

Abstract
Liver X receptors (LXRs) are nuclear receptors previously identified to be important in lipid metabolism. Recent reports suggest that LXR agonists also exhibit anti-inflammatory properties in mouse models of atherosclerosis and contact dermatitis. In the present study, we investigated the effects of LXR agonists on mouse microglia and astrocytes. When chronically activated, these resident-CNS glia have been implicated in the pathology of neuroinflammatory disorders including multiple sclerosis (MS). Our studies demonstrated for the first time that LXR agonists inhibited the production of nitric oxide, the pro-inflammatory cytokines IL-1β and IL-6 and the chemokine MCP-1 from LPS-stimulated microglia and astrocytes. Furthermore, LXR agonists inhibited LPS-induction of nuclear factor-kappa B (NF-κB) DNA-binding activity. These agonists also blocked LPS-induction of IκB-α protein degradation in microglia, suggesting a mechanism by which these agonists modulate NF-κB DNA-binding activity. These studies suggest that LXR agonists suppress the production of pro-inflammatory molecules by CNS glia, at least in part, by modulating NF-κB-signaling pathways. Retinoid X receptors (RXRs) physically interact with LXR receptors, and the resulting obligate heterodimer regulates the expression of LXR-responsive genes. Interestingly, a combination of LXR and RXR agonists additively suppressed the production of NO by microglia and astrocytes. Collectively, these studies suggest that LXR agonists may be effective in the treatment of neuroinflammatory diseases including MS.
Keywords: LXR, Microglia, Astrocytes, Cytokine, Chemokine, Multiple Sclerosis
1. INTRODUCTION
Multiple sclerosis (MS) affects approximately 400,000 people in the United States (Anderson et al., 1992). This disorder commonly strikes young adults and can profoundly affect quality of life (McFarlin and McFarland, 1982a; McFarlin and McFarland, 1982b). Multiple factors have been proposed to contribute to the development of MS, including viral infections, environmental factors and genetic background (Gilden, 2005; Marrie, 2004). MS is thought to be an autoimmune disease characterized by immune mediated myelin destruction and axonal transection. Currently available treatments for MS are modestly efficacious, and there is no cure (Cohen et al., 1999; Wekerle, 2002). Therefore, a great need exists for developing novel MS therapies.
Liver X receptors (LXRs) are nuclear receptors involved in the regulation of lipid metabolism (Cao et al., 2004; Edwards et al., 2002). Two LXR receptor subtypes exist; LXR-α, which is expressed on cells including macrophages and hepatocytes, and LXR-β, which is ubiquitously expressed (Janowski et al., 1999). Recent studies indicate that LXR agonists also modulate inflammatory responses. For example, LXR agonists suppressed inflammation in mouse models of atherosclerosis and contact dermatitis (Fowler et al., 2003; Joseph et al., 2003b). This suggests that LXR agonists may be effective in the treatment of neuroinflammatory disorders including MS.
Microglia are immune cells that reside in the CNS. They are antigen presenting cells and upon activation are capable of phagocytosis and the production of various pro-inflammatory molecules such as nitric oxide (NO) and interleukin-1β (IL-1β) (Benveniste, 1997). These molecules are capable of destroying pathogens, but can also be toxic to neurons and myelin-producing oligodendrocytes, cells which are compromised in MS (Drew and Chavis, 2001a). The secretion of C-X-C and C-C chemokines by activated microglia may also contribute to the recruitment of leukocytes from the periphery, as well as to the migration of microglia to sites of CNS inflammation (Aarum et al., 2003). Like microglia, chronically activated astrocytes are believed to contribute to MS through production of NO and various pro-inflammatory cytokines and chemokines (De Keyser et al., 2003; Holley et al., 2003; Raine, 1997; Storer et al., 2005a; Trapp et al., 1998; Zeinstra et al., 2003). Therefore, agents that block the activation of microglia and astrocytes may be effective in the treatment of MS.
The current studies demonstrate that LXR agonists alone, or in combination with RXR agonists, suppress the production of various pro-inflammatory mediators by activated microglia and astrocytes. Furthermore, these studies demonstrate that LXR agonists block the degradation of I-κB-α and suppress NF-κB DNA-binding activity in LPS-stimulated microglia. This suggests that LXR agonists may modulate the production of pro-inflammatory mediators, at least in part, through effects on NF-κB signaling pathways. These results support the growing potential of LXR agonists in the treatment of MS and other neuroinflammatory diseases.
2. MATERIALS AND METHODS
2.1 Reagents
The LXR agonist TO-901317 was obtained from Cayman Chemical Company (Ann Arbor, MI) and acetyl-podocarpic dimer (APD) was synthesized at Merck (Whitehouse Station, NJ). The RXR agonist 9-cis-retinoic acid, lipopolysaccharide, and L-Leucine methyl ester hydrochloride (L-LME) were obtained from Sigma (St. Louis MO). Lectin, Griffonia simplicifolia, was obtained from Sigma and anti-glial fibrilary acidic protein (GFAP) antibody was obtained from Dako (Carpinteria, CA). DMEM media, glutamine, trypsin, and antibiotics used for tissue culture were obtained from BioWhittaker (Walkersville, MD). OPI medium supplement was obtained from Sigma (St Louis, MO). GM-CSF was obtained from BD Pharmingen (San Diego, CA). Fetal bovine serum (FBS) from Hyclone (Logan, UT) was used for primary cultures and FBS used for N9 microglia cultures was obtained from Sigma. C57BL/6 mice were obtained from Harlan (Indianapolis, IN), and bred in house.
2.2 Cell Culture
Primary mouse microglia were obtained through a modification of the McCarthy and deVellis (McCarthy and de Vellis, 1980) protocol. Briefly, cerebral cortices were excised from 1–3 day-old C57BL/6 mice and the meninges removed. Cortices were minced into small pieces and cells were separated by trypsinization and trituration of cortical tissue, and cellular debris was removed by filtration through a 70-μm cell strainer. Cells were plated into tissue culture flasks and allowed to grow to confluence (~1 week) in complete DMEM media containing 10% FBS, 1.4 m L-glutamine, 100 U/mL penicillin, 0.1 mg/mL streptomycin, OPI medium supplement, and GM-CSF. Flasks were shaken overnight (200 rpm at 37°C) in a temperature-controlled shaker to loosen microglia and oligodendrocytes from the more adherent astrocytes. These non-adherent cells were plated for 2 hours and then lightly shaken to separate oligodendrocytes from the relatively more adherent microglia. Primary astrocytes were prepared as described above, with the exception that GM-CSF was omitted from the culture media. These panning procedures were repeated, as necessary, to obtain primary microglia or astrocytes of greater than 95% purity as determined by immunocytochemical staining with lectin or anti-glial fibrilary acidic protein (GFAP) antibody, respectively. The N9 microglial cell line is derived from myc-immortalized mouse microglia (Corradin et al., 1993), and was graciously provided by P. Ricciardi-Castagnoli (U. Milan, Italy). N9 cells were cultured in MEM medium containing 10% FBS, 1.4 mM glutamine, and 20 μM 2-mercaptoethanol. Cells were seeded into 96-well plates (5 X 104 cells/well) and incubated overnight at 37°C /5% CO2. Cells were treated with the LXR agonist TO-91317 or APD, or LXR agonist in combination with the RXR agonist, 9-cis-retinoic acid for 1 hour, and then stimulated with LPS for 24 hours. Finally, tissue culture supernatants and cells were collected for nitrite assay, enzyme-linked immunosorbent assay (ELISA) and MTT reduction assay.
2.3 Nitric oxide production
Levels of the NO derivative nitrite were determined in the culture medium by Griess reaction as described previously (Drew and Chavis, 2001). Optical densities were determined using a Spectromax 190 microplate reader (Molecular Devices, Sunnyvale, CA) at 550 nm. A standard curve using NaNO2 was generated for each experiment for quantitation.
2.4 Cell viability assays
Cell viability was determined by MTT reduction assay as described previously (Drew and Chavis, 2001). Optical densities were determined using a Spectromax 190 microplate reader (Molecular Devices, Sunnyvale, CA) at 570 nm. Results were reported as percent viability relative to untreated cultures.
2.5 Enzyme-linked immunosorbent assay (ELISA)
Cytokine (IL-1β and IL-6) and chemokine (MCP-1) levels in tissue culture media were determined by ELISA as described by the manufacturer (OptEIA Sets, Pharmingen, San Diego, CA). Optical densities were determined using a Spectromax 190 microplate reader (Molecular Devices, Sunnyvale, CA) at 450 nm. Cytokine and chemokine concentrations in media were determined from standards containing known concentrations of the proteins.
2.6 Western blot analysis
Total cellular protein was isolated from untreated, LPS stimulated, and TO-91317 pre-treated then LPS stimulated cells plated in 6-well plates (2 X 106 cells/well). Protein was then quantitated by BCA Protein Assay as described by the manufacturer (Pierce, Rockford, IL, USA). Cellular proteins (20 μg) were separated electrophoretically on 4–15%-HCl Ready-gels obtained from Bio-Rad (Hercules, CA) containing the denaturant sodium dodecyl sulfate (SDS). Proteins were transferred electrophoretically to nitrocellulose membranes (NitroBind; MSI, Westborough, MA, USA) and then incubated with polyclonal rabbit anti-human IκB-α antibody (Calbiochem, La Jolla, CA, USA) for 1 h at room tempearature at a 1:750 dilution. Blots were then incubated with horseradish peroxidase conjugated goat anti-rabbit IgG (Santa Cruz Biotechnology, Santa Cruz, CA, USA) for 1 h at room temperature at a 1:5000 dilution. IκB-α protein was detected by ChemiGlow Western Blotting Detection Reagents as described by the manufacturer (Alpha Innotech Corporation, San Leandro, CA, USA), followed by exposure in BioRad ChemiImager.
2.7 Electrophoretic Mobility Shift Assay (EMSA)
Nuclear extracts were prepared from untreated cells, cells stimulated with LPS, or cells treated with 60 μM TO-91317 for 1 hour then stimulated with LPS for 24 h as previously described (Drew et al., 1993). A combination of the protease inhibitors, 0.5mM dithiothreitol, 10μg/ml leupeptin & aprotinin, 10μg/ml soy bean trypsin inhibitor, and 200mM PMSF & pepstatin A were used during preparation of nuclear extracts. Oligonucleotide probes used in gel mobility shift assay were end-labeled using T4 polynucleotide kinase (Invitrogen, Calsbad, CA, USA) as described previously (Drew et al., 1993). Binding assays were performed in a volume of 20μl containing 20mM Tris-HCl (pH 7.5), 0.2mM EDTA, 5% glycerol, 1mM MgCl2, 1mM dithiothreitol, 100mM NaCl, 4μg poly dI-dC, 4μg nuclear extracts and 105cpm of γ32P-ATP (Perkin Elmer, Wellesley, CA, USA) labeled double stranded oligonucleotide probe. Double-stranded oligonucleotide probes used in the analysis were the NF-κB element of the mutated HIV promoter (5′-AGTTGAGGGGACTTTCCAGGC-3′) and the murine leukemia virus UCR element (5’-CTGCAGTAACGCCATTTTGCAAGGCATGAA-3’). The latter is bound by a ubiquitously expressed factor that generally is unaltered by experimental treatment and thus was used to control for equal loading and integrity of individual nuclear protein extracts as previously described (Dhib-Jalbut et al., 1995; Drew et al., 1993). Radiolabled DNA-protein complexes were separated from free probes by electrophoresis through 6% polyacrylamide gels in the presence of 0.4% tris-borate-EDTA for 1.5 hour at 150V. The gels were dried and visualized by autoradiography. For competition assays, the indicated unlabeled oligonucleotides were included in 100-fold molar excess relative to radiolabeled probe in the binding assays.
2.8 Statistics
Data were analyzed by one-way ANOVA followed by a Bonferroni post-hoc test to determine the significance of difference.
3. RESULTS
3.1 Effect of LXR agonists on NO production by N9 microglial cells
Activated microglia produce NO that may be toxic to oligodendrocytes and neurons, cells which are compromised in MS. In the present studies we demonstrated that LPS increased nitrite production by N9 mouse microglia. The LXR agonists, TO-901317 (Figure 1A) and APD (Figure 1C), both inhibited LPS-induced nitrite production in a dose-dependent manner. The LXR agonists were not toxic to the cells at the concentrations examined as demonstrated by MTT reduction assays (Figure 1B and D). Thus, TO-901317 and APD inhibition of NO production was not due to effects on cell viability.
Figure 1. Effects of LXR agonists on LPS induction of NO in N9 microglia.

Cells were pre-treated for 1 h with the indicated concentrations (μM) of LXR agonists (APD; TO-901317 = T09). Cells were then stimulated by LPS (1μg/ml) and 24 h later, the level of nitrite in the culture medium was determined by Griess reaction (A and C). Cell viability was determined by MTT assay (B and D). Representative results from a single experiment are shown and at least three independent experiments were performed. Values represent the mean ± s.d. from one experiment performed in triplicate. *p<0.05 and ***p<0.001 vs. LPS-treated cultures.
3.2 Effect of LXR agonists on NO production by primary mouse microglia
Untreated primary microglia expressed undetectable levels of NO, and LPS significantly increased NO production by these cells (Figure 2). The LXR agonists TO-901317 (Figure 2A) and APD (data not shown) significantly suppressed LPS-induction of NO by primary microglia but did no effect cell viability (Figure 2B). Interestingly, slightly higher concentrations of TO-901317 were required to inhibit LPS induction of NO production in primary microglia than N9 microglial cells.
Figure 2. Effects of LXR agonist on LPS induction of NO in mouse primary microglia.

Cells were pre-treated for 1 h with the indicated concentrations of LXR agonist TO-901317. Cells were then stimulated by LPS (1μg/ml) and 24 h later, the level of nitrite in the culture medium was determined by Griess reaction (A). Cell viability was determined by MTT assay (B). Representative results from a single experiment are shown and at least three independent experiments were performed. Values represent the mean ± s.d. from one experiment performed in triplicate. **p<0.01 and ***p<0.001 vs. LPS-treated cultures.
3.3 Effect of LXR agonists on cytokine and chemokine production by microglia
Activated microglia produce a variety of inflammatory cytokines and chemokines, which may modulate MS. In the present study, LPS induced the secretion of cytokines IL-1β and IL-6, and the chemokine MCP-1 (Figure 3). Pre-treatment with TO-901317 suppressed the production of IL-1β (Figure 3A), IL-6 (Figure 3B) and MCP-1 (Figure 3C) in a dose-dependent manner.
Figure 3. Effects of LXR agonist on cytokine and chemokine production by mouse primary microglia.

Cells were pre-treated for 1 h with the indicated concentrations of LXR agonist, TO-901317. LPS (1μg/ml) was added as indicated, and 24 h later, IL-1β (A), IL-6 (B), and MCP-1 (C) levels were measured in the culture medium by ELISA. Representative results from a single experiment are shown and at least three independent experiments were performed. Values represent the mean ± s.d. from one experiment performed in triplicate. **p<0.01 and ***p<0.001 vs. LPS-treated cultures.
3.4 Combined effect of LXR and RXR agonists on microglia function
LXR and RXR are capable of physically interacting to form heterodimers. These heterodimers bind liver X response elements (LXREs) present in the promoter elements of LXR responsive genes. The present studies demonstrate that LXR agonist TO-901317 and the RXR agonist 9-cis RA independently inhibited NO production by LPS-stimulated microglia. In addition, these agonists in combination inhibited NO production more strongly than the RXR or the LXR agonist alone (Figure 4A). These agonists did not significantly decrease microglia cell viability in these studies (Figure 4B). These studies suggest that LXR and RXR agonists act cooperatively in inhibiting NO production by activated microglia.
Figure 4. Combined effects of LXR and RXR agonists on nitrite production by mouse primary microglia.

Cells were pre-treated for 1 h with TO-901317 (60μM), 9-cis RA (0.75μM), or their combination. LPS (1μg/ml) was added as indicated, and 24 h later, the concentration of nitrite in the culture medium was determined (A). Cell viability was determined by MTT assay (B). Representative results from a single experiment are shown and at least three independent experiments were performed. Values represent the mean ± s.d. from one experiment performed in triplicate. ***p<0.001 and**p<0.01 vs. LPS-treated cultures. ###p<0.001 vs. LPS plus TO-91317 and vs. LPS plus 9-cis RA-treated cultures.
3.5 Effect of LXR agonist on NF-κB DNA-binding activity
NF-κB plays an important role in the activation of a variety of genes which encode pro-inflammatory proteins. In the present study, we demonstrated that NF-κB DNA-binding activity was increased following stimulation of microglia with LPS. Importantly, the LXR agonist TO-901317 significantly inhibited LPS-induction of NF-κB binding activity in these cells (Figure 5). This suggests that LXR agonists may inhibit production of pro-inflammatory molecules through a mechanism involving inhibition of NF-κB binding to the promoters of genes which encode these molecules.
Figure 5. Effects of LXR agonist on NF-κB binding activity in primary mouse microglia.
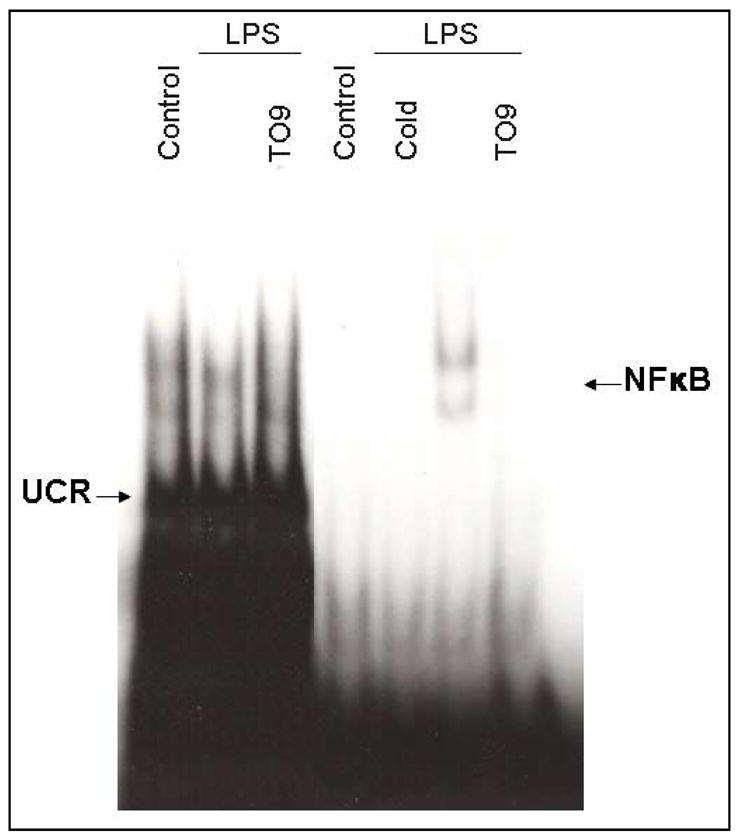
Cells were pre-treated for 1 h with T0-901317 (60μM), then LPS (1μg/ml) was added, and 24 h later, nuclear extracts were isolated. The effect of TO-901317 on the DNA binding activity of NF-κB was determined by EMSA. A 100-fold excess of unlabeled (cold) NF-κB probe was added to the EMSA binding mixture as a control for non-specific DNA binding. The EMSA figure represents a typical example from five independent experiments. Similar results were observed when cells were stimulated with LPS for 6 h instead of 24 h (data not shown).
3.6 Effect of LXR agonist on IκB-α expression
Inhibitor-κB (IκB) binds to NF-κB masking its nuclear localization signal and thus sequestering this transcription factor complex in the cytoplasm. A variety of stimuli have been demonstrated to phosphorylate IκB resulting in the release and nuclear translocation of NF-κB. In the present study, we demonstrated that LPS treatment of primary microglia decreased the expression of IκB-α and that the LXR agonist TO-901317 counteracted the effect of LPS. This suggests that TO-91317 prevented the degradation of IκB levels in microglia, thus blocking NF-κB translocation to the nucleus and LPS induction of NF-κB genes including iNOS which regulates the production of NO, as well as genes that encode pro-inflammatory cytokines and chemokines. Collectively, these results suggest a mechanism by which the LXR agonist TO-901317 inhibits LPS induction of NF-κB DNA-binding activity in primary microglia.
3.7 Effect of LXR agonist on NO production by primary astrocytes
Like microglia, activated astrocytes can also produce NO which can be toxic to oligodendrocytes thus contributing to the demyelination seen in MS patients. In the present study, the synthetic LXR agonist, TO-901317, was demonstrated to significantly reduce the production of NO by primary astrocytes activated by LPS in a dose-dependent manner (Figure 7A). This agonist was not toxic to astrocytes at the concentrations examined as determined by the MTT reduction assay (Figure 7B). Thus TO-901317 inhibition of NO production by astrocytes was not due to cell toxicity.
Figure 7. Effects of LXR agonist on LPS induction of NO in mouse primary astrocytes.

Cells were pre-treated 1 h with the indicated concentration of LXR agonist TO-901317. LPS (2μg/ml) was added as indicated, and 24 h later, the concentration of nitrite in the culture medium was determined (A). Cell viability was determined by MTT assay (B). Values represent the mean ± s.d. for a single experiment performed in triplicate. At least three independent experiments were conducted. ***p<0.001 vs. LPS-treated cultures.
3.8 Effect of LXR agonists on cytokine and chemokine production by astrocytes
Like microglia, activated astrocytes produce a variety of inflammatory cytokines and chemokines which may modulate MS. In the present study, LPS induced the secretion of the cytokines IL-1β and IL-6 and the chemokine MCP-1 (Figure 8). Pre-treatment with TO-901317 suppressed the production IL-1β (Figure 8A), IL-6 (Figure 8B) and MCP-1 (Figure 8C) in a dose-dependent manner.
Figure 8. Effects of LXR agonist on cytokine and chemokine production by mouse primary astrocytes.

Cells were pre-treated for 1 h with the indicated concentrations of LXR agonist, TO-901317. LPS (2μg/ml) was added as indicated, and 24 h later, IL-1β (A), IL-6 (B), and MCP-1 (C) levels were determined in culture medium by ELISA. Values represent the mean ± s.d. from a representative experiment performed in triplicate. At least three independent experiments were conducted. *p<0.05 and ***p<0.001 vs. LPS-treated cultures.
3.9 Combined effect of LXR and RXR agonists on astrocyte function
The LXR agonist TO-901317 and the RXR agonist 9-cis RA inhibited NO production by LPS-stimulated astrocytes. In addition, a combination of these agonists more potently inhibited NO production in these cells than either agonist alone (Figure 9A). These agonists did not significantly decrease astrocyte viability (Figure 9B). The studies suggest that LXR and RXR agonists act cooperatively in inhibiting NO production by activated astrocytes.
Figure 9. Combined effects of LXR and RXR agonists on nitrite production by mouse primary astrocytes.

Cells were pre-treated for 1 h with the indicated concentration of T0-91317, 9-cis RA, or their combination. LPS (2μg/ml) was added as indicated, and 24 h later, the concentration of nitrite in the culture medium was determined (A). Cell viability was determined by MTT assay (B). Values represent the mean ± s.d. from a representative experiment performed in triplicate. At least three independent experiments were conducted. ***p<0.001 vs. LPS-treated cultures. ###p<0.001 vs. LPS plus TO-91317 and vs. LPS plus 9-cis RA-treated cultures.
4. DISCUSSION
MS is a chronic neuroinflammatory disease thought to be initiated by activated T cells that recognize self antigen (Martin and McFarland, 1995; Xiao and Link, 1999). These activated T cells are capable of migrating into the CNS where they trigger the activation of CNS glia. Upon activation, glia, including microglia and astrocytes, produce a variety of cytokines and chemokines which may contribute to the pathology associated with MS. For example, NO is abundantly produced by activated glia. In addition, cytokines including IL-1β, and IL-6, and the chemokine MCP-1 are produced by activated glia. These pro-inflammatory molecules are associated with the induction and effector phase of EAE, an animal model of MS (Wang et al., 2002). Therefore, agents that suppress the production of these molecules may be effective in the treatment of MS.
LXRs, like PPARs, are nuclear receptors that play an important role in lipid and glucose metabolism. Previously, we and others have demonstrated that PPARs also are important modulators of inflammation (Kielian and Drew, 2003b; Ricote et al., 1998). For example, PPAR-γ agonists including the naturally occurring prostaglandin metabolite 15d-PGJ2 and synthetic ligands including thiazolidinediones which are used commonly for the treatment of type II diabetes, suppressed immune function of cells including macrophages T cells, and CNS glia (Clark et al., 2000; Drew and Chavis, 2001a; Jiang et al., 1998; Ricote et al., 1998; Storer et al., 2005a; Storer et al., 2005b). PPAR-γ ligands also suppress inflammation and pathology in animal models of atherosclerosis, arthritis, inflammatory bowel disease and MS (Diab et al., 2002; Feinstein et al., 2002; Kawahito et al., 2000; Li et al., 2000; Natarajan and Bright, 2002; Niino et al., 2001; Su et al., 1999). Like PPAR-γ agonists, PPAR-α agonists suppress the function of T cells and CNS glia in vitro (Lovett-Racke et al., 2004; Xu et al., 2006a; Xu et al., 2005b). In addition, these agonists are effective in the treatment of EAE (Lovett-Racke et al., 2004). Relatively little is known concerning the role of LXR agonists in modulating inflammation. Recently, however, these agonists were demonstrated to suppress the activation of macrophages in vitro and mouse models of atherosclerosis and contact dermatitis in vivo (Fowler et al., 2003; Joseph et al., 2004a; Joseph et al., 2003b; Joseph et al., 2002c). The present studies indicate that LXR agonists suppress the production of NO, as well as the pro-inflammatory cytokines IL-1β and IL-6, and the chemokine MCP-1. These molecules are believed to contribute to the development of EAE and MS. Thus, these studies suggest that LXR agonists may be effective in the treatment of these disorders.
Our studies demonstrated that the LXR agonist TO-091317 blocked LPS induction of NF-κB DNA-binding activity in microglia. NF-κB is an important modulator of a variety of genes which respond to immune stimuli, including iNOS, as well as many of the genes encoding the cytokines and chemokines examined in the current studies. We further demonstrated that TO-901317 inhibits NF-κB DNA binding activity, at least in part, by suppressing LPS-induced degradation of IκB, which blocks the nuclear translocation of NF-κB. LPS is known to stimulate NF-κB by engaging the toll-like receptor TLR4 through a MyD88-dependent pathway (Kielian, 2006a). Future studies will investigate additional potential mechanisms by which LXR agonists modulate MyD88-dependent signaling and the activation of NF-κB.
The current studies demonstrated that RXR agonist 9-cis RA and the LXR agonist TO-091317 act cooperatively to inhibit the production of NO by primary mouse microglia and astrocytes. LXRs are known to physically interact with RXRs to form heterdimers. Maximal receptor activity requires ligand binding by both of these receptors, suggesting a mechanism for the combined effects of LXR and RXR agonists on microglia and astrocyte function in the present study. Interestingly, RXRs also physically interact with PPAR ligands. We have previously demonstrated that PPAR and RXR ligands act cooperatively in suppressing inflammatory responses (Lovett-Racke et al., 2004; Xu et al., 2006a; Xu et al., 2005b). Future studies will compare the profile of genes regulated by PPAR relative to LXR agonists. The role of specific co-activators and co-repressors that regulate gene expression in response to PPAR and LXR agonists will also be evaluated.
In summary, we have demonstrated that LXR agonists were capable of blocking the production of NO by LPS-stimulated primary mouse microglia and astrocytes. These ligands also inhibited the production of pro-inflammatory cytokines IL-1β and IL-6 and the chemokine MCP-1. Furthermore, we demonstrate that LXR agonists inhibit LPS-induced degradation of IκB-α and block NF-κB DNA-binding activity. This suggests that LXR agonists may suppress the expression of these pro-inflammatory molecules, at least in part, by altering NF-κB signaling pathways. In addition, the RXR agonist 9-cis RA and the LXR agonist TO-091317 acted cooperatively to inhibit NO production by both astrocytes and microglia. These studies suggest that LXR agonists, perhaps in combination with RXR agonists, may be effective in the treatment of neuroinflammatory disorders including MS.
Figure 6. Effects of LXR agonist on IκB-α protein expression.

Primary mouse microglia were untreated, stimulated with LPS (1μg/ml), or pre-treated with TO-901317 (60μM) for 1 h prior to LPS simulation. Following 45 min of LPS stimulation, nuclear protein was isolated, and IκB-α protein levels were determined by western blot analysis. Representative results from a single experiment are shown and at least three independent experiments were performed.
Acknowledgments
This work was supported by grants from the National Institutes of Health (NS42860 and NS047546) and from the Arkansas Biosciences Institute. The authors thank Dr. JiHong Xu for critically reviewing the manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aarum J, Sandberg K, Haeberlein SL, Persson MA. Migration and differentiation of neural precursor cells can be directed by microglia. Proc Natl Acad Sci U S A. 2003;100:15983–15988. doi: 10.1073/pnas.2237050100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson DW, Ellenberg JH, Leventhal CM, Reingold SC, Rodriguez M, Silberberg DH. Revised estimate of the prevalence of multiple sclerosis in the United States. Ann Neurol. 1992;31:333–336. doi: 10.1002/ana.410310317. [DOI] [PubMed] [Google Scholar]
- Benveniste EN. Role of macrophages/microglia in multiple sclerosis and experimental allergic encephalomyelitis. J Mol Med. 1997;75:165–173. doi: 10.1007/s001090050101. [DOI] [PubMed] [Google Scholar]
- Cao G, Liang Y, Jiang XC, Eacho PI. Liver X receptors as potential therapeutic targets for multiple diseases. Drug News Perspect. 2004;17:35–41. doi: 10.1358/dnp.2004.17.1.829024. [DOI] [PubMed] [Google Scholar]
- Clark RB, Bishop-Bailey D, Estrada-Hernandez T, Hla T, Puddington L, Padula SJ. The nuclear receptor PPAR gamma and immunoregulation: PPAR gamma mediates inhibition of helper T cell responses. J Immunol. 2000;164:1364–1371. doi: 10.4049/jimmunol.164.3.1364. [DOI] [PubMed] [Google Scholar]
- Cohen JA, Carter JL, Kinkel RP, Schwid SR. Therapy of relapsing multiple sclerosis. Treatment approaches for nonresponders. J Neuroimmunol. 1999;98:29–36. doi: 10.1016/s0165-5728(99)00078-8. [DOI] [PubMed] [Google Scholar]
- Corradin SB, Mauel J, Donini SD, Quattrocchi E, Ricciardi-Castagnoli P. Inducible nitric oxide synthase activity of cloned murine microglial cells. Glia. 1993;7:255–262. doi: 10.1002/glia.440070309. [DOI] [PubMed] [Google Scholar]
- De Keyser J, Zeinstra E, Frohman E. Are astrocytes central players in the pathophysiology of multiple sclerosis? Arch Neurol. 2003;60:132–136. doi: 10.1001/archneur.60.1.132. [DOI] [PubMed] [Google Scholar]
- Dhib-Jalbut SS, Xia Q, Drew PD, Swoveland PT. Differential up-regulation of HLA class I molecules on neuronal and glial cell lines by virus infection correlates with differential induction of IFN-beta. J Immunol. 1995;155:2096–2108. [PubMed] [Google Scholar]
- Diab A, Deng C, Smith JD, Hussain RZ, Phanavanh B, Lovett-Racke AE, Drew PD, Racke MK. Peroxisome proliferator-activated receptor-gamma agonist 15-deoxy-Delta(12,14)-prostaglandin J(2) ameliorates experimental autoimmune encephalomyelitis. J Immunol. 2002;168:2508–2515. doi: 10.4049/jimmunol.168.5.2508. [DOI] [PubMed] [Google Scholar]
- Drew PD, Chavis JA. The cyclopentone prostaglandin 15-deoxy-Delta(12,14) prostaglandin J2 represses nitric oxide, TNF-alpha, and IL-12 production by microglial cells. J Neuroimmunol. 2001a;115:28–35. doi: 10.1016/s0165-5728(01)00267-3. [DOI] [PubMed] [Google Scholar]
- Drew PD, Lonergan M, Goldstein ME, Lampson LA, Ozato K, McFarlin DE. Regulation of MHC class I and beta 2-microglobulin gene expression in human neuronal cells. Factor binding to conserved cis-acting regulatory sequences correlates with expression of the genes. J Immunol. 1993b;150:3300–3310. [PubMed] [Google Scholar]
- Edwards PA, Kast HR, Anisfeld AM. BAREing it all: the adoption of LXR and FXR and their roles in lipid homeostasis. J Lipid Res. 2002;43:2–12. [PubMed] [Google Scholar]
- Feinstein DL, Galea E, Gavrilyuk V, Brosnan CF, Whitacre CC, Dumitrescu-Ozimek L, Landreth GE, Pershadsingh HA, Weinberg G, Heneka MT. Peroxisome proliferator-activated receptor-gamma agonists prevent experimental autoimmune encephalomyelitis. Ann Neurol. 2002;51:694–702. doi: 10.1002/ana.10206. [DOI] [PubMed] [Google Scholar]
- Fowler AJ, Sheu MY, Schmuth M, Kao J, Fluhr JW, Rhein L, Collins JL, Willson TM, Mangelsdorf DJ, Elias PM, Feingold KR. Liver X receptor activators display anti-inflammatory activity in irritant and allergic contact dermatitis models: liver-X-receptor-specific inhibition of inflammation and primary cytokine production. J Invest Dermatol. 2003;120:246–255. doi: 10.1046/j.1523-1747.2003.12033.x. [DOI] [PubMed] [Google Scholar]
- Gilden DH. Infectious causes of multiple sclerosis. Lancet Neurol. 2005;4:195–202. doi: 10.1016/S1474-4422(05)01017-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holley JE, Gveric D, Newcombe J, Cuzner ML, Gutowski NJ. Astrocyte characterization in the multiple sclerosis glial scar. Neuropathol Appl Neurobiol. 2003;29:434–444. doi: 10.1046/j.1365-2990.2003.00491.x. [DOI] [PubMed] [Google Scholar]
- Janowski BA, Grogan MJ, Jones SA, Wisely GB, Kliewer SA, Corey EJ, Mangelsdorf DJ. Structural requirements of ligands for the oxysterol liver X receptors LXRalpha and LXRbeta. Proc Natl Acad Sci U S A. 1999;96:266–271. doi: 10.1073/pnas.96.1.266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang C, Ting AT, Seed B. PPAR-gamma agonists inhibit production of monocyte inflammatory cytokines. Nature. 1998;391:82–86. doi: 10.1038/34184. [DOI] [PubMed] [Google Scholar]
- Joseph SB, Bradley MN, Castrillo A, Bruhn KW, Mak PA, Pei L, Hogenesch J, O'Connell RM, Cheng G, Saez E, Miller JF, Tontonoz P. LXR-dependent gene expression is important for macrophage survival and the innate immune response. Cell. 2004a;119:299–309. doi: 10.1016/j.cell.2004.09.032. [DOI] [PubMed] [Google Scholar]
- Joseph SB, Castrillo A, Laffitte BA, Mangelsdorf DJ, Tontonoz P. Reciprocal regulation of inflammation and lipid metabolism by liver X receptors. Nature Medicine. 2003b;9:213–219. doi: 10.1038/nm820. [see comment] [DOI] [PubMed] [Google Scholar]
- Joseph SB, McKilligin E, Pei L, Watson MA, Collins AR, Laffitte BA, Chen M, Noh G, Goodman J, Hagger GN, Tran J, Tippin TK, Wang X, Lusis AJ, Hsueh WA, Law RE, Collins JL, Willson TM, Tontonoz P. Synthetic LXR ligand inhibits the development of atherosclerosis in mice. Proc Natl Acad Sci U S A. 2002c;99:7604–7609. doi: 10.1073/pnas.112059299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawahito Y, Kondo M, Tsubouchi Y, Hashiramoto A, Bishop-Bailey D, Inoue K, Kohno M, Yamada R, Hla T, Sano H. 15-deoxy-delta(12,14)-PGJ(2) induces synoviocyte apoptosis and suppresses adjuvant-induced arthritis in rats. J Clin Invest. 2000;106:189–197. doi: 10.1172/JCI9652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kielian T. Toll-like receptors in central nervous system glial inflammation and homeostasis. J Neurosci Res. 2006a;83:711–730. doi: 10.1002/jnr.20767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kielian T, Drew PD. Effects of peroxisome proliferator-activated receptor-gamma agonists on central nervous system inflammation. J Neurosci Res. 2003b;71:315–325. doi: 10.1002/jnr.10501. [DOI] [PubMed] [Google Scholar]
- Li AC, Brown KK, Silvestre MJ, Willson TM, Palinski W, Glass CK. Peroxisome proliferator-activated receptor gamma ligands inhibit development of atherosclerosis in LDL receptor-deficient mice. J Clin Invest. 2000;106:523–531. doi: 10.1172/JCI10370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovett-Racke AE, Hussain RZ, Northrop S, Choy J, Rocchini A, Matthes L, Chavis JA, Diab A, Drew PD, Racke MK. Peroxisome proliferator-activated receptor alpha agonists as therapy for autoimmune disease. J Immunol. 2004;172:5790–5798. doi: 10.4049/jimmunol.172.9.5790. [DOI] [PubMed] [Google Scholar]
- Marrie RA. Environmental risk factors in multiple sclerosis aetiology. Lancet Neurol. 2004;3:709–718. doi: 10.1016/S1474-4422(04)00933-0. [DOI] [PubMed] [Google Scholar]
- Martin R, McFarland HF. Immunological aspects of experimental allergic encephalomyelitis and multiple sclerosis. Crit Rev Clin Lab Sci. 1995;32:121–182. doi: 10.3109/10408369509084683. [DOI] [PubMed] [Google Scholar]
- McCarthy KD, de Vellis J. Preparation of separate astroglial and oligodendroglial cell cultures from rat cerebral tissue. Journal of Cell Biology. 1980;85:890–902. doi: 10.1083/jcb.85.3.890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McFarlin DE, McFarland HF. Multiple sclerosis (first of two parts) N Engl J Med. 1982a;307:1183–1188. doi: 10.1056/NEJM198211043071905. [DOI] [PubMed] [Google Scholar]
- McFarlin DE, McFarland HF. Multiple sclerosis (second of two parts) N Engl J Med. 1982b;307:1246–1251. doi: 10.1056/NEJM198211113072005. [DOI] [PubMed] [Google Scholar]
- Natarajan C, Bright JJ. Peroxisome proliferator-activated receptor-gamma agonists inhibit experimental allergic encephalomyelitis by blocking IL-12 production, IL-12 signaling and Th1 differentiation. Genes Immun. 2002;3:59–70. doi: 10.1038/sj.gene.6363832. [DOI] [PubMed] [Google Scholar]
- Niino M, Iwabuchi K, Kikuchi S, Ato M, Morohashi T, Ogata A, Tashiro K, Onoe K. Amelioration of experimental autoimmune encephalomyelitis in C57BL/6 mice by an agonist of peroxisome proliferator-activated receptor-gamma. J Neuroimmunol. 2001;116:40–48. doi: 10.1016/s0165-5728(01)00285-5. [DOI] [PubMed] [Google Scholar]
- Raine CS. The Norton Lecture: a review of the oligodendrocyte in the multiple sclerosis lesion. J Neuroimmunol. 1997;77:135–152. doi: 10.1016/s0165-5728(97)00073-8. [DOI] [PubMed] [Google Scholar]
- Ricote M, Li AC, Willson TM, Kelly CJ, Glass CK. The peroxisome proliferator-activated receptor-gamma is a negative regulator of macrophage activation. Nature. 1998;391:79–82. doi: 10.1038/34178. [DOI] [PubMed] [Google Scholar]
- Storer PD, Xu J, Chavis J, Drew PD. Peroxisome proliferator-activated receptor-gamma agonists inhibit the activation of microglia and astrocytes: implications for multiple sclerosis. J Neuroimmunol. 2005a;161:113–122. doi: 10.1016/j.jneuroim.2004.12.015. [DOI] [PubMed] [Google Scholar]
- Storer PD, Xu J, Chavis JA, Drew PD. Cyclopentenone prostaglandins PGA2 and 15-deoxy-delta12,14 PGJ2 suppress activation of murine microglia and astrocytes: implications for multiple sclerosis. J Neurosci Res. 2005b;80:66–74. doi: 10.1002/jnr.20413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su CG, Wen X, Bailey ST, Jiang W, Rangwala SM, Keilbaugh SA, Flanigan A, Murthy S, Lazar MA, Wu GD. A novel therapy for colitis utilizing PPAR-gamma ligands to inhibit the epithelial inflammatory response. J Clin Invest. 1999;104:383–389. doi: 10.1172/JCI7145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapp BD, Peterson J, Ransohoff RM, Rudick R, Mork S, Bo L. Axonal transection in the lesions of multiple sclerosis. N Engl J Med. 1998;338:278–285. doi: 10.1056/NEJM199801293380502. [DOI] [PubMed] [Google Scholar]
- Wang J, Asensio VC, Campbell IL. Cytokines and chemokines as mediators of protection and injury in the central nervous system assessed in transgenic mice. Curr Top Microbiol Immunol. 2002;265:23–48. doi: 10.1007/978-3-662-09525-6_2. [DOI] [PubMed] [Google Scholar]
- Wekerle H. Tackling multiple sclerosis. Nature. 2002;420:39–40. doi: 10.1038/420039a. [DOI] [PubMed] [Google Scholar]
- Xiao BG, Link H. Antigen-specific T cells in autoimmune diseases with a focus on multiple sclerosis and experimental allergic encephalomyelitis. Cell Mol Life Sci. 1999;56:5–21. doi: 10.1007/s000180050002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Chavis JA, Racke MK, Drew PD. Peroxisome proliferator-activated receptor-alpha and retinoid X receptor agonists inhibit inflammatory responses of astrocytes. J Neuroimmunol. 2006a;176:95–105. doi: 10.1016/j.jneuroim.2006.04.019. [DOI] [PubMed] [Google Scholar]
- Xu J, Storer PD, Chavis JA, Racke MK, Drew PD. Agonists for the peroxisome proliferator-activated receptor-alpha and the retinoid X receptor inhibit inflammatory responses of microglia. J Neurosci Res. 2005b;81:403–411. doi: 10.1002/jnr.20518. [DOI] [PubMed] [Google Scholar]
- Zeinstra E, Wilczak N, De Keyser J. Reactive astrocytes in chronic active lesions of multiple sclerosis express co-stimulatory molecules B7-1 and B7-2. J Neuroimmunol. 2003;135:166–171. doi: 10.1016/s0165-5728(02)00462-9. [DOI] [PubMed] [Google Scholar]