

Abstract
Here we describe preparatory techniques adapted for the study of proteins of inner ear tissues and fluids that have allowed us to apply state-of-the-art analytical techniques in spite of the minute size and anatomical complexities of this organ. Illustrative examples address unresolved issues of functional and clinical significance. First, we demonstrate how quick-freezing and freeze drying prevents artifacts that arise from sampling endolymphatic sac (ES) content in the liquid state. This set the stage for the generation of the first protein profile of the ES. Identification of crucial proteins will help elucidate mechanisms of endolymph volume regulation and pathogenesis of Meniere’s disease. Second, we show how a unique situation allowed identification of otoconial proteins by mass spectrometric analysis without prior separation and we discuss possible roles for these minor otoconins in otoconial development and prevention of degenerative diseases that affect balance. Finally, we demonstrate techniques for the precise dissection of organ of Corti and its substructures, while preserving their near normal chemical state. We extended an earlier study in which we identified a novel calcium-binding protein by IEF, oncomodulin, localized in the outer hair cells and show here the applicability of prefractionation for the screening of calcium-binding proteins of organ of Corti. These studies demonstrate how advanced preparatory and analytical techniques can be applied to studies of the inner ear.
Keywords: Ca-binding proteins, Inner ear proteins, Meniere’s disease, Minor otoconial proteins, Vertigo
1 Introduction
Professor Righetti, one of the great pioneers in the field of separation science, has made a significant impact on inner ear research. In this report we present several applications of these techniques to elucidate unresolved issues of inner ear research. As microanalytical techniques are being refined, sensitivity and specificity of the assays are not the over-riding obstacle for the inner ear researcher. While the small amounts of tissue available frequently require adjustments in the volume for the steps preceding analysis, such as fractionation, the preparatory techniques required for satisfactory spatial and temporal resolution can be the primary stumbling blocks. It is not necessarily the microscopic size of the substructures of the inner ear that poses the principal problems, but its complex anatomy, multitude of fragile cell types, and close association with contiguous fluids. Moreover, the inner ear houses both the cochlear and vestibular labyrinths and is encapsulated within a hard bony shell [1, 2].
We have overcome these problems to a large extent by quick freezing and freeze drying specimens in a cold surround system. The freeze-dried inner ear structures are well preserved in their 3-D state. After removal of the loose dry remnants of the adjacent fluids, the substructures can be readily identified and precisely dissected into substructures. Apart from the high degree of spatial resolution the specimens maintain a near normal metabolic state, or reflect the state that exists as a consequence of experimental perturbations.
We present here three examples of functionally and/or clinically highly significant inner ear issues to illustrate the elaborate preparatory and analytical techniques that had to be optimized to the special situation of the inner ear. The focus is on adaptation of techniques and represents work in progress.
1.1 Endolymphatic sac – physiological significance and role in Meniere’s disease
This section illustrates a novel preparatory approach for the harvesting of the contents of the endolymphatic sac (ES), which avoids the prohibitive technical problems and inevitable contamination of sampling in the liquid state. The physiological significance of the ES is highlighted by its presumptive role in the pathogenesis of Meniere’s disease, perhaps the most widely known inner ear disorder. Meniere’s disease is characterized by a triad of symptoms: attacks of vertigo, fluctuating loss of hearing, and tinnitus [1, 2]. The impetus for this study was the recent realization, that changes in certain macromolecules contained in the ES may play a key role in endolymph volume regulation and in the pathogenesis of Meniere’s disease.
Endolymph, which occupies the interior fluid compartment of the inner ear and is in intimate contact with the sensory structures, forms a closed system extending from the cochlea and vestibule in the periphery to the intracranially located and minute ES (Fig. 1A). According to current consensus the ES is not only responsible for resorptive activities, including disposal of waste products, but also involved in secretion and regulation of endolymph volume. Post mortem studies by Dix and Hallpike [3] established a marked swelling of the endolymphatic system – called endolymphatic hydrops – as the pathological substrate of Meniere’s disease. Subsequently it was demonstrated that experimental blockage of the ES and duct in the guinea pig resulted in development of a typical endolymphatic hydrops [4]. This seemed to confirm the long-held belief of a “back-up” of endolymph, due to impaired reabsorption by a dysfunctional ES of endolymph, which continuously flows in longitudinal direction toward the ES from its site of secretion in cochlea and vestibule. This mechanistic explanation had to be abandoned, when Salt and DeMott [5] demonstrated the absence of a standing endolymph flow under normal physiological conditions. Transient volume flow could, however, be induced by experimentally changing the volume of endolymph. Injection of and/or withdrawal of small fluid volumes resulted in volume flow directed either toward or away from the ES, in an apparent attempt to restore the original volume. This suggests that volume control of the endolymphatic system is subject to auto-regulation with the ES, the presumed regulatory center. The latter view was strengthened by the observation of histological/histochemical changes of the contents of the ES in response to experimental manipulation of endolymph volume. Volume expansion led to a marked decrease of the density of the “homogeneous substance”, believed to consist of glycoproteins, whereas endolymph contraction had the opposite effect. As a result the authors [6] proposed that the macromolecules in question may play an essential role in endolymph volume regulation, and may be instrumental in the pathogenesis of Meniere’s disease.
Figure 1.
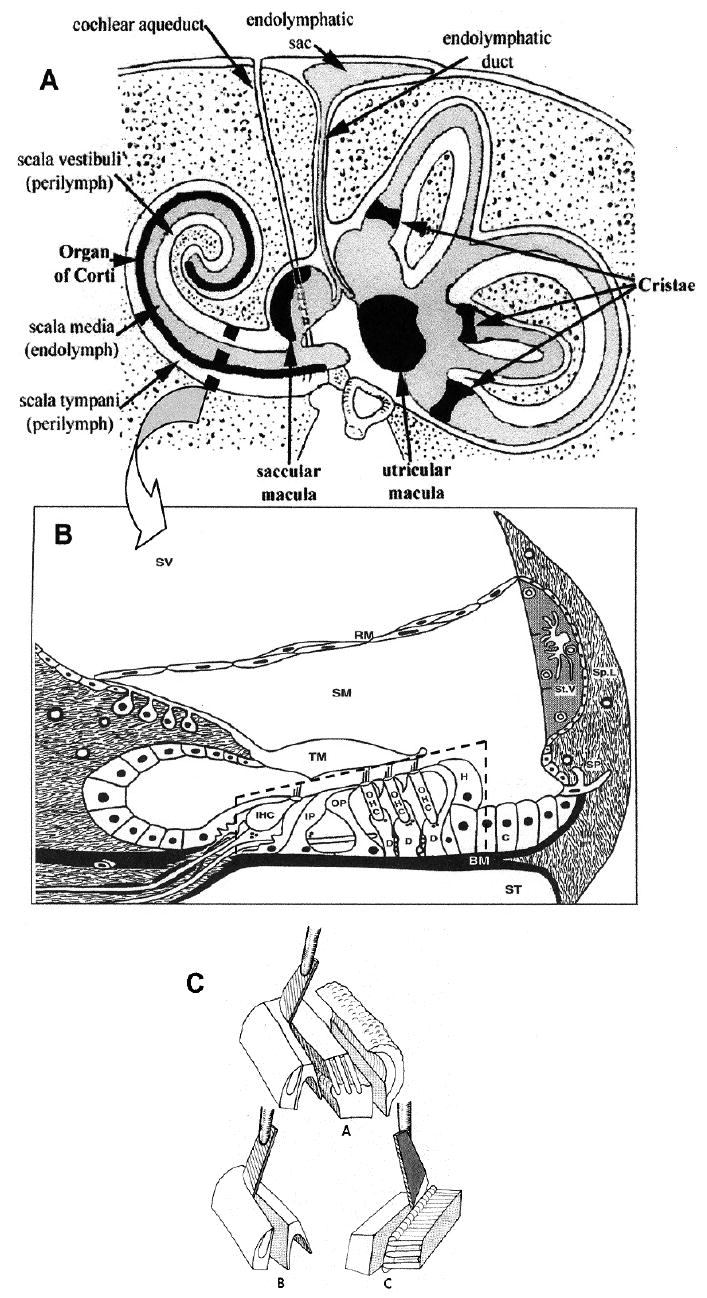
(A) Diagram of the human inner ear and temporal bone in cross-section. Reproduced with permission from [37]. Arrow points to an enlarged diagram of a cross-section of a cochlear turn (B). Organ of Corti (broken-line box) consisting of sensory cells (IHC – inner hair cells; OHC – three rows of outer hair cells) and supporting cells (IP – inner pillar; OP – outer pillar; D – Deiters; H – Hensen; C – Claudius) rests on the basilar membrane (BM), and is covered by the tectorial membrane (TM). There are three fluid compartments: scala vestibuli (SV) and scala tympani (ST), containing perilymph, and scala media (SM) containing endolymph. Reproduced with permission from [38]. (C) Schematic representation of the method for the separation of the organ of Corti into cell layers. A – organ of Corti divided into inner layer (left) and outer layer (middle). B – isolation of inner hair cell layer. C – isolation of outer hair cell layer.
We realized an opportunity to attack an important physiological and clinical issue by first establishing protein profiles of the fluid of the ES. We overcame serious problems inherent in sampling in the fluid state by resorting to freeze drying. Because of the high protein content of ES fluid (see Section 3), freeze drying results in the formation of firm casts of endolymph solids, which can readily be removed.
1.2 Otoconial proteins – identification and clinical significance
The unusually small number of otoconial matrix proteins revealed in preliminary studies allowed direct mass spectrometric analysis of digests of otoconial extracts, without prior separation procedures, such as 2-DE, thereby greatly enhancing the sensitivity and reliability of the assay. High sensitivity was essential because we had to resort to the use of mouse otoconia as explained below.
Otoconia are minute biomineral particles, thousands of which are imbedded into the gelatinous membrane that overlies the vestibular gravity receptor cells (Fig. 2). The high density of the otoconial mass markedly increases the sensitivity for the perception of linear acceleration and gravity. The inorganic shell of otoconia is made up of CaCO3 crystals, while the organic matrix consists of protein. Because of the low endolymphatic levels of the main inorganic components of otoconia, i.e. calcium and carbonate ions, efficient concentrating mechanisms are required for crystal nucleation and growth. These processes are mediated predominantly by glycoproteins.
Figure 2.

Composite diagram summarizing the organization of the filaments in the otoconial mass and gelatinous matrix. Internally, the otoconia contain a central core of densely organized filaments. Matrix of the supporting gelatinous membrane consists of a cross-linked, isotropic 3-D network of filaments. A loose filament network forms the interotoconial matrix. Reproduced with permission from [39].
Alteration of otoconial function leads to impairment of balance and orientation in space. Slow, progressive degeneration of otoconia is part of the normal aging process, and results in significant loss of balance and falls in a large percentage of elderly patients [7, 8]. Benign paroxysmal positional vertigo (BPPV), another prominent vestibular disorder, is caused by displacement of otoconia into the semicircular canals, which may lead to attacks of vertigo upon changes in head position [9]. In spite of the demonstrated significant morbidity, little is known about the molecular processes involved in otoconial development, maintenance, and pathology.
The first matrix protein selected for characterization was Otoconin90, the principal matrix protein, estimated to account for 90% of the total protein of the otoconial core [10-12]. Figure 3 demonstrates a typical example of 2-DE separation of otoconial matrix proteins from guinea pig. The strongly staining highly acidic protein represents Otoconin90. The partial amino acid sequence obtained from the excised Otoconin90 spot indicated near identity with an orphan cDNA clone isolated from a human tumor cell line [13]. The corresponding protein exhibits high homology to secretory PLA2, the main constituent of snake venom and pancreatic fluid. Cloning in the mouse confirmed these findings and enabled generation of antibodies and in situ probes [12, 14]. Subsequent histochemical studies in the developing otocyst provided first insights into the molecular processes involved in otoconial development.
Figure 3.
Silver-stained 2-DE separation of proteins extracted from guinea pig otoconia. (A) Protein derived from 302 μg (dry weight) of otoconia from eight ears (equaling approximately 15 μg total protein) were extracted with EDTA. Gel was spiked with molecular mass markers (carbonic anhydrase, ovalbumin, and β-galactosidase: 29, 45, and 116 kDa, respectively; arrows). (B) Molecular mass markers only.
Several approaches could be taken to identify other factors essential for otoconial development and morphogenesis. We have chosen to proceed with the characterization of the remaining matrix proteins, often referred to as the “minor” otoconins. Although preliminary 2-DE examinations had been performed in the guinea pig (Fig. 3), subsequent mass spectrometric studies were carried out in the anatomically less suitable mouse. The availability of complete gene and gene product databases outweighed the difficulties in harvesting sufficient amounts of otoconial material for analysis.
1.3 Ca-binding proteins: physiological significance in the organ of Corti
Two examples that illustrate unusual approaches in the discovery of as yet unidentified calcium-binding proteins of the organ of Corti are presented. In the first example (subproject 3.3.1) we demonstrate the power of IEF for studies at the microscale. Using exclusively IEF we discovered a novel calcium-binding protein, oncomodulin, characterized by an extremely low p/ (outside the usual range of 2-DE) in the organ of Corti, heretofore not to believed to be expressed in normal human cells (Fig. 4) [15]. It has become a much used marker for outer hair cells.
Figure 4.

Nondenaturing 1-D IEF of graded concentrations of oncomodulin standard, ranging from 62.5 to 3.9 ng (lanes 1–6) and organ of Corti (2 μg total protein) (lane 7). For details see [35].
The second example (subproject 3.3.2) shows the power of prefractionation which allowed us to detect several Ca-binding proteins, not previously demonstrated to be expressed in the organ of Corti.
In contrast to the preceding topic, which considered calcium in its role in carbonate-based mineralization, this section deals with calcium in its capacity as universal second messenger and how this mechanism is related to the proper function of the inner and outer hair cells of the organ of Corti (Fig. 1B). The transduction current flowing through the hair cells is predominantly carried by K+, and is powered by the unusually steep electrochemical gradient between endolymph and the interior of the hair cells. Cochlear endolymph has very high levels of K+ (~150 mM) and very low levels of Na+ (~1 mM). Moreover, endolymph is characterized by a positive resting potential of about 80–100 mV, called endolymphatic potential. Since the transducer channels of the hair cells are nonselective, a small proportion of the transducer current is carried by Ca++, even though its level in endolymph is extremely low (~20 μM) [16]. Nevertheless, the minute amount of the Ca++ entering the hair cells through these nonselective channels will disable the normal Ca++-mediated signal transduction mechanisms. This can be remedied only by efficient Ca++ extrusion mechanisms and/or sequestration or buffering by Ca-binding proteins. The functions of the inner and outer hair cells are fundamentally different from each other: The outer hair cell is a modified motor cell, whereas the inner hair cell is a bona fide sensory cell. A far higher transduction current passes through the outer hair cell placing particularly high demands on Ca++-clearing mechanisms. Consequently we would expect significant differences in the endowment with Ca++-binding proteins and possibly the expression of substances specifically adapted to the high demands of the outer hair cells.
Subproject 3.3.1 is presented to demonstrate the power of IEF as method of choice for identification and characterization of a highly acidic protein, especially in work at the microscale. The characterization of oncomodulin serves, moreover, to exemplify the technique of subdivision of the freeze-dried organ of Corti into fragments, largely representative of individual cell types. A schematic representation of this technique is shown in Fig. 1C.
Subproject 3.3.2. A cDNA library of mouse organ of Corti suggests possible expression of some 100 calcium-binding proteins (http://neibank.nei.nih.gov/cgi-bin/showDataTable.cgi?lib=NbLib0053&level=1&role=Calcium%20ion%20binding). The purpose of the present project was to determine which of these potential Ca-binding proteins are actually expressed in the organ of Corti, and whether they are localized to either type of hair cell. We implemented a prefractionation technique that does not depend on the use of 45-Ca, and which is potentially more sensitive.
2 Materials and methods
2.1 General sample preparation
Guinea pigs and mice were euthanized with compressed CO2 in a precharged container, and decapitated. The temporal bones were rapidly removed from the skull, cleared of soft tissues, and immersed in 2-methyl butane chilled to its melting point with liquid nitrogen and freeze-dried at −40°C for 3 days. Working in a humidity-controlled environment at 18°C the freeze-dried tissues to be analyzed were dissected under a stereomicroscope by means of fine microblades and hair points. Samples were weighed on quartz-fiber balances, or a Cahn electrobalance. Procedural details are described in [17].
2.2 Sample preparation for ES contents
Because of prohibitive obstacles of sampling of ES content in the liquid state (inter alia serious contamination with plasma) we resorted to freeze-drying. Following decapitation, the skull was quickly opened and portions of the brain were removed, exposing the lateral skull base, including the region of the ES. The exposed area was rapidly frozen by pouring large amounts of coolant over it, followed by immersion of the whole head into liquid nitrogen. The region of the ES, including the temporal bone, was separated from the skull in the frozen state at −40°C in a cryostat, and freeze-dried in toto as described.
Microdissection was carried out in a humidity-controlled environment at 18°C. After removal of an overlying thin bony shelf, the ES was exposed and opened. The white cast of freeze-dried endolymph becomes visible and was carefully removed. The dry specimens were weighed on quartz-fiber balances. After application of empirically derived correction factors a realistic estimate of the corresponding endolymph volume can be calculated [17].
2.3 Sample preparation for otoconial proteins
Guinea pig. Following freeze-drying of the temporal bone, the vestibulum is opened and the gravity receptor organs are exposed. The otoconial mass is lifted from the gelatinous layer. Remnants of adherent microfibrillar material are separated from the otoconia by three cycles of suspension in 0.1% SDS/NaAc, pH 7.4, followed by sonication for 3 min, and centrifugation at 10 000×g for 1 min. The matrix proteins were then extracted from the clean otoconia with 100 mM EDTA, pH 7 for a minimum of 1 h at 4°C. EDTA was removed by dialyzing against 0.1 M MgCl2 for at least 1h and against water at 4°C overnight. The samples were subsequently biodried. The mouse required a modified isolation technique. Following freeze drying, the otoconial material was elevated in toto from the underlying sensory epithelium. The gelatinous membrane was removed by brief treatment with undiluted bleach, leaving the otoconia perfectly clean. Otoconial proteins were then extracted as in guinea pig.
2.4 Dissection of organ of Corti and separation into substructures
Following freeze-drying the osseous capsule of the cochlea is chipped away by means of a small stainless steel watchmaker’s forceps. The spiral ligament is cautiously severed from the basilar membrane leaving the organ of Corti resting on it intact. The tectorial membrane is lifted off the organ of Corti with the aid of hair points and forceps. Short segments of the whole organ of Corti are then separated intact from the basilar membrane using a razor blade knife as lever. Dissection of substructures of the organ of Corti is preferably carried out in the guinea pig using razor blade knifes. A schematic representation of the initial steps in the subdivision of the organ of Corti into components cell layers is shown in Fig. 1C.
2.5 IEF
IEF was performed under nondenaturing conditions on a Hoefer SE200 Mighty Small Mini-Vertical Unit. Gel composition: 11% polyacrylamide, 1% piperazine di-acrylamide, 10% glycerol, carrier ampholytes (1.66% of p/ 3.5–10 and 0.33% of p/ 2.5–4). The gels were pre-focused for 15 min at 5 W at 15°C using 0.02 M acetic acid and 0.02 M NaOH as anolyte and catholyte, respectively. After the samples were loaded, the gels were run at 5 W until the dye front reached the bottom (~1.5 h). Proteins were visualized with the silver staining procedure described by Oakley et al. [18]. An oncomodulin dilution curve was included in each gel. Gels were scanned, and the concentration of the oncomodulin band (identified previously by antibodies and sequencing [19], was determined by integration and comparison with the oncomodulin standard curve using software from IO Informatics (http://www.io-informatics.com/).
2.6 2-D PAGE (2-DE)
Freeze-dried samples were solubilized directly in lysis buffer containing 8 M urea, 4% CHAPS, 65 mM dithioerythritol, 40 mM Tris-HCl, pH 9.5, 2 M thiourea, and a trace of bromophenol blue. 2-DE was carried out using IPG strips from Pharmacia in the first dimension, and the DALT technique in the second dimension. The gel gradient was poured with the aid of a computerized gradient maker (Angelique, Large Scale Biology, Rockville MD). Proteins were visualized by silver staining [18]. Gels were analyzed using the IO Informatics software.
2.7 Pretreatment with deglycosylation enzymes
To denature the sample 10 uL of 1% SDS were added to the freeze-dried samples of the contents of the ES, followed by boiling for 2 min which significantly increases the rate of deglycosylation. Ninety microliters of 0.02 M sodium phosphate buffer, pH 7.2, containing a second detergent, Nonidet (0.5%; five-fold to ten-fold in excess compared to SDS) was then added to the denatured sample before 0.4 U and/or 2 mU N-glycosidase F and neuraminidase are added, respectively. The samples were incubated 15–20 min at 37°C. Gel patterns of treated and untreated samples were compared.
2.8 Prefractionation of calcium-binding proteins
Calcium-binding proteins were separated according to the method of Moore [20]. The method takes advantage of the fact that Ca-binding proteins undergo a conformational change when calcium is bound, which exposes one or more hydrophobic regions. A column (1 cm id; 20 cm length) was packed with a thick slurry of phenyl sepharose CL4B (Sigma). The freeze-dried pooled samples of organ of Corti were solubilized in buffer containing: 20 mM Tris-HCl, pH 7.5; 50 mM NaCl; 0.1 mM EGTA. The tissue was first depleted of those proteins which bind to Sepharose in the absence of calcium at low salt concentration, and the nonabsorbed fraction was collected. The unspecifically bound proteins were completely removed from the absorbant by elution with 50% ethylene glycol; this fraction was discarded. The column was then equilibrated with the above buffer containing in addition 1 mM calcium, and the nonabsorbed fraction obtained in the previous step was added. The small fraction bound to the column (= calcium-binding fraction) was eluted with buffer containing 3 mM EGTA. Elution was followed by absorbance at 225 or 280 nm. The calcium-binding proteins which come off in two peaks were pooled and dialyzed against 10 mM MgCl2, and then water. The sample was subsequently dried and solubilized in lysis buffer.
2.9 MS
The biodried otoconial proteins extracted from mouse otoconia were analyzed by MS at the Washington University Proteomics Center. The data were collected by two mass spectrometers, a 7-T LTQ-FT mass spectrometer (Thermo, Bremen, Germany), and an electrosprayquadrupole-TOF mass spectrometer (Q-STAR XL, Applied Biosystems). Both instruments were interfaced to a low-flow (20–200 nL/min) liquid chromatograph (Eksigent nano-LC, Eksigent, Livermore, CA) using an Endurance autosampler (Spark, Plainsboro, NJ). The nanocapillary source was in both cases the PicoView system from New Objective (Woburn, MA).
Sample injection volume was 5 μL. The mass spectrometer was operated in the data-dependent mode to automatically switch between MS and MS/MS. Survey full scan MS spectra (from m/z 350 to 1500) were acquired for both the Q-STAR and the LTQ-FT; however, in the case of the LTQ-FT the full scan was acquired with resolution of r = 100 000). General mass spectrometric conditions for the LTQ-FT were as follows: spray voltage, 2.8 kV; capillary temperature, 200°C; normalized collision energy 35 using wide band activation mode.
The Q-STAR was utilized using the following parameters: The spray voltage was set to 2700 V with a curtain gas setting of 20, a nebulizer gas setting of 15. The declustering potential was 90 and the focusing potential was 300. Collision energies are calculated by the Analyst QS software according to the following function with dependence on the m/z value of the parent ion: collision energy = 0.058×m/z − 2.
In both cases the LC system was operated with a flow rate of 200 nL/min. The gradient consisted of HPLC-grade water (Fisher Scientific, Pittsburgh) in 1% formic acid (Sigma-Aldrich, St. Louis), solvent A, and ACN (Burdick & Johnson) in 1% formic acid, solvent B. Column equilibration and sample injection were performed at 10% A for 10 min each, followed by an increase of solvent B by 1%/min. The column was a C-18 PicoFrit column from New Objective with an inner diameter of 75 μm, a pore size of 5 μm, and a length of 10 cm.
3 Results and discussion
3.1 ES
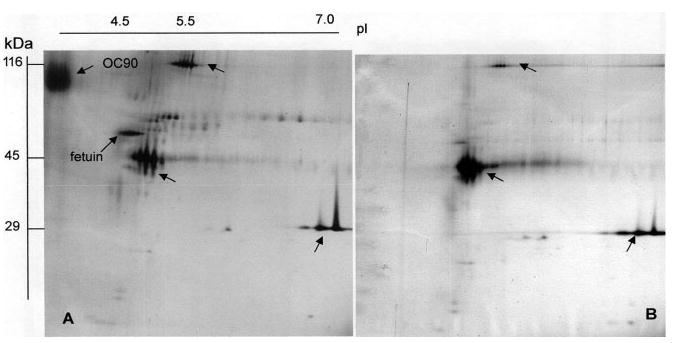
The total protein content of endolymph of the ES is extremely high – approximately 1625 mg/100 mL, which is about 40-fold higher than that of cochlear endolymph (38.4 mg/100 mL). Figure 5A shows the protein profile obtained by 2-DE of a 450 nL aliquot of ES content, pooled from five ears (note that in the guinea pig the average volume of endolymph of the ES is only 170 nL vs. 1.5 μL in the cochlea). In view of the small sample volume it is obvious that only proteins of relatively high abundance are detectable by silver staining. The profile clearly shows the major plasma proteins (60–80 kDa range) with their characteristic charge trains, which are suggestive of glycosylated proteins (for comparison see inset of Fig. 5A and [21]). To confirm the presence of glycoproteins, susceptibility to deglycosylating enzymes was tested. One aliquot of 450 nL ES endolymph was pretreated with N-glycosidase F and neuraminidase (Fig. 5B). Several prominent proteins visible in Fig. 5A and identified previously in cochlear endolymph (Fig. 5D) and plasma [22] such as α-antichymotrypsin, α2-HS-glycoprotein (fetuin A), α-1 antitrypsin, transferrin, and Apo D were no longer visible in the treated sample (Fig. 5B), confirming that they are glycoproteins.
Figure 5.

Silver-stained 2-DE electrophoretic separation of proteins of guinea pig of: (A) contents only of five ESs (corresponding to approx. 450 nL); insert shows a typical gel of guinea pig plasma, reduced in size; (B) same as A but pretreated with the deglycosylating enzymes N-glycosidase F and neuraminidase; (C) ES tissue plus its contents; and (D) 10 μL of cochlear endolymph obtained in the fluid state from ten ears. (1) α-chymotrypsin; (2) α-antitrypsin; (3) α-HS-glycoprotein; (4) transferrin; (5) Apo D; (6) Apo J; (7) fetuin. Sample volume of D was adjusted for easy comparison with corresponding proteins in A and B.
Figure 5D shows the 2-DE protein profile of a 10 uL sample of pooled cochlearendolymph obtained from ten guinea pig ears (obtained by sampling in the liquid state). The most striking difference between the cochlear and ES endolymph profiles is the absence of Apo D in the latter. Apo D was found to be one of the major proteins in cochlear endolymph, perilymph, and cerebrospinal fluid (CSF), and present in plasma in exceedingly low concentration [21]. The 2-DE pattern of the ES tissue plus content was, as expected, dominated by cellular proteins (Fig. 5C), but in addition contained the commensurate ES/plasma proteins. In summary, the total protein content of endolymph of the ES is approximately 40% that of plasma (4169 mg/100 mL), and shows a rather similar protein pattern. Cochlear endolymph (38.4 mg/100 mL) shows an approximately 40-fold lower total protein content and significant amounts of Apo D. This protein is also present in high relative concentrations in perilymph and CSF, but extremely low relative concentrations in plasma (Figs. 5A and C) [21].
In view of these enormous endolymph protein gradients between different segments of the same fluid compartment, it is necessary to postulate the existence of marked regional differences in the mode and rate of endolymph protein regulation. Gradients of this magnitude are incompatible with the traditional concept of longitudinal endolymph flow from a source in the cochlea to a sink in the ES [23]. Maintenance of the protein gradient in question is more consistent with the fact that endolymph volume flow is absent under normal conditions [5].
A promising strategy for the detection and/or identification of the macromolecule(s) corresponding to the “homogeneous substance” would be to search for substances that exhibit a significant increase and/or decrease in density in response to experimental contraction and/or expansion of endolymph volume [6, 24]. To facilitate evaluation, we are in the process of implementing 2-D differential gel electrophoretic analysis based on Cy3 and Cy5 dye labeling for the two conditions. It is important to note in this context that the size of the ES lumen does not change significantly as a consequence of experimental manipulation of endolymph volume (A. Salt, personal communication).
3.2 Otoconial proteins
The list of proteins extracted from mouse otoconia and identified by mass spectroscopy is shown in Table 1. The following proteins were identified.
Table 1.
Mouse otoconial matrix proteins identified by MS
| Otoconial proteins identified by MS | |
|---|---|
| Mouse | gi number |
| Otoconin-95 | 4176764 |
| Otoconin-90 | 4092677 |
| Myosin regulatory light polypeptide 9 | 51706911 |
| Sparc-like protein 1 | 1498641 |
| Osteopontin precursor | 200158 |
| Alpha fetuin | 2511777 |
| Laminin alpha 3 | 51770789 |
| Countertrypin | 407619 |
| Countertrypin | 407613 |
3.2.1 Osteopontin
Osteopontin is a sialoprotein, also referred to as secreted phosphoprotein 1, and is known to be incorporated into matrix ofbone and other calcified tissue. It has multiple roles in normal physiology and development including cell adhesion and protein binding in the extracellular matrix. Osteopontin has previously been suggested to be involved in the synthesis of otoconia [25, 26]. Sakagami [26] showed that Osteopontin mRNA was present in vestibular sensory hair cells and nonsensory dark cells, but was able to identify immunoreactivity for osteopontin only in mature otoconia.
3.2.2 Fetuin
This is a known inhibitor of calcium phosphate-based calcification and is abundant in plasma. In fetuin deficiency, such as uremia, vessels are massively calcified. It is as a rule synthesized in the liver, but is occasionally produced also in other structures during development. Fetuin may serve as a modulator of calcium carbonate crystal growth during otoconial synthesis. It is present also at relatively high levels in endolymph (Fig. 5A), where it may serve as inhibitor of calcification of mature otoconia. Recent studies have shown that fetuin is actively internalized and concentrated in intracellular vesicles of calcifying vascular smooth muscle cells. It is then secreted through the release of apoptotic bodies or matrix vesicles from viable cells where it significantly inhibits the ability of matrix vesicles to nucleate calcium phosphate [27]. The fetuin knockout shows no obvious vestibular dysfunction [28] (Jahnen-Dechent, personal communication). However, we might predict that these animals may have an altered otoconial morphology that might represent increased calcium phosphate deposition in the otoconial mass and throughout the inner ear. Changes of this kind would only be detected with SEM and spectroscopy on isolated vestibular samples. Alternatively, countertrypin, a calcium-binding protein and fetuin family member, also found in endolymph and the otoconial core, may serve a redundant role with fetuin in otoconial development and maintenance.
3.2.3 Other minor proteins
The remaining identified minor otoconins are characterized by either having putative calcium-binding domains and/or being involved in the extracellular matrix. For example: myosin regulatory light polypeptide 9 contains two EF-hand domains and binds calcium. Extracellular matrix-associated protein, or SC1 (also known as “hevin,”) is a member of the SPARC family of matricellular proteins, which mediate interactions between cells and their extracellular matrices. The SC1 knockout mouse has no obvious phenotype [29], and shows no obvious vestibular dysfunction (S. Funk and H. Sage, personal communication). SC1 has two-calcium-binding EF-hand motifs and the N-terminus binds five to eight calcium ions and interacts with hydroxyapatite. SC1 inhibits endothelial cell spreading in culture and plays a role in cell adhesion, shape, and motility. Laminin alpha 3 is a component of basal lamina and is involved in attaching cells to the extracellular matrix via interactions with integrins. Laminin alpha 3 may play a role in anchoring developing otoconial particles to the otoconial membrane.
It will be essential to look for mRNA and protein expression in the developing vestibular epithelia and/or gelatinous membrane. In particular, it will be interesting to determine if proteins such as fetuin, primarily a serum protein, are synthesized in the inner ear, or must be transported from plasma via perilymph into the endolymphatic space.
3.2.4 Calbindin
Calbindin was not detectable in the mammalian otoconial matrix, in contradistinction to the avian and reptilian otoconial matrix as reported on the basis of immunohistochemical evidence [30-32]. Immunohistochemistry for calbindin D28K in the mouse inner ear revealed reactivity only in a subset of vestibular hair cells of the utricular and saccular maculae (I. Hughes, unpublished observation).
3.2.5 Possible role of minor otoconins in maintenance of mature otoconia
Currently we know very little about organic processes involved in the maintenance of mature otoconia, and reports on regeneration of mammalian otoconia have been difficult to reconcile with the majority of the literature and biochemical data [14]. We have already shown that Otoconin90 expression decreases dramatically after development, suggesting the lack of mechanisms for turnover of the principal protein of the otoconial core. Examination of the expression of the minor otoconins throughout development and adulthood may point to mechanisms for maintaining the structure of otoconia through the turnover of the minor otoconins.
3.3 Calcium-binding proteins
Subproject 3.3.1: Oncomodulin
The protein in question, later identified as oncomodulin, had originally escaped detection because of its high acidity (p/ 3.1), which was beyond the range covered in our 2-DE studies of the organ of Corti. The rather conspicuous Ca-binding spot was detected during unrelated experimentation based on IEF, in combination with 45-Ca labeling, but could not be identified on the basis of existing databases. Because of the potential functional significance of this conspicuous Ca-binding protein we resorted to amino acid sequencing of the excised spot, which indicated identity with oncomodulin, a novel Ca-binding protein and Ca sensor, representing the β-parvalbumin isotype of parvalbumin, heretofore not believed to be expressed in normal mammalian tissue (Fig. 4) [33].
The subsequent work aimed at cellular localization and quantitation of oncomodulin was again accomplished by exclusive reliance upon IEF. By first subdividing the organ of Corti into inner and outer layers, and ultimately into individual cell layers (Fig. 1C) we were able to localize oncomodulin expression exclusively to the outer hair cells later confirmed by immunohistochemistry [34]. Using the same techniques we were able to quantify the protein in the outer hair cells [35].
Subsequently the same techniques, in combination with in situ hybridization and immunochemical studies, were used to follow the development of the expression of oncomodulin in outer hair cells, with that of α-parvalbumin in the inner hair cells, and their interrelation with the emerging cholinergic efferent innervation [36]. Currently it is believed that oncomodulin is instrumental in the intracellular mediation of cholinergic effects upon the outer hair cells.
Subproject 3.3.2
The results of prefractionation of the whole organ of Corti on a phenyl sepharose column are shown in Figs. 6A and B. Figure 6A shows some of the most prominent non-Ca-binding proteins of the organ of Corti, among them OCP1 and OCP2, the latter of which contains EF hands motifs that failed to indicate binding of 45-Ca also in previous experimentation. Figure 6B shows the 2-DE pattern of the Ca-binding fraction. Several known Ca-binding proteins, such as calmodulin and calbindin, are identified. Oncomodulin is not apparent due to its low p/ 3.1. Prior to attempting identification of the remaining Ca-binding spots, it appears indicated to determine whether they are preferentially expressed in either of the two hair cell layers. In order to detect additional Ca-binding proteins two approaches are available: (i) increase of sample size and (ii) increase of analytical sensitivity by implementation of differential Cy3 and Cy5 techniques.
Figure 6.
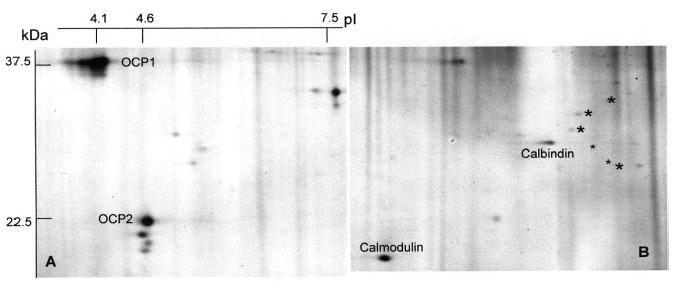
Silver-stained 2-DE separation of Ca-binding and nonbinding proteins of guinea pig organ of Corti. Freeze-dried organ of Corti was separated into Ca-binding and non-Ca-binding proteins on a phenyl Sepharose CL4B column as described in Section 2. Note that the two most prominent proteins of the organ of Corti, OCP1 and OCP2, do not bind calcium, while calmodulin, calbindin, and several weaker proteins (asterisks) do bind calcium.
4 Concluding remarks
It is hoped that this somewhat untraditional presentation provides a bird’s eye view to researchers in separation science of how this entity, which has revolutionized biology and medicine, can be applied to this minute, complex, and unique sensory organ.
Acknowledgments
This research was supported by NIDCD Grant DC01414 (IT), DC06974 (IH), and DC02236 (DMO&RT). The mass spectroscopy analysis was carried out by Dr. Julia Gross at the Washington University Proteomics Center (Dr. Reid Townsend, Director).
Abbreviation
- ES
endolymphatic sac
References
- 1.Haybach PJ. Meniere’s Disease. What You Need to Know. Vestibular Disorder Association; Portland, OR: 1998. [Google Scholar]
- 2.Harris JP. Meniere’s Disease. Kugler Publications; The Hague, The Netherlands: 1999. [Google Scholar]
- 3.Dix MR, Hallpike CS. Ann Otol Rhinol Laryngol. 1952;61:987–1016. doi: 10.1177/000348945206100403. [DOI] [PubMed] [Google Scholar]
- 4.Kimura RS. Otolaryngol Clin North Am. 1968;1:457–471. [Google Scholar]
- 5.Salt AN, DeMott JE. Hear Res. 1997;107:29–40. doi: 10.1016/s0378-5955(97)00018-x. [DOI] [PubMed] [Google Scholar]
- 6.Rask-Andersen H, DeMott JW, Bagger-Sjöbäck D, Salt AN. Hear Res. 1999;149:46–54. doi: 10.1016/s0378-5955(99)00153-7. [DOI] [PubMed] [Google Scholar]
- 7.Ross MD, Peacor D, Johnsson LB, Allard LF. Ann Otol Rhinol Laryngol. 1976;85:310–326. doi: 10.1177/000348947608500302. [DOI] [PubMed] [Google Scholar]
- 8.Oghalai JS, Manolidis S, Barth JL, Stewart MG, et al. Otolaryngol Head Neck Surg. 2000;122:630–634. doi: 10.1016/S0194-5998(00)70187-2. [DOI] [PubMed] [Google Scholar]
- 9.Parnes LS, Agrawal SK, Atlas J. Can Med J. 2003;169:681–693. [PMC free article] [PubMed] [Google Scholar]
- 10.Pote KG, Ross MD. Comp Biochem Physiol B. 1991;98:287–295. doi: 10.1016/0305-0491(91)90181-c. [DOI] [PubMed] [Google Scholar]
- 11.Ornitz DM, Bohne BA, Thalmann I, Harding GW, et al. Hear Res. 1998;122:60–70. doi: 10.1016/s0378-5955(98)00080-x. [DOI] [PubMed] [Google Scholar]
- 12.Wang Y, Kowalski PE, Thalmann I, Ornitz DM, et al. Proc Natl Acad Sci USA. 1998;95:15345–15350. doi: 10.1073/pnas.95.26.15345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kowalski PE, Freeman JD, Nelson DT, Mager DL. Genomics. 1997;39:38–46. doi: 10.1006/geno.1996.4471. [DOI] [PubMed] [Google Scholar]
- 14.Thalmann R, Ignatova E, Kachar B, Ornitz DM, et al. Ann N Y Acad Sci. 2001;942:162–178. doi: 10.1111/j.1749-6632.2001.tb03743.x. [DOI] [PubMed] [Google Scholar]
- 15.Thalmann I, Shibasaki O, Comegys TH, Henzl MT, et al. Biochem Biophys Res Commun. 1995;215:142–147. doi: 10.1006/bbrc.1995.2444. [DOI] [PubMed] [Google Scholar]
- 16.Salt AN, Inamura N, Thalmann R, Vora A. Am J Otolaryngol. 1989;10:371–375. doi: 10.1016/0196-0709(89)90030-6. [DOI] [PubMed] [Google Scholar]
- 17.Thalmann R, Smith CA, Vernon JA, editors. The Handbook of Auditory and Vestibular Research Methods. CC Thomas; Springfield IL: 1976. pp. 359–419. [Google Scholar]
- 18.Oakley BR, Kirsch DR, Morris N. Anal Biochem. 1980;105:361–363. doi: 10.1016/0003-2697(80)90470-4. [DOI] [PubMed] [Google Scholar]
- 19.Henzl MT, Shibasaki O, Comegys TH, Thalmann I, et al. Hear Res. 1997;106:105–111. doi: 10.1016/s0378-5955(97)00005-1. [DOI] [PubMed] [Google Scholar]
- 20.Moore BW. Neurochem Res. 1988;13:693–697. doi: 10.1007/BF00971590. [DOI] [PubMed] [Google Scholar]
- 21.Thalmann I, Kohut RI, Ryu JH, Thalmann R. Amer J Otol. 1995;16:153–157. [PubMed] [Google Scholar]
- 22.Thalmann I, Comegys TH, Liu SZ, Ito Z, et al. Hear Res. 1992;63:37–42. doi: 10.1016/0378-5955(92)90071-t. [DOI] [PubMed] [Google Scholar]
- 23.Guild SR. Am J Anat. 1927;39:57–81. [Google Scholar]
- 24.Takumida M, Bagger-Sjöbäck D, Rask-Andersen H. Hear Res. 1989;40:1–16. doi: 10.1016/0378-5955(89)90094-4. [DOI] [PubMed] [Google Scholar]
- 25.Takemura T, Sakagami M, Nakase T, Kubo T, et al. Hear Res. 1994;79:99–104. doi: 10.1016/0378-5955(94)90131-7. [DOI] [PubMed] [Google Scholar]
- 26.Sakagami M. Med Electron Microsc. 2000;33:3–10. doi: 10.1007/s007950000001. [DOI] [PubMed] [Google Scholar]
- 27.Reynolds JL, Skepper JN, McNair R, Kasama T, et al. J Am Soc Nephrol. 2005;16:2920–2930. doi: 10.1681/ASN.2004100895. [DOI] [PubMed] [Google Scholar]
- 28.Jahnen-Dechent W, Schinke T, Trindl A, Muller-Esterl W, et al. J Biol Chem. 1997;272:31496–31503. doi: 10.1074/jbc.272.50.31496. [DOI] [PubMed] [Google Scholar]
- 29.McKinnon PJ, McLaughlin SK, Kapsetaki M, Margolskee RF. Mol Cell Biol. 2000;20:656–660. doi: 10.1128/mcb.20.2.656-660.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Baird RA, Steyger PS, Schuff NR. Hear Res. 1997;103:85–100. doi: 10.1016/s0378-5955(96)00167-0. [DOI] [PubMed] [Google Scholar]
- 31.Balsamo G, Avallone B, Del Genio F, Trapani S, et al. Hear Res. 2000;148:1–8. doi: 10.1016/s0378-5955(00)00094-0. [DOI] [PubMed] [Google Scholar]
- 32.Piscopo M, Balsamo G, Mutone R, Avallone B, et al. Hear Res. 2003;178:89–94. doi: 10.1016/s0378-5955(03)00053-4. [DOI] [PubMed] [Google Scholar]
- 33.Brewer LM, MacManus JP. Placenta. 1987;8:351–363. doi: 10.1016/0143-4004(87)90063-4. [DOI] [PubMed] [Google Scholar]
- 34.Sakaguchi N, Henzl MT, Thalmann I, Thalmann R, et al. J Histochem Cytochem. 1998;46:29–39. doi: 10.1177/002215549804600105. [DOI] [PubMed] [Google Scholar]
- 35.Thalmann I, Thalmann R, Henzl MT. Prim Sens Neuron. 1998;2:283–296. [Google Scholar]
- 36.Yang D, Thalmann I, Thalmann R, Simmons DD. J Neurobiol. 2004;58:479–492. doi: 10.1002/neu.10289. [DOI] [PubMed] [Google Scholar]
- 37.Forge A, Wright T. Brit Med J. 2002;63:5–24. doi: 10.1093/bmb/63.1.5. [DOI] [PubMed] [Google Scholar]
- 38.Slepecky N. In: The Cochlea. Dallos P, Popper AN, Fay RR, editors. Springer; New York: 1996. pp. 44–129. [Google Scholar]
- 39.Lins U, Farina M, Kurc M, Riordan G, et al. J Struct Biol. 2000;131:67–78. doi: 10.1006/jsbi.2000.4260. [DOI] [PubMed] [Google Scholar]