds M, Peterson E, Quievryn G, Zhitkovich A. Human nucleotide excision repair efficiently removes chromium-DNA phosphate adducts and protects cells against chromate toxicity. J Biol Chem. 2004;279:30419–30424. doi: 10.1074/jbc.M402486200. [DOI] [PubMed] [Google Scholar]
- 81.O’Brien TJ, Fornsaglio JL, Ceryak S, Patierno SR. Effects of hexavalent chromium on the survival and cell cycle distribution of DNA repair-deficient S. cerevisiae. DNA Repair. 2002;1:617–627. doi: 10.1016/s1568-7864(02)00078-2. [DOI] [PubMed] [Google Scholar]
- 82.Sheela S, Barrett JC. Degradation of type IV collagen by neoplastic human skin fibroblasts. Carcinogenesis. 1985;6:173–179. doi: 10.1093/carcin/6.2.173. [DOI] [PubMed] [Google Scholar]
- 83.Tsujimoto Y, Shimizu S. VDAC regulation by the Bcl-2 family of proteins: Review [73 refs] Cell Death Differ. 2000;7:1174–1181. doi: 10.1038/sj.cdd.4400780. [DOI] [PubMed] [Google Scholar]
- 84.Vieira HL, Haouzi D, El Hamel C, Jacotot E, Belzacq AS, Brenner C, Kroemer G. Permeabilization of the mitochondrial inner membrane during apoptosis: Impact of the adenine nucleotide translocator. Cell Death Differ. 2000;7:1146–1154. doi: 10.1038/sj.cdd.4400778. [DOI] [PubMed] [Google Scholar]
- 85.Halestrap AP, McStay GP, Clarke SJ. The permeability transition pore complex: Another view: review [86 refs] Biochimie. 2002;84:153–166. doi: 10.1016/s0300-9084(02)01375-5. [DOI] [PubMed] [Google Scholar]
- 86.Harris MH, Thompson CB. The role of the Bcl-2 family in the regulation of outer mitochondrial membrane permeability: review [92 refs] Cell Death Differ. 2000;7:1182–1191. doi: 10.1038/sj.cdd.4400781. [DOI] [PubMed] [Google Scholar]
- 87.Pritchard DE, Singh J, Carlisle DL, Patierno SR. Cyclosporin A inhibits chromium(VI)-induced apoptosis and mitochondrial cytochrome c release and restores clonogenic survival in CHO cells. Carcinogenesis. 2000;21:2027–2033. doi: 10.1093/carcin/21.11.2027. [DOI] [PubMed] [Google Scholar]