

The pathogenesis of neuropathic pain is incompletely understood and treatments are often inadequate. Cytoplasmic Ca2+ regulates numerous cellular processes in neurons. This review therefore examines the pathogenic contribution of altered inward Ca2+ flux (ICa) through voltage-gated Ca2+ channels in sensory neurons after peripheral nerve injury. We reviewed studies that recorded membrane currents through intracellular and patch-clamp techniques, as well as intracellular Ca2+ levels using fluorimetric indicators, and performed behavioral analysis of rodent nerve injury models. Following nerve injury by partial ligation, a response characterized by sustained lifting, shaking, and licking of the paw after sharp mechanical stimulation is a reliable indicator or neuropathic pain. Primary sensory neurons isolated from animals with this behavior show a decrease in high-voltage activated ICa by approximately one third. Low voltage-activated ICa is nearly eliminated by peripheral nerve injury. Loss of ICa leads to decreased activation of Ca2+-activated K+ currents, which are also directly reduced in traumatized neurons. As a result of these changes in membrane currents, membrane voltage recordings show increased action potential duration and diminished afterhyperpolarization. Excitability is elevated, as indicated by resting membrane potential depolarization and a decreased current threshold for action potential initiation. Traumatized nociceptive neurons develop increased repetitive firing during sustained depolarization after axotomy. Concurrently, cytoplasmic Ca2+ transients are diminished. In conclusions, axotomized neurons, especially pain-conducting ones, develop instability and elevated excitability after peripheral injury. Treatment of neuronal ICa loss at the level of injury of the dorsal root ganglion may provide a novel therapeutic pathway.
Neuropathic pain, sensory neurons, and calcium
Nerve injury is a dominant or contributing factor in a wide variety of painful conditions, including persistent pain following thoracic, breast, and amputation surgery, radiculopathy from disc disease, nerve invasion by cancer, trauma, metabolic injury from ischemia or diabetes, infectious conditions such as herpes zoster and AIDS, and complex regional pain syndrome (CRPS) following minor injury. Neuropathic pain is often disabling because of its intensity, lancinating quality, and resistance to currently available treatments.
The pathogenic mechanisms generating hypersensitivity and spontaneous pain following injury of a peripheral nerve are anatomically distributed and complex. Altered neural structure and function have been identified at the peripheral tissues, the dorsal horn of the spinal cord (1-6), and the brain (7). In addition, the primary afferent neuron itself is an important site of changes generating pain. Increased neuronal excitability is evident at the injury site and also in the somata of the injured neurons (8,9). Persistence of membrane instability in the dorsal root ganglion (DRG) neurons isolated from injured animals (10,11) demonstrates that aberrant excitability is intrinsic to the soma of primary afferents. Prolonged afterdischarge following stimulation (5,12) and altered patterns of provoked activity (13) further characterize injured sensory neurons.
Major aspects of neuronal function are regulated by Ca2+, including neurotransmitter release, excitability, neuron growth, differentiation, and death (14,15), as well as the development of plasticity and gene expression (16). A similarly important role for Ca2+ has been established in processes of signaling pain, especially in facilitated pain states. Also, voltage-gated Ca2+ channels (VGCC) modulate pain in clinical and experimental settings (17-24). Due to the importance of this disease condition and the relatively recent attention to membrane Ca2+ currents (ICa) in its pathogenesis, this paper reviews recent studies examining the effects of nerve injury on directly measured ICa and cytoplasmic Ca2+ levels ([Ca2+]c) in primary afferent neurons.
Neuropathic models and sensory testing
Examination of the pathogenesis of neuropathic pain has been accelerated by the introduction of rodent models of nerve injury that produce behavior indicative of spontaneous and inducible pain (25). A complete section of a nerve produces spontaneous pain but also an anesthetic limb. Partial injury retains a subset of afferent fibers and results in altered sensory function, including more intense pain during noxious stimulation (hyperalgesia) and pain after normally innocuous stimuli (allodynia). The first widely used model of incomplete peripheral nerve injury was chronic constriction injury (CCI, Figure 1), in which four ligatures of chromic gut suture produce axotomy (transection of the neuronal axon), ischemia, or inflammation (26-28). Within 10 days of injury, animals may demonstrate hyperalgesia and allodynia induced by mechanical and thermal stimuli (29). These amplified behavioral responses are mediated by a subgroup of surviving afferent fibers, as demonstrated by similar findings after incomplete sciatic transection (30).
Figure 1.

Neuropathic pain models involving peripheral nerve injury in the rat. (A) In chronic constriction injury (Bennett et al, 26), four chronic gut ligatures are placed around the distal sciatic nerve. They tighten and create inflammation with time, and additionally cause ischemia and axonal discontinuity. (B) The spinal nerve ligation model (Kim et al, 33) is produced by ligating and sectioning the fifth lumbar (L5) and L6 spinal nerves, so that the sciatic nerve is supplied only by the neurons of L4. This allows anatomic segregation of the axotomized (L5) and intact (L4) elements. Contributions to abnormal pain may arise from the axotomized neurons (a), or from degeneration of distal segments (b) that initiates inflammation and irritation of intact fibers (c).
After partial nerve injury, axotomized sensory neurons develop substantial phenotypic shifts, including altered membrane channels and receptors, and new sensitivities to chemical stimulation by catecholamines, cytokines, bradykinin, and neurotrophins at both the injury site and proximally in the DRG (10,31). Surviving intact fibers are also exposed to abnormal conditions, due to production of inflammatory mediators from degeneration of disconnected fibers of axotomized neurons sharing the same nerve fascicles (32). The spinal nerve ligation (SNL) mode (33), in which the fifth lumbar (L5) and L6 spinal nerve components of the sciatic nerve are ligated but L4 remains intact, permits separate evaluation of the anatomically distinct axotomized neurons of the L5 DRG and the neighboring L4 neurons (Figure 1).
As in human clinical conditions, animal subjects show variability in the sensory and behavioral consequences of incomplete nerve injury. It is therefore important to distinguish those animals that successfully develop the desired phenotype. Since pain is “an unpleasant sensory and emotional experience” (34), the best we can do in animal experimentation is to record behavior and infer the experience. When a pin is applied to the footpad of a rat with only enough pressure to indent but not puncture the skin, the response is either a brief reflex withdrawal or a hyperalgesic reaction characterized by sustained lifting, shaking, and licking of the paw (35). As the latter response occurs only after true SNL but not sham exposure of the nerve alone, and only on the side ipsilateral to the injury (Figure 2), this may be accepted as an indication of a neuropathic pain state. Other commonly used measures, such as threshold testing for withdrawal from low intensity mechanical stimulation with von Frey fibers or from thermal stimuli, are altered after sham surgery without nerve section and also contralateral to the nerve section (35). Accordingly, we have adopted the complex and sustained hyperalgesia response as an indicator of neuropathic pain.
Figure 2.

Measurement of mechanical hyperalgesia. Touch of a 22-gauge spinal needle to the plantar surface of the hind paw may elicit a prolonged lifting, shaking, and chewing of the paw. This is rare in control animals and those having sham surgery to expose but not cut the spinal nerves. However, after spinal nerve ligation (SNL), there is an ipsilateral (circles) increase in incidence of this behavior when tested 4, 11, and 18 days after baseline (BL). # – different from baseline; * – different from contralateral (squares). From Hogan et al (35), with permission.
ICa in injured sensory neurons
DRG neurons express a variety of VGCCs. High voltage activated (HVA) currents are present in a variety of subtypes (L, N, P/Q, R) that are distinguished by their voltage dependency, kinetics, and pharmacology. The specific roles of these subtypes in DRG cells are not fully established, but currents through N and possibly P/Q channels initiate neurotransmitter release. Low voltage activated (LVA) currents (36), or T currents, inactivate rapidly during sustained depolarization but close (deactivate) slowly after repolarization of the membrane. Because of these features, T-currents account for up to 50% of Ca2+ entry (37-40). DRG neurons show definite heterogeneity with respect to HVA Ca2+ channels. L-type currents contribute substantially to total ICa in small neurons, N-type current is present in all sizes of neurons, and non-L/non-N current (presumptive P/Q and R) is prominent in large (≥40µm) and medium neurons (41). DRG somata are particularly heterogeneous in their expression of T-currents, which are most evident in medium sized (30-40 μm) neurons (42-45).
The importance of ICa to the functioning of neurons makes it likely that altered ICa may contribute to functional abnormalities that accompany neuropathic pain. Also, analgesia from intrathecal administration of Ca2+-channel blockers (17-24) raises the possibility that the primary disorder includes overexpression of ICa. Our initial observations in neurons dissociated from hyperalgesic rats after CCI (46) show that peak ICa density is in fact diminished by injury (from 3.1 ± 0.3 to 2.2 ± 0.3 pS/pF in medium neurons, and from 3.9 ± 0.4 to 3.0 ± 0.4 pS/pF in large neurons) using standard patch clamp whole cell recording (47) (Figure 3). The medium-sized neuronal population includes cells that transmit both nociceptive and low threshold sensations, while the large neurons are specific for low threshold sensory modality.
Figure 3.

Inward high-voltage activated Ca2+ currents measured by patch-clamp recording in dissociated sensory neurons decrease after chronic constriction injury. (A) Sample currents elicited by square-wave voltage commands (bottom) are decreased in an injured neuron (middle) compared to a neuron from a control animal (top). (B) Current-voltage plot of average data for medium sized neurons shows a loss of peak current in injured neurons from animals with neuropathic pain. From Hogan et al (46), with permission.
The CCI model does not allow a distinction between direct effects of axotomy and indirect inflammatory mechanisms. Examination of ICa specifically in axotomized neurons (L5 after SNL) (48) shows that current loss is present in all neuronal size groups, including the nociceptive small diameter population. Current loss is also evident in the adjacent L4 neurons, but only in the large cell category. These recordings were performed with sustained square wave currents, which lack the kinetic complexity of an actual action potential (AP). Additional recordings of the neuronal current response to voltage commands in the form of an AP (Figure 4) show comparable findings, which assure the pathophysiologic validity of the ICa loss.
Figure 4.

Inward low-voltage activated Ca2+ currents measured by patch-clamp recording in dissociated sensory neurons decrease after chronic constriction injury. (A) Current-voltage plot of average data for medium sized-neurons shows a loss of peak current in injured neurons from animals with neuropathic pain, and particularly a loss of current at low voltages. (B) Presentation of a voltage command in the from of an action potential (top) produces current through low-voltage activated Ca2+ channels (bottom) that is substantially reduced in an injured neuron compared with a control neuron. From McCallum et al (49), with permission.
LVA channels that generate the T-type current have an undefined role in sensory neurons, but may be substantially diminished by injury (49). The peak T-type ICa, isolated by the elimination of other ICa with relevant toxins, is reduced by 60% after CCI, and total LVA Ca2+ influx is reduced by 80%. The mechanism is a depolarizing shift in the voltage dependence of activation and an increase in the rate of channel deactivation and inactivation.
Together, these findings indicate that nerve injury, particularly axotomy, results in a loss of ICa in primary sensory neurons that is present after different types of injury, affects the full range of neuron sizes, and includes both LVA and HVA current types. Although this was not our expected result, various other observations indicate a role of decreased ICa in generating pain. Norepinephrine, which produces pain when applied to injured nerves, reduces ICa and increases excitability of rat DRG somata injured by axotomy (50,51). Also, intrathecal Ca2+ administration is antinociceptive (52). Agents that induce pain, including bradykinin and capsaicin, inhibit DRG ICa (53-55), although this is not necessarily their principal algesic mechanism. Axonal injury in other systems (56,57) depresses ICa. Some of our findings have been confirmed (58), although these authors failed to identify a loss in LVA currents.
Biophysical response of intact neurons to injury
Although patch-clamp recording is ideal for evaluation of the performance of a specific membrane channel, dialysis of the cytoplasm by the pipette solution alters the natural internal conditions of the cell and blockade of other currents precludes the natural interactions that dictate the generation of APs. To determine neuronal function in a setting that less disrupts function, we used the intracellular microelectrode technique in intact DRGs, which further avoids disruption by cell dissociation and permits characterization of the neurons by conduction velocity (CV).
Pronounced electrophysiological changes were seen only in L5 neurons following SNL (59). Both Aα/β (fast CV) and Aδ (slow CV) myelinated neuron types show increased AP duration, and decreased afterhyperpolarization (AHP) amplitude and duration (Figure 5A). The AHP duration in neurons with C fibers (CV<1.5m/s) shortens after axotomy. In contrast to the axotomized L5 neurons, neighboring L4 neurons develop no changes in AP duration or AHP dimensions. These parameters are of particular importance in considering mechanisms of elevated pain sensitivity, as a prolonged AP duration may release more neurotransmitter at the first synapse in the spinal cord dorsal horn (60), while reduced AHP dimensions results in increased burst firing and elevated maximal firing rate (61,62).
Figure 5.

Action potential (AP) dimensions in control neurons and after spinal nerve ligation (SNL), by intracellular microelectrode recording from intact dorsal root ganglia. (A) Three sample recordings from control and from the fifth lumber (L5) ganglion after SNL show the loss of afterhyperpolarization and increase in AP duration after injury. CV – conduction velocity. Resting membrane potential is indicated by the dotted lines. (B) Average data for the incidence of repetitive firing during sustained depolarization of neurons in the control group (C), after sham injury (Sh), and in the L4 and L5 ganglia after SNL. Small numbers in the columns indicate number of neurons recorded. Brackets indicate significant differences by post hoc testing. From Sapunar et al (59), with permission.
After axotomy, both Aα/β low-threshold neurons and Aδ presumed nociceptive neurons show resting membrane potential depolarization and a decreased current threshold for AP initiation. Importantly, axotomized Aδ neurons develop increased repetitive firing during sustained depolarization after axotomy (Figure 5B), whereas Aα/β neurons do not. Thus, axotomized neurons, especially pain-conducting ones, develop instability and elevated excitability. Additionally, in the L5 ganglion after axotomy, a novel set of neurons (24% of total) have fast CVs characteristic of myelinated neurons, despite exhibiting long AP durations typical of slowly conducting C-type fibers (Figure 6). The histologic counterpart of these cells may have recently been identified by Hammond et al (63), who described the emergence exclusively in the L5 ganglion after SNL of a novel group of very small neurons that label with N52 antibody, which identifies myelinated neurons. These cells might thus exhibit the AP features of small neurons but have accelerated CV due to myelination. Overall, our findings indicate a clear pathogenic distinction between the substantially altered axotomized neurons and the less affected neighboring ones.
Figure 6.

Relationship of conduction velocity (CV) and action potential (AP) duration for sensory neurons recorded by intracellular microelectrode, in control neurons and fifth lumbar (L5) neurons after spinal nerve ligation (SNL). (A) Control neurons show an inverse relation between CV and AP duration. (B) After injury, there is general prolongation of AP duration, but also the appearance of a new population with very prolonged AP despite a CV characteristic of nociceptive C-type fibers that have CV less than 1.5m/s (vertical line). The horizontal line indicates AP duration of 2 ms. From Sapunar et al (59), with permission.
Functional consequence of decreased ICa
ICa is an inward current, so the direct effect of its loss should stabilize the membrane and shorten the AP. However, there are secondary effects through the operation of the Ca2+ admitted through the VGCCs upon Ca2+-sensitive K+ channels, which generate IK(Ca). These release K+ from the cell when the [Ca2+]c in their immediate vicinity cause them to enter the open state. In operation, they contribute to the repolarization to the AP and the generation of the AHP (64). The end result of ICa loss will thus be determined by the balance of the direct effect of decreased ICa and the indirect effect of lost stimulation of IK(Ca). We examined the effect of selective ICa blockers on dissociated neurons to determine the combined effect of these two processes (48). The form of a triggered AP was measured before and after ablation of ICa by combined application of toxins. Likewise, presentation of a voltage command in the form an AP was used to record the provoked currents with ICa intact and after ablation (Figure 7). We observed that ICa loss was associated with a prolongation of AP duration and loss of inward current during depolarization, but also loss of outward current during repolarization and the AHP. Thus, loss of ICa, as occurs with injury, results in a substantial decrease in outward current. Others have similarly observed a predominant effect of Ca2+ entry in producing outward current (65).
Figure 7.
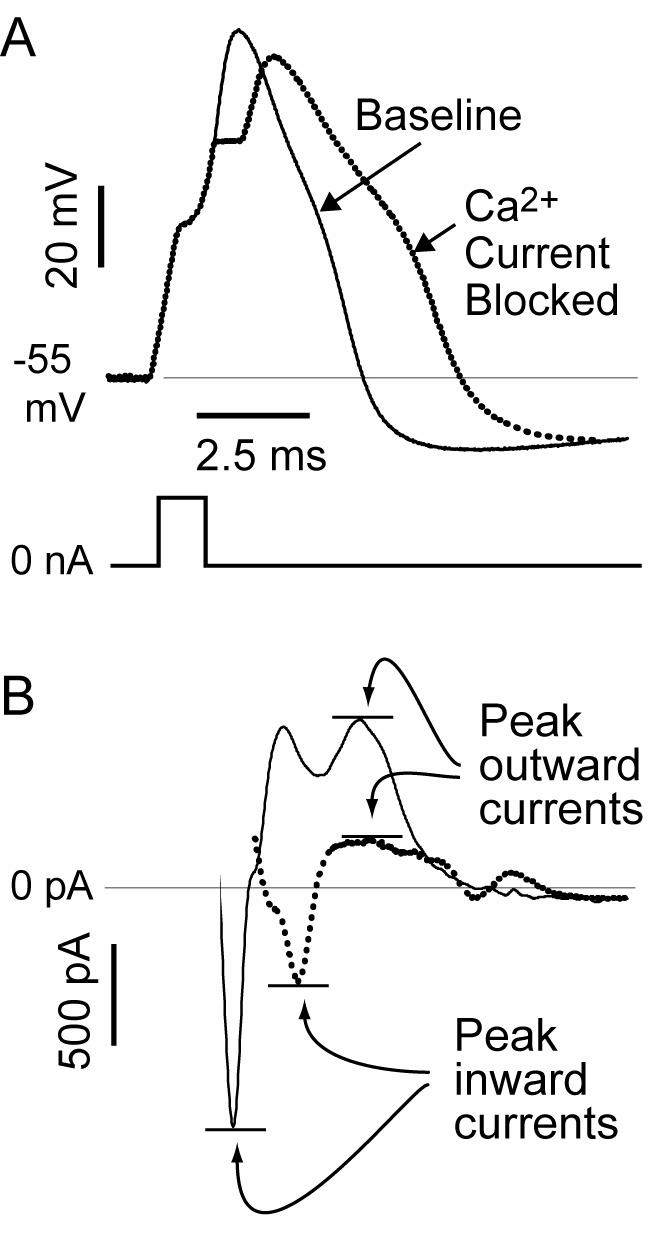
Consequence of loss on Ca2+ current on membrane function. (A) Recordings from a dissociated sensory neuron by the patch-clamp technique show the action potential (AP) voltage traces (top) produced by a current pulse (bottom) at baseline and after Ca2+ current is blocked by selective toxins. (B) Using the baseline AP trace as a voltage command (not shown), the maximal inward component of the total current is reduced, but the later outward current is also diminished, accounting for the prolongation of the AP and diminished afterhyperpolarization evident in the voltage traces. From McCallum et al (48), with permission.
Neuronal function is dictated by the complex interplay of the entire ensemble of membrane channels, which interact with each other through their effect on transmembrane voltage and on [Ca2+]c. Using non-dissociated intact DRGs to limit artifact, we therefore examined the role of ICa by manipulating conditions to limit or enhance ICa, during intracellular recording (66). Our preliminary results confirm that suppressing ICa with cadmium application in the superfusate, Ca2+ withdrawal, or intracellular EDTA delivery decreases AHP dimensions and increases repetitive firing during sustained depolarization (Figure 8). Selective VGCC subtype toxins similarly reduced AHP dimensions and also prolonged the AP duration in the neurons of intact ganglia. Reciprocally, elevating Ca2+ entry with high bath Ca2+ concentrations or application of a Ca2+ ionophore increases the AHP and suppresses repetitive firing. These findings clearly indicate the excitatory consequences of decreased ICa in sensory neurons and support the loss of ICa as a mechanism causing increased excitability after injury.
Figure 8.

Regulation of firing properties of sensory neurons by Ca2+ current. During intracellular recording from an intact dorsal root ganglion under baseline conditions (A) only a single action potential (AP) is triggered by injection of progressively larger depolarizing currents C, After lowering the Ca2+ concentration in the bath (B) with reciprocal elevation of Mg2+ concentration, multiple APs are initiated by identical current injection.
A model of the role of ICa loss in nerve injury
K(Ca) channels help regulate neuronal excitability by hyperpolarizing the membrane after the AP and by decreasing membrane resistance, making the membrane less excitable in response to depolarizing currents (67). In the normal state (Figure 9), Ca2+ entry through VGCCs during the AP stimulates IK(Ca) and assures a normal level of excitability. In most neurons, sustained depolarizations result in only a single AP. After injury, decreased ICa results in less IK(Ca) and therefore burst firing.
Figure 9.

Influence of injury on neuronal excitability. In the normal condition, a depolarizing stimulus opens voltage-gated Ca2+ channels. The resulting of Ca2+ activates Ca2+-sensitive K+ channels, which regulate the afterhyperpolarization (AHP) of the action potential. By controlling the membrane potential and resistance, the AHP in turn regulates repetitive firing. After injury, decreased Ca2+ influx causes a diminished outward K+ current and less of AHP, such that the same stimulus results in repetitive firing.
The frequency-encoded sensory signal is filtered at points of impedance mismatch along the axon, especially at the site in the DRG where the afferent neuron splits into the dorsal root fiber and the T-branch that leads to the neuronal soma (68,69). Since the ability of a neuron to conduct repetitive spikes through this particular site is regulated by Ca2+-sensitive processes (70), we hypothesized that injury and the loss of ICa would modulate spike conduction failure. We found (71) that axotomy causes low threshold neurons, identified by their lack of an inflection on the AP, to develop a prolonged refractory period during paired spike stimulation and a decreased maximal following frequency of tetanic bursts. In contrast, axotomy of nociceptive neurons limited AHP amplification during an impulse train and increased the frequency at which these neurons can fire repetitively during tetanic stimulation. As a result, axotomy increases the ability of putative nociceptors to conduct high frequency trains of APs, whereas injury increases filtering of non-nociceptive afferent sensory traffic. This is important since high frequency bursts are transmitted with increased synaptic reliability while tonic discharge with the same average rate of firing may not successfully induce activity in the postsynaptic neuron (72). Also, burst discharge is particularly effective in producing dorsal horn neuronal plasticity (73), which may play a critical role supporting chronic pain states (74,75).
A direct of effect of injury upon Ca2+-activated K+ channels
Altered function of other ionic membrane channels contributes to the disordered membrane biophysics observed after nerve injury, including substantial changes in voltage-gated Na+ and K+ channels (76,77). It is therefore possible that the post-injury shift in membrane function includes alteration of K(Ca), not just reduced Ca2+ stimulation of the channels. We tested this by maximally stimulating dissociated neurons during patch-clamp recording with high intracellular [Ca2+]c and identifying currents sensitive to selective IK(Ca) blockers (78). This revealed IK(Ca) with components sensitive to apamin, clotrimazole, and iberiotoxin, indicative of SK, IK, and BK subtypes of IK(Ca). SNL decreases total IK(Ca) in axotomized (L5) neurons, but increases total IK(Ca) in adjacent (L4) DRG neurons. All IK(Ca) subtypes are decreased by axotomy, but iberiotoxin-sensitive and clotrimazole-sensitive current densities are increased in adjacent L4 neurons after SNL. Thus, the injury has divergent effects on axotomized neurons and adjacent intact neurons, and direct effects on the K(Ca) channel amplify the action of ICa loss.
Effect of injury on intracellular Ca2+ signaling
The major signal downstream from AP-induced inward Ca2+ flux is the critical second messenger [Ca2+]c (79,80). AP trains trigger an elevation of [Ca2+]c (the Ca2+ transient) in the primary afferent neuron that persists seconds to minutes after the membrane activity (81), and thereby provides an integrative and memory process very early in the anatomic pathway of somatic sensory signaling. Virtually all aspects of neuronal function are controlled by the Ca2+ regulatory pathway, including synaptic transmission, enzymatic activity, membrane currents, cellular energetics, gene expression, cell differentiation, and death (16,82). All these events have been implicated in the reaction to nerve trauma but the effect of peripheral nerve injury on intracellular Ca2+ regulation has not been previously examined.
Intracellular Ca2+ level is controlled by balanced interactions of Ca2+ storage, release, and extrusion. Most of the Ca2+ that enters through VGCC or receptor-operated channels is buffered in the cytoplasm. Free Ca2+ in peripheral sensory neurons is tightly regulated to approximately 100 nM, or about 20 000-fold less than the extracellular concentration. However, after SNL injury (Figure 10), axotomized neurons show a depressed resting level for both small nociceptive and large non-nociceptive categories (83). Only non-nociceptive neurons develop a depressed resting [Ca2+]c in the adjacent L4 population. Decreased [Ca2+]c may precipitate cell loss, including programmed cell death by apoptosis (84-86), as has been noted after axotomy (87,88). Resting [Ca2+]c also regulates receptor-triggered calcium signaling (89-91).
Figure 10.

Injury reduces resting level of cytoplasmic Ca2+ ([Ca2+]c). Spinal nerve ligation injury reduces [Ca2+]c in axotomized fifth lumbar (L5) neurons of both sizes, but only in large neurons from the adjacent L4 population. Small numbers in the bars indicate number of neurons. From Fuchs et al (83), with permission.
A complex system of Ca2+ sequestration, release, and extrusion regulates [Ca2+]c and shapes the Ca2+ transient that follows neuronal activation. Ca2+ is pumped out of the cell by plasma membrane Ca2+-ATPases (PMCAs) and is sequestered into endoplasmic reticulum (ER) stores by sarcoplasmic-endoplasmic reticulum Ca2+-ATPase (SERCA) channels. The ER also functions as a source that releases Ca2+ into the cytoplasm through channels sensitive to inositol 1,4,5-triphosphate (IP3) and others sensitive to the plant alkaloid ryanodine, thereby known as ryanodine receptors (RyRs). Cytoplasmic accumulation of Ca2+ activates RyRs as well as IP3 channels, accounting for the phenomenon of Ca2+-induced Ca2+ release (CICR). Depletion of the ER Ca2+ pool activates a voltage-independent plasma membrane channel (store-operated Ca2+ channel, SOCC) that conducts an inward Ca2+ flux, a process termed capacitative Ca2+ entry (CCE), which serves to refill Ca2+ stores (92). Our initial data (93) demonstrates that the activity-induced Ca2+ transient is markedly altered in injured neurons (Figure 11). Specifically, after axotomy, the transient duration and amplitude in nociceptive neurons are significantly diminished, whereas amplitude is increased in axotomized non-nociceptors. In the adjacent L4 neurons, only the transient amplitude is diminished only in nociceptors. These effects are likely attributable to combined effects of decreased Ca2+ load entering the neurons and injury-related disruption of the processes regulating [Ca2+]c.
Figure 11.

Schematic traces of Ca2+ transients indicated by fluorescence ratio of Fura-2 (R340/380), summarizing injury-induced alterations. The bottom two panels show traces for axotomized neurons from the 5th lumbar (L5) ganglion after spinal nerve ligation, while the upper two panels show traces for neighboring neurons from the L4 ganglion. For both, the left panels show traces from large and capsaicin-insensitive neurons that convey non-nociceptive sensory information, while the right panels show traces from small and capsaicin-sensitive nociceptive neurons. In each case, the dotted line represents traces from control neurons and the solid line represents traces from neurons after injury.
What causes pain after peripheral nerve injury?
While the processes that produce hyperalgesia after nerve injury are diverse, Figure 12 attempts to explain the potential contributions that follow loss of ICa in primary sensory neurons. Increased burst firing from decreased AHP inflicts greater nociceptive traffic on the secondary neurons of the dorsal horn, where prolonged AP duration results in greater excitatory neurotransmitter release. Although axotomized neurons (L5 ganglion after SNL) are disconnected from sensory fields, they are nonetheless activated through direct mechanical stimulation during movement, depolarization by circulating and local algogens and inflammatory mediators, and sympathetic activity (8,10,31,94,95). Furthermore, the process of cross-excitation spreads activity among adjacent neurons in the DRG (96). This is critical since most clinical nerve injuries are partial. Afferent traffic from these sources is effectively amplified by the reduced signal filtering at the T-branch of injured neurons. The intense bursts of activity along this L5 pathway sensitize the spinal dorsal horn to input along intact (eg, SNL L4) pathways (1,73), so that natural stimuli are perceived as more intense. Other processes that do not involve reduced ICa in the axotomized neurons include irritation of intact fibers by inflammation induced by adjacent degenerating neuron segments distal to the axotomy, altered channel and receptor expression in the intact fibers, and anatomic changes in connectivity in the dorsal horn.
Figure 12.

Schematic diagram depicting the contribution of reduced sensory neuron Ca2+ current (ICa) to neuropathic pain. Injured nerve tissue distal to the spinal nerve ligation (SNL) undergoes Wallerian degeneration (dotted line), while neurons from the fifth lumbar (L5) dorsal root ganglion (DRG) are activated by movement, catecholamines, other algogenic agents and cross-excitation from adjacent intact neurons. Reduced ICa shortens afterhyperpolarizations (AHPs), which contributes to burst firing. Loss of ICa impairs natural signal filtering at the T-branch, where the stem axon splits into spinal nerve and dorsal root branches. Reduced ICa also prolongs action potential (AP) duration, which may increase excitatory synaptic transmission in the dorsal horn (DH). Spared nerves transmit activity evoked by stimulation in the receptive field, and these signals encounter DH neurons that are sensitized by L5 input. From McCallum et al (48), with permission.
Relief of pain in animal models by intrathecal administration of ICa blockers (18,97) may appear to contradict our model and findings. However, the function of Ca2+ signaling in different tissues is distinct, and this analgesia is explained by a blockade of neurotransmitter release in the spinal cord, which is not applicable at peripheral sites. VGCC blockers applied at the SNL injury site have no analgesic effect (97), although topical ICa blockers do decrease pain behavior in other models (98) that have a large inflammatory component. Interpretation of gene knockout studies (99-101) is severely limited by the simultaneous effects of current loss at multiple sites that have divergent roles for ICa.
The DRG is an undeveloped site for chronic pain treatment
There is a great need for more effective treatment of neuropathic pain that follows peripheral nerve injury. Although critical changes leading to chronic pain reside in the DRG, no treatments for chronic pain after peripheral nerve injury have been devised that target the DRG. Drugs that elevate [Ca2+]c, for instance Ca2+ ionophores, might be administered directly to the DRG, optimizing drug potency at the effector site while minimizing undesirable CNS and systemic effects. Further, stable genetic transfer via viral vectors may allow regulated expression of VGCC. Clinical methods for administering drugs directly to the selected DRGs are well established (102). Therefore, it can be hoped that better understanding of the peripheral pathophysiology of neuropathic pain might ultimately translate into pain therapy through targeted delivery of drugs or genes to selected DRGs.
Acknowledgment
This work was supported in part by grant No. NS-42150 from the National Institutes of Health, Bethesda, Maryland, USA.
References
- 1.Coderre TJ, Katz J, Vaccarino AL, Melzack R. Contribution of central neuroplasticity to pathological pain: review of clinical and experimental evidence. Pain. 1993;52:259–85. doi: 10.1016/0304-3959(93)90161-H. [DOI] [PubMed] [Google Scholar]
- 2.Tal M, Bennett GJ. Extra-territorial pain in rats with a peripheral mononeuropathy: mechano-hyperalgesia and mechano-allodynia in the territory of an uninjured nerve. Pain. 1994;57:375–82. doi: 10.1016/0304-3959(94)90013-2. [DOI] [PubMed] [Google Scholar]
- 3.Sugimoto T, Bennett GJ, Kajander KC. Transsynaptic degeneration in the superficial dorsal horn after sciatic nerve injury: effects of a chronic constriction injury, transection, and strychnine. Pain. 1990;42:205–13. doi: 10.1016/0304-3959(90)91164-E. [DOI] [PubMed] [Google Scholar]
- 4.Mao J, Price DD, Coghill RC, Mayer DJ, Hayes RL. Spatial patterns of spinal cord [14C]-2-deoxyglucose metabolic activity in a rat model of painful peripheral mononeuropathy. Pain. 1992;50:89–100. doi: 10.1016/0304-3959(92)90116-S. [DOI] [PubMed] [Google Scholar]
- 5.Laird JM, Bennett GJ. An electrophysiological study of dorsal horn neurons in the spinal cord of rats with an experimental peripheral neuropathy. J Neurophysiol. 1993;69:2072–85. doi: 10.1152/jn.1993.69.6.2072. [DOI] [PubMed] [Google Scholar]
- 6.Mao J, Mayer DJ, Hayes RL, Price DD. Spatial patterns of increased spinal cord membrane-bound protein kinase C and their relation to increases in 14C-2-deoxyglucose metabolic activity in rats with painful peripheral mononeuropathy. J Neurophysiol. 1993;70:470–81. doi: 10.1152/jn.1993.70.2.470. [DOI] [PubMed] [Google Scholar]
- 7.Ignatowski TA, Covey WC, Knight PR, Severin CM, Nickola TJ, Spengler RN. Brain-derived TNFalpha mediates neuropathic pain. Brain Res. 1999;841:70–7. doi: 10.1016/s0006-8993(99)01782-5. [DOI] [PubMed] [Google Scholar]
- 8.Wall PD, Devor M. Sensory afferent impulses originate from dorsal root ganglia as well as from the periphery in normal and nerve injured rats. Pain. 1983;17:321–39. doi: 10.1016/0304-3959(83)90164-1. [DOI] [PubMed] [Google Scholar]
- 9.Zhang JM, Song XJ, LaMotte RH. An in vitro study of ectopic discharge generation and adrenergic sensitivity in the intact, nerve-injured rat dorsal root ganglion. Pain. 1997;72:51–7. doi: 10.1016/s0304-3959(97)00013-4. [DOI] [PubMed] [Google Scholar]
- 10.Petersen M, Zhang J, Zhang JM, LaMotte RH. Abnormal spontaneous activity and responses to norepinephrine in dissociated dorsal root ganglion cells after chronic nerve constriction. Pain. 1996;67:391–7. doi: 10.1016/0304-3959(96)03146-6. [DOI] [PubMed] [Google Scholar]
- 11.Study RE, Kral MG. Spontaneous action potential activity in isolated dorsal root ganglion neurons from rats with a painful neuropathy. Pain. 1996;65:235–42. doi: 10.1016/0304-3959(95)00216-2. [DOI] [PubMed] [Google Scholar]
- 12.Babbedge RC, Soper AJ, Gentry CT, Hood VC, Campbell EA, Urban L. In vitro characterization of a peripheral afferent pathway of the rat after chronic sciatic nerve section. J Neurophysiol. 1996;76:3169–77. doi: 10.1152/jn.1996.76.5.3169. [DOI] [PubMed] [Google Scholar]
- 13.Na HS, Leem JW, Chung JM. Abnormalities of mechanoreceptors in a rat model of neuropathic pain: possible involvement in mediating mechanical allodynia. J Neurophysiol. 1993;70:522–8. doi: 10.1152/jn.1993.70.2.522. [DOI] [PubMed] [Google Scholar]
- 14.Anglister L, Farber IC, Shahar A, Grinvald A. Localization of voltage-sensitive calcium channels along developing neurites: their possible role in regulating neurite elongation. Dev Biol. 1982;94:351–65. doi: 10.1016/0012-1606(82)90353-0. [DOI] [PubMed] [Google Scholar]
- 15.Ekstrom PA. Neurones and glial cells of the mouse sciatic nerve undergo apoptosis after injury in vivo and in vitro. Neuroreport. 1995;6:1029–32. doi: 10.1097/00001756-199505090-00020. [DOI] [PubMed] [Google Scholar]
- 16.Ghosh A, Greenberg ME. Calcium signaling in neurons: molecular mechanisms and cellular consequences. Science. 1995;268:239–47. doi: 10.1126/science.7716515. [DOI] [PubMed] [Google Scholar]
- 17.Diaz A, Dickenson AH. Blockade of spinal N- and P-type, but not L-type, calcium channels inhibits the excitability of rat dorsal horn neurones produced by subcutaneous formalin inflammation. Pain. 1997;69:93–100. doi: 10.1016/s0304-3959(96)03271-x. [DOI] [PubMed] [Google Scholar]
- 18.Malmberg AB, Yaksh TL. Voltage-sensitive calcium channels in spinal nociceptive processing: blockade of N- and P-type channels inhibits formalin-induced nociception. J Neurosci. 1994;14:4882–90. doi: 10.1523/JNEUROSCI.14-08-04882.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Malmberg AB, Yaksh TL. Effect of continuous intrathecal infusion of omega-conopeptides, N-type calcium-channel blockers, on behavior and antinociception in the formalin and hot-plate tests in rats. Pain. 1995;60:83–90. doi: 10.1016/0304-3959(94)00094-U. [DOI] [PubMed] [Google Scholar]
- 20.Miranda HF, Bustamante D, Kramer V, Pelissier T, Saavedra H, Paeile C, et al. Antinociceptive effects of Ca2+ channel blockers. Eur J Pharmacol. 1992;217:137–41. doi: 10.1016/0014-2999(92)90833-p. [DOI] [PubMed] [Google Scholar]
- 21.Wong CH, Dey P, Yarmush J, Wu WH, Zbuzek VK. Nifedipine-induced analgesia after epidural injection in rats. Anesth Analg. 1994;79:303–6. doi: 10.1213/00000539-199408000-00018. [DOI] [PubMed] [Google Scholar]
- 22.Staats PS, Yearwood T, Charapata SG, Presley RW, Wallace MS, Byas-Smith M, et al. Intrathecal ziconotide in the treatment of refractory pain in patients with cancer or AIDS: a randomized controlled trial. JAMA. 2004;291:63–70. doi: 10.1001/jama.291.1.63. [DOI] [PubMed] [Google Scholar]
- 23.Atanassoff PG, Hartmannsgruber MW, Thrasher J, Wermeling D, Longton W, Gaeta R, et al. Ziconotide, a new N-type calcium channel blocker, administered intrathecally for acute postoperative pain. Reg Anesth Pain Med. 2000;25:274–8. doi: 10.1016/s1098-7339(00)90010-5. [DOI] [PubMed] [Google Scholar]
- 24.Wermeling D, Drass M, Ellis D, Mayo M, McGuire D, O'Connell DJ, et al. Pharmacokinetics and pharmacodynamics of intrathecal ziconotide in chronic pain patients. J Clin Pharmacol. 2003;43:624–36. [PubMed] [Google Scholar]
- 25.Hogan Q. Animal pain models. Reg Anesth Pain Med. 2002;27:385–401. doi: 10.1053/rapm.2002.33630. [DOI] [PubMed] [Google Scholar]
- 26.Bennett GJ, Xie YK. A peripheral mononeuropathy in rat that produces disorders of pain sensation like those seen in man. Pain. 1988;33:87–107. doi: 10.1016/0304-3959(88)90209-6. [DOI] [PubMed] [Google Scholar]
- 27.Maves TJ, Pechman PS, Gebhart GF, Meller ST. Possible chemical contribution from chromic gut sutures produces disorders of pain sensation like those seen in man. Pain. 1993;54:57–69. doi: 10.1016/0304-3959(93)90100-4. [DOI] [PubMed] [Google Scholar]
- 28.Myers RR, Yamamoto T, Yaksh TL, Powell HC. The role of focal nerve ischemia and Wallerian degeneration in peripheral nerve injury producing hyperesthesia. Anesthesiology. 1993;78:308–16. doi: 10.1097/00000542-199302000-00015. [DOI] [PubMed] [Google Scholar]
- 29.Attal N, Jazat F, Kayser V, Guilbaud G. Further evidence for 'pain-related' behaviours in a model of unilateral peripheral mononeuropathy. Pain. 1990;41:235–51. doi: 10.1016/0304-3959(90)90022-6. [DOI] [PubMed] [Google Scholar]
- 30.Seltzer Z, Dubner R, Shir Y. A novel behavioral model of neuropathic pain disorders produced in rats by partial sciatic nerve injury. Pain. 1990;43:205–18. doi: 10.1016/0304-3959(90)91074-S. [DOI] [PubMed] [Google Scholar]
- 31.Petersen M, Eckert AS, Segond von Banchet G, Heppelmann B, Klusch A, Kniffki KD. Plasticity in the expression of bradykinin binding sites in sensory neurons after mechanical nerve injury. Neuroscience. 1998;83:949–59. doi: 10.1016/s0306-4522(97)00465-x. [DOI] [PubMed] [Google Scholar]
- 32.DeLeo JA, Yezierski RP. The role of neuroinflammation and neuroimmune activation in persistent pain. Pain. 2001;90:1–6. doi: 10.1016/s0304-3959(00)00490-5. [DOI] [PubMed] [Google Scholar]
- 33.Kim SH, Chung JM. An experimental model for peripheral neuropathy produced by segmental spinal nerve ligation in the rat. Pain. 1992;50:355–63. doi: 10.1016/0304-3959(92)90041-9. [DOI] [PubMed] [Google Scholar]
- 34.Pain terms: a list with definitions and notes on usage. Recommended by the IASP Subcommittee on Taxonomy. Pain. 1986;6:247–52. [PubMed] [Google Scholar]
- 35.Hogan Q, Sapunar D, Modric-Jednacak K, McCallum JB. Detection of neuropathic pain in a rat model of peripheral nerve injury. Anesthesiology. 2004;101:476–87. doi: 10.1097/00000542-200408000-00030. [DOI] [PubMed] [Google Scholar]
- 36.Randall AD, Tsien RW. Contrasting biophysical and pharmacological properties of T-type and R-type calcium channels. Neuropharmacology. 1997;36:879–93. doi: 10.1016/s0028-3908(97)00086-5. [DOI] [PubMed] [Google Scholar]
- 37.White G, Lovinger DM, Weight FF. Transient low-threshold Ca2+ current triggers burst firing through an afterdepolarizing potential in an adult mammalian neuron. Proc Natl Acad Sci U S A. 1989;86:6802–6. doi: 10.1073/pnas.86.17.6802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lovinger DM, White G. Post-natal development of burst firing behavior and the low-threshold transient calcium current examined using freshly isolated neurons from rat dorsal root ganglia. Neurosci Lett. 1989;102:50–7. doi: 10.1016/0304-3940(89)90306-6. [DOI] [PubMed] [Google Scholar]
- 39.Huguenard JR. Low-threshold calcium currents in central nervous system neurons. Annu Rev Physiol. 1996;58:329–48. doi: 10.1146/annurev.ph.58.030196.001553. [DOI] [PubMed] [Google Scholar]
- 40.Lambert RC, McKenna F, Maulet Y, Talley EM, Bayliss DA, Cribbs LL, et al. Low-voltage-activated Ca2+ currents are generated by members of the CavT subunit family (alpha1G/H) in rat primary sensory neurons. J Neurosci. 1998;18:8605–13. doi: 10.1523/JNEUROSCI.18-21-08605.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Scroggs RS, Fox AP. Calcium current variation between acutely isolated adult rat dorsal root ganglion neurons of different size. J Physiol. 1992;445:639–58. doi: 10.1113/jphysiol.1992.sp018944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schroeder JE, Fischbach PS, McCleskey EW. T-type calcium channels: heterogeneous expression in rat sensory neurons and selective modulation by phorbol esters. J Neurosci. 1990;10:947–51. doi: 10.1523/JNEUROSCI.10-03-00947.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cribbs LL, Lee JH, Yang J, Satin J, Zhang Y, Daud A, et al. Cloning and characterization of alpha1H from human heart, a member of the T-type Ca2+ channel gene family. Circ Res. 1998;83:103–9. doi: 10.1161/01.res.83.1.103. [DOI] [PubMed] [Google Scholar]
- 44.Perez-Reyes E, Cribbs LL, Daud A, Lacerda AE, Barclay J, Williamson MP, et al. Molecular characterization of a neuronal low-voltage-activated T-type calcium channel. Nature. 1998;391:896–900. doi: 10.1038/36110. [DOI] [PubMed] [Google Scholar]
- 45.Talley EM, Cribbs LL, Lee JH, Daud A, Perez-Reyes E, Bayliss DA. Differential distribution of three members of a gene family encoding low voltage-activated (T-type) calcium channels. J Neurosci. 1999;19:1895–911. doi: 10.1523/JNEUROSCI.19-06-01895.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hogan QH, McCallum JB, Sarantopoulos C, Aason M, Mynlieff M, Kwok WM, et al. Painful neuropathy decreases membrane calcium current in mammalian primary afferent neurons. Pain. 2000;86:43–53. doi: 10.1016/s0304-3959(99)00313-9. [DOI] [PubMed] [Google Scholar]
- 47.Hamill OP, Marty A, Neher E, Sakmann B, Sigworth FJ. Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflugers Arch. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- 48.McCallum JB, Kwok WM, Sapunar D, Fuchs A, Hogan QH. Painful peripheral nerve injury decreases calcium current in axotomized sensory neurons. Anesthesiology. 2006;105:160–8. doi: 10.1097/00000542-200607000-00026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.McCallum JB, Kwok WM, Mynlieff M, Bosnjak ZJ, Hogan QH. Loss of T-type calcium current in sensory neurons of rats with neuropathic pain. Anesthesiology. 2003;98:209–16. doi: 10.1097/00000542-200301000-00032. [DOI] [PubMed] [Google Scholar]
- 50.Abdulla FA, Smith PA. Ectopic alpha2-adrenoceptors couple to N-type Ca2+ channels in axotomized rat sensory neurons. J Neurosci. 1997;17:1633–41. doi: 10.1523/JNEUROSCI.17-05-01633.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Holz GG, IV, Rane SG, Dunlap K. GTP-binding proteins mediate transmitter inhibition of voltage-dependent calcium channels. Nature. 1986;319:670–2. doi: 10.1038/319670a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Welch SP, Stevens DL, Dewey WL. A proposed mechanism of action for the antinociceptive effect of intrathecally administered calcium in the mouse. J Pharmacol Exp Ther. 1992;260:117–27. [PubMed] [Google Scholar]
- 53.Bleakman D, Brorson JR, Miller RJ. The effect of capsaicin on voltage-gated calcium currents and calcium signals in cultured dorsal root ganglion cells. Br J Pharmacol. 1990;101:423–31. doi: 10.1111/j.1476-5381.1990.tb12725.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bleakman D, Thayer SA, Glaum SR, Miller RJ. Bradykinin-induced modulation of calcium signals in rat dorsal root ganglion neurons in vitro. Mol Pharmacol. 1990;38:785–96. [PubMed] [Google Scholar]
- 55.Kusano K, Gainer H. Modulation of voltage-activated Ca currents by pain-inducing agents in a dorsal root ganglion neuronal line, F-11. J Neurosci Res. 1993;34:158–69. doi: 10.1002/jnr.490340203. [DOI] [PubMed] [Google Scholar]
- 56.Jassar BS, Pennefather PS, Smith PA. Changes in sodium and calcium channel activity following axotomy of B-cells in bullfrog sympathetic ganglion. J Physiol. 1993;472:203–31. doi: 10.1113/jphysiol.1993.sp019943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Laiwand R, Werman R, Yarom Y. Electrophysiology of degenerating neurones in the vagal motor nucleus of the guinea-pig following axotomy. J Physiol. 1988;404:749–66. doi: 10.1113/jphysiol.1988.sp017317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Baccei ML, Kocsis JD. Voltage-gated calcium currents in axotomized adult rat cutaneous afferent neurons. J Neurophysiol. 2000;83:2227–38. doi: 10.1152/jn.2000.83.4.2227. [DOI] [PubMed] [Google Scholar]
- 59.Sapunar D, Ljubkovic M, Lirk P, McCallum JB, Hogan QH. Distinct membrane effects of spinal nerve ligation on injured and adjacent dorsal root ganglion neurons in rats. Anesthesiology. 2005;103:360–76. doi: 10.1097/00000542-200508000-00020. [DOI] [PubMed] [Google Scholar]
- 60.Sabatini BL, Regehr WG. Control of neurotransmitter release by presynaptic waveform at the granule cell to Purkinje cell synapse. J Neurosci. 1997;17:3425–35. doi: 10.1523/JNEUROSCI.17-10-03425.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Amir R, Devor M. Spike-evoked suppression and burst patterning in dorsal root ganglion neurons of the rat. J Physiol. 1997;501:183–96. doi: 10.1111/j.1469-7793.1997.183bo.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Waddell PJ, Lawson SN. Electrophysiological properties of subpopulations of rat dorsal root ganglion neurons in vitro. Neuroscience. 1990;36:811–22. doi: 10.1016/0306-4522(90)90024-x. [DOI] [PubMed] [Google Scholar]
- 63.Hammond DL, Ackerman L, Holdsworth R, Elzey B. Effects of spinal nerve ligation on immunohistochemically identified neurons in the L4 and L5 dorsal root ganglia of the rat. J Comp Neurol. 2004;475:575–89. doi: 10.1002/cne.20209. [DOI] [PubMed] [Google Scholar]
- 64.Sah P. Ca(2+)-activated K+ currents in neurones: types, physiological roles and modulation. Trends Neurosci. 1996;19:150–4. doi: 10.1016/s0166-2236(96)80026-9. [DOI] [PubMed] [Google Scholar]
- 65.Blair NT, Bean BP. Roles of tetrodotoxin (TTX)-sensitive Na+ current, TTX-resistant Na+ current, and Ca2+ current in the action potentials of nociceptive sensory neurons. J Neurosci. 2002;22:10277–90. doi: 10.1523/JNEUROSCI.22-23-10277.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hogan Q, McCallum JB, Seagard J. Blockade of Ca2+ current excites dorsal root ganglion (DRG) neurons. Abstr Soc Neurosci. Available from: http://sfn.scholarone.com/itin2000/main.html?new_page_id=76&abstract_id=18774&is_tech=0 Accessed: February 1, 2007.
- 67.Engel J, Schultens HA, Schild D. Small conductance potassium channels cause an activity-dependent spike frequency adaptation and make the transfer function of neurons logarithmic. Biophys J. 1999;76:1310–9. doi: 10.1016/S0006-3495(99)77293-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Stoney SD., Jr Limitations on impulse conduction at the branch point of afferent axons in frog dorsal root ganglion. Exp Brain Res. 1990;80:512–24. doi: 10.1007/BF00227992. [DOI] [PubMed] [Google Scholar]
- 69.Luscher C, Streit J, Quadroni R, Luscher HR. Action potential propagation through embryonic dorsal root ganglion cells in culture. I. Influence of the cell morphology on propagation properties. J Neurophysiol. 1994;72:622–33. doi: 10.1152/jn.1994.72.2.622. [DOI] [PubMed] [Google Scholar]
- 70.Luscher C, Lipp P, Luscher HR, Niggli E. Control of action potential propagation by intracellular Ca2+ in cultured rat dorsal root ganglion cells. J Physiol. 1996;490:319–24. doi: 10.1113/jphysiol.1996.sp021146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lirk P, Sapunar D, McCallum JB, Hogan WH. Effects of spinal nerve ligation upon repetitive firing behavior. Abstr Soc Neurosci. Available from: http://sfn.scholarone.com/itin2004/main.html?new_page_id=126&abstract_id=16731&p_num=748.8&is_tech=0. Accessed: February 1, 2007.
- 72.Krahe R, Gabbiani F. Burst firing in sensory systems. Nat Rev Neurosci. 2004;5:13–23. doi: 10.1038/nrn1296. [DOI] [PubMed] [Google Scholar]
- 73.Lisman JE. Bursts as a unit of neural information: making unreliable synapses reliable. Trends Neurosci. 1997;20:38–43. doi: 10.1016/S0166-2236(96)10070-9. [DOI] [PubMed] [Google Scholar]
- 74.Galan A, Laird JM, Cervero F. In vivo recruitment by painful stimuli of AMPA receptor subunits to the plasma membrane of spinal cord neurons. Pain. 2004;112:315–23. doi: 10.1016/j.pain.2004.09.011. [DOI] [PubMed] [Google Scholar]
- 75.Fang L, Wu J, Lin Q, Willis WD. Calcium-calmodulin-dependent protein kinase II contributes to spinal cord central sensitization. J Neurosci. 2002;22:4196–204. doi: 10.1523/JNEUROSCI.22-10-04196.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Waxman SG, Dib-Hajj S, Cummins TR, Black JA. Sodium channels and pain. Proc Natl Acad Sci U S A. 1999;96:7635–9. doi: 10.1073/pnas.96.14.7635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Everill B, Kocsis JD. Reduction in potassium currents in identified cutaneous afferent dorsal root ganglion neurons after axotomy. J Neurophysiol. 1999;82:700–8. doi: 10.1152/jn.1999.82.2.700. [DOI] [PubMed] [Google Scholar]
- 78.Sarantopoulos CJ, Kwok WM, Hogan QH. Calcium-activated potassium currents in mammalian primary afferent neurons: the effect of neuropathic injury after spinal nerve ligation (SNL). Society for Neuroscience Abstracts. Available from: http://sfn.scholarone.com/itin2005/main.html?new_page_id=126&abstract_id=20269&p_num=513.9&is_tech=0 Accessed: February 1, 2007.
- 79.Berridge MJ. Neuronal calcium signaling. Neuron. 1998;21:13–26. doi: 10.1016/s0896-6273(00)80510-3. [DOI] [PubMed] [Google Scholar]
- 80.Verkhratsky AJ, Petersen OH. Neuronal calcium stores. Cell Calcium. 1998;24:333–43. doi: 10.1016/s0143-4160(98)90057-4. [DOI] [PubMed] [Google Scholar]
- 81.Werth JL, Usachev YM, Thayer SA. Modulation of calcium efflux from cultured rat dorsal root ganglion neurons. J Neurosci. 1996;16:1008–15. doi: 10.1523/JNEUROSCI.16-03-01008.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Paschen W. Dependence of vital cell function on endoplasmic reticulum calcium levels: implications for the mechanisms underlying neuronal cell injury in different pathological states. Cell Calcium. 2001;29:1–11. doi: 10.1054/ceca.2000.0162. [DOI] [PubMed] [Google Scholar]
- 83.Fuchs A, Lirk P, Stucky C, Abram SE, Hogan QH. Painful nerve injury decreases resting cytosolic calcium concentrations in sensory neurons of rats. Anesthesiology. 2005;102:1217–25. doi: 10.1097/00000542-200506000-00023. [DOI] [PubMed] [Google Scholar]
- 84.Galli C, Meucci O, Scorziello A, Werge TM, Calissano P, Schettini G. Apoptosis in cerebellar granule cells is blocked by high KCl, forskolin, and IGF-1 through distinct mechanisms of action: the involvement of intracellular calcium and RNA synthesis. J Neurosci. 1995;15:1172–9. doi: 10.1523/JNEUROSCI.15-02-01172.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Collins F, Schmidt MF, Guthrie PB, Kater SB. Sustained increase in intracellular calcium promotes neuronal survival. J Neurosci. 1991;11:2582–7. doi: 10.1523/JNEUROSCI.11-08-02582.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Johnson EM, Jr, Koike T, Franklin JA. “calcium set-point hypothesis” of neuronal dependence on neurotrophic factor. Exp Neurol. 1992;115:163–6. doi: 10.1016/0014-4886(92)90242-i. [DOI] [PubMed] [Google Scholar]
- 87.Schmalbruch H. Loss of sensory neurons after sciatic nerve section in the rat. Anat Rec. 1987;219:323–9. doi: 10.1002/ar.1092190314. [DOI] [PubMed] [Google Scholar]
- 88.Vestergaard S, Tandrup T, Jakobsen J. Effect of permanent axotomy on number and volume of dorsal root ganglion cell bodies. J Comp Neurol. 1997;388:307–12. [PubMed] [Google Scholar]
- 89.Healy JI, Dolmetsch RE, Timmerman LA, Cyster JG, Thomas ML, Crabtree GR, et al. Different nuclear signals are activated by the B cell receptor during positive versus negative signaling. Immunity. 1997;6:419–28. doi: 10.1016/s1074-7613(00)80285-x. [DOI] [PubMed] [Google Scholar]
- 90.Hoffmann A, Kann O, Ohlemeyer C, Hanisch UK, Kettenmann H. Elevation of basal intracellular calcium as a central element in the activation of brain macrophages (microglia): suppression of receptor-evoked calcium signaling and control of release function. J Neurosci. 2003;23:4410–9. doi: 10.1523/JNEUROSCI.23-11-04410.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Caterina MJ, Schumacher MA, Tominaga M, Rosen TA, Levine JD, Julius D. The capsaicin receptor: a heat-activated ion channel in the pain pathway. Nature. 1997;389:816–24. doi: 10.1038/39807. [DOI] [PubMed] [Google Scholar]
- 92.Putney JW., Jr Capacitative calcium entry revisited. Cell Calcium. 1990;11:611–24. doi: 10.1016/0143-4160(90)90016-n. [DOI] [PubMed] [Google Scholar]
- 93.Fuchs A, Lirk P, Hogan QH. Calcium transients in injured sensory neurons. Society for Neuroscience Abstracts. Available from: http://sfn.scholarone.com/itin2005/main.html?new_page_id=126&abstract_id=2957&p_num=860.22&is_tech=0. Accessed: February 1, 2007.
- 94.Devor M, Janig W, Michaelis M. Modulation of activity in dorsal root ganglion neurons by sympathetic activation in nerve-injured rats. J Neurophysiol. 1994;71:38–47. doi: 10.1152/jn.1994.71.1.38. [DOI] [PubMed] [Google Scholar]
- 95.Schafers M, Lee DH, Brors D, Yaksh TL, Sorkin LS. Increased sensitivity of injured and adjacent uninjured rat primary sensory neurons to exogenous tumor necrosis factor-alpha after spinal nerve ligation. J Neurosci. 2003;23:3028–38. doi: 10.1523/JNEUROSCI.23-07-03028.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Devor M, Wall PD. Cross-excitation in dorsal root ganglia of nerve-injured and intact rats. J Neurophysiol. 1990;64:1733–46. doi: 10.1152/jn.1990.64.6.1733. [DOI] [PubMed] [Google Scholar]
- 97.Chaplan SR, Pogrel JW, Yaksh TL. Role of voltage-dependent calcium channel subtypes in experimental tactile allodynia. J Pharmacol Exp Ther. 1994;269:1117–23. [PubMed] [Google Scholar]
- 98.Xiao WH, Bennett GJ. Synthetic omega-conopeptides applied to the site of nerve injury suppress neuropathic pains in rats. J Pharmacol Exp Ther. 1995;274:666–72. [PubMed] [Google Scholar]
- 99.Clark NC, Nagano N, Kuenzi FM, Jarolimek W, Huber I, Walter D, et al. Neurological phenotype and synaptic function in mice lacking the CaV1.3 alpha subunit of neuronal L-type voltage-dependent Ca2+ channels. Neuroscience. 2003;120:435–42. doi: 10.1016/s0306-4522(03)00329-4. [DOI] [PubMed] [Google Scholar]
- 100.Saegusa H, Kurihara T, Zong S, Minowa O, Kazuno A, Han W, et al. Altered pain responses in mice lacking alpha 1E subunit of the voltage-dependent Ca2+ channel. Proc Natl Acad Sci U S A. 2000;97:6132–7. doi: 10.1073/pnas.100124197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Saegusa H, Kurihara T, Zong S, Kazuno A, Matsuda Y, Nonaka T, et al. Suppression of inflammatory and neuropathic pain symptoms in mice lacking the N-type Ca2+ channel. EMBO J. 2001;20:2349–56. doi: 10.1093/emboj/20.10.2349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Hogan QH. Injection for diagnosis and therapy of back disease. In: An HS, editor. Principles and techniques of spine surgery. Baltimore (MD): Williams and Wilkins; 1998, p. 707-29. [Google Scholar]