

Abstract
We describe the genome sequence of the protist Trichomonas vaginalis, a sexually transmitted human pathogen. Repeats and transposable elements comprise about two-thirds of the ~160-megabase genome, reflecting a recent massive expansion of genetic material. This expansion, in conjunction with the shaping of metabolic pathways that likely transpired through lateral gene transfer from bacteria, and amplification of specific gene families implicated in pathogenesis and phagocytosis of host proteins may exemplify adaptations of the parasite during its transition to a urogenital environment. The genome sequence predicts previously unknown functions for the hydrogenosome, which support a common evolutionary origin of this unusual organelle with mitochondria.
Trichomonas vaginalis is a flagellated protist that causes trichomoniasis, a common but overlooked sexually transmitted human infection, with ~170 million cases occurring annually worldwide (1). The extracellular parasite resides in the urogenital tract of both sexes and can cause vaginitis in women and urethritis and prostatitis in men. Acute infections are associated with pelvic inflammatory disease, increased risk of human immunodeficiency virus 1 (HIV-1) infection, and adverse pregnancy outcomes. T. vaginalis is a member of the parabasilid lineage of microaerophilic eukaryotes that lack mitochondria and peroxisomes but contain unusual organelles called hydrogenosomes. Although previously considered to be one of the earliest branching eukaryotic lineages, recent analyses leave the evolutionary relationship of parabasalids to other major eukaryotic groups unresolved (2, 3). In this article, we report the draft sequence of T. vaginalis, the first parabasalid genome to be described.
Genome structure, RNA processing, and lateral gene transfer
The T. vaginalis genome sequence, generated using whole-genome shotgun methodology, contains 1.4 million shotgun reads assembled into 17,290 scaffolds at ~7.2× coverage (4). At least 65% of the T. vaginalis genome is repetitive (table S1). Despite several procedures developed to improve the assembly (4), the superabundance of repeats resulted in a highly fragmented sequence, preventing investigation of T. vaginalis genome architecture. The repeat sequences also hampered measurement of genome size, but we estimate it to be ~160 Mb (4). A core set of ~60,000 protein-coding genes was identified (Table 1), endowing T. vaginalis with one of the highest coding capacities among eukaryotes (table S2). Introns were identified in 65 genes, including the ~20 previously documented (5). Transfer RNAs (tRNAs) for all 20 amino acids were found, and ~250 ribosomal DNA (rDNA) units were identified on small contigs and localized to one of the six T. vaginalis chromosomes (Fig. 1).
Table 1.
Summary of the T. vaginalis genome sequence data. Assembly size (bp, base pairs) includes all contigs and differs from estimated genome size of ~160 Mb (4). The scaffold size is the minimum scaffold length, such that more than half the genome is contained in scaffolds of at least that length. The number of predicted genes may include low-complexity repeats or novel transposable elements rather than true T. vaginalis genes, but in the absence of decisive evidence these remain in the gene set. The number of evidence-supported genes includes those with either similarity to a known protein (E < 1 × 10−10, >25% length of protein) or similarity to an expressed sequence tag (>95% identity over >90% length of the gene). A total of 763 rDNA fragments (258 copies of 28S, 254 copies of 18S, and 251 copies of 5.8S) were identified.
| Feature | Value |
|---|---|
| Genome | |
| Size of assembly (bp) | 176,441,227 |
| G+C content (%) | 32.7 |
| No. of scaffolds | 17,290 |
| N50 scaffold size (bp) | 68,338 |
| Protein-coding genes | |
| No. of predicted genes | 59,681 |
| No. of evidence-supported genes | 25,949 |
| No. of genes with introns | 65 |
| Mean gene length (bp) | 928.6 |
| Gene G+C content (%) | 35.5 |
| Gene density (bp) | 2956 |
| Mean length of intergenic regions (bp) | 1165.4 |
| Intergenic G+C content (%) | 28.8 |
| Non–protein-coding genes | |
| Predicted tRNA genes | 479 |
| Predicted 5.8S, 18S, and 28S rDNA units | ~250 |
Fig. 1.

Karyotype and fluorescent in situ hybridization (FISH) analysis of T. vaginalis chromosomes. (A) Metaphase chromosome squashes of T. vaginalis reveal six chromosomes (I to VI). (B) FISH analysis using an 18S rDNA probe shows that all ~250 rDNA units localize to a single chromosome. (C) In contrast, the Tvmar1 transposable element (8) is dispersed throughout the genome.
The Inr promoter element was found in ~75% of 5′ untranslated region (UTR) sequences (4), supporting its central role in gene expression (6). Intriguingly, the eukaryotic transcription machinery of T. vaginalis appears more metazoan than protistan (table S3 to S5). The presence of a T. vaginalis Dicer-like gene, two Argonaute genes, and 41 transcriptionally active DEAD-DEAH-box helicase genes suggests the existence of an RNA interference (RNAi) pathway (fig. S1). Identification of these components raises the possibility of using RNAi technology to manipulate T. vaginalis gene expression.
During genome annotation, we identified 152 cases of possible prokaryote-to-eukaryote lateral gene transfer (LGT) [tables S6 and S7 and Supporting Online Material (SOM) text], augmenting previous reports of conflicting phylogenetic relationships among several enzymes (7). The putative functions of these genes are diverse, affecting various metabolic pathways (fig. S2) and strongly influencing the evolution of the T. vaginalis metabolome. A majority (65%) of the 152 LGT genes encode metabolic enzymes, more than a third of which are involved in carbohydrate or amino acid metabolism (Fig. 2). Several LGT genes may have been acquired from Bacteroidetes-related bacteria, which are abundant among vertebrate intestinal flora (fig. S3).
Fig. 2.

Schematic of T. vaginalis amino acid metabolism. A complete description of enzymatic reactions (represented as numbers) is given in the SOM text. Broken lines represent enzymes for which no gene was identified in the genome sequence, although the activity would appear to be required. Green boxes indicate enzymes encoded by candidate LGT genes.
Repeats, transposable elements, and genome expansion
The most common 59 repeat families identified in the assembly (4) constitute ~39 Mb of the genome and can be classified as (i) virus-like; (ii) transposon-like, including ~1000 copies of the first mariner element identified outside animals (8); (iii) retrotransposon-like; and (iv) unclassified (Table 2). Most of the 59 repeats are present in hundreds of copies (average copy number ~660) located on small (1- to 5-kb) contigs, and each repeat family is extraordinarily homogenous, with an average polymorphism of ~2.5%.
Table 2.
Summary of highly repetitive sequences in the genome of T. vaginalis. The 31 unclassified repeat families have been collapsed into one group.
| Repeat name | Putative identity | Copy no. | Length (bp) | Cumulative length (bp) | Average pairwise difference (%) |
|---|---|---|---|---|---|
| Viral | |||||
| R128b | Poxvirus D5 protein | 203 | 2348 | 370,658 | 1.2 |
| R169a | Phage tail fiber prot | 903 | 752 | 607,929 | 3.9 |
| R1794 | Hypothetical | 2243 | 1037 | 1,879,762 | 3.0 |
| R299a | KilA-N terminal domain | 867 | 873 | 655,984 | 6.6 |
| R3a | Poxvirus D5 protein | 954 | 2637 | 1,721,187 | 4.6 |
| R9a | KilA-N terminal domain | 831 | 663 | 503,959 | 3.6 |
| R947a | KilA-N terminal domain | 518 | 669 | 298,778 | 3.1 |
| Average (total) | 931 | 1283 | (6,038,257) | 3.7 | |
| Transposon | |||||
| R8 | Mariner transposase | 982 | 1304 | 1,158,473 | 0.5 |
| R107 | Integrase | 384 | 2246 | 518,936 | 0.9 |
| R119 | Mutator-like profile | 282 | 2954 | 303,256 | 1.1 |
| R11b | Integrase | 1842 | 981 | 1,634,764 | 2.1 |
| R128a | HNH endonuclease | 200 | 800 | 131,491 | 1.5 |
| R130a | Mutator-like profile | 173 | 1129 | 137,526 | 0.7 |
| R165 | Mutator-like profile | 365 | 2410 | 368,152 | 1.1 |
| R178 | Integrase | 580 | 2433 | 636,901 | 1.5 |
| R204 | Integrase | 51 | 1323 | 51,185 | 1.6 |
| R210 | Mutator-like profile | 75 | 2127 | 118,581 | 0.9 |
| R2375 | Endonuclease | 566 | 765 | 329,717 | 5.4 |
| R242b | Integrase | 49 | 1151 | 45,564 | 2.5 |
| R26b | Integrase | 1148 | 1141 | 1,175,921 | 2.6 |
| R289a | Integrase | 54 | 1250 | 53,674 | 2.2 |
| R309b | Integrase | 56 | 1601 | 73,737 | 1.4 |
| R414a | Integrase | 37 | 909 | 27,708 | 1.4 |
| R41b | Integrase | 927 | 1184 | 807,711 | 2.5 |
| R473 | Integrase | 19 | 1124 | 15,254 | 1.3 |
| Integ.a95313 | Integrase | 68 | 1301 | 22,023 | 2.3 |
| Average (total) | 414 | 1481 | (7,610,574) | 1.8 | |
| Retrotransposon | |||||
| R1407.RT | Reverse transcriptase | 6 | 2349 | 6,833 | 1.0 |
| copia.a38393 | Copia-like profile | 7 | 11001 | 16,504 | 7.5 |
| Average (total) | 6.5 | 6675 | (23,337) | 4.3 | |
| 31 unclassified families | |||||
| Average (total) | 793 | 1131 | (24,617,704) | 2.6 | |
| Total all | 38,289,872 | ||||
| Average all | 660 | 1450 | 2.5 | ||
The lack of a strong correlation between copy number and average pairwise difference between copies (fig. S4) suggests that a sudden expansion of the repeat families had occurred. To estimate the time of expansion, we compared the degree of polymorphism among T. vaginalis repeats to the divergence between T. vaginalis and its sister taxon T. tenax, a trichomonad of the oral cavity (9), for several protein-coding loci (4). Our results indicate that repeat family amplification occurred after the two species split (table S8). Several families have also undergone multiple expansions, as implied by bi- or trimodal distribution of pairwise distances between copies (fig. S5). T. vaginalis repeat families appear to be absent in T. tenax but are present in geographically diverse T. vaginalis (4), consistent with the expansion having occurred after speciation but before diversification of T. vaginalis.
The large genome size, high repeat copy number, low repeat polymorphism, and evidence of repeat expansion after T. vaginalis and T. tenax diverged suggest that T. vaginalis has undergone a very recent and substantial increase in genome size. To determine whether the genome underwent any large-scale duplication event(s), we analyzed age distributions of gene families with five or fewer members (4). A peak in the age distribution histogram of pairs of gene families was observed (fig. S6), indicating that the genome underwent a period of increased duplication, and possibly one or more large-scale genome duplication events.
Metabolism, oxidative stress, and transport
T. vaginalis uses carbohydrate as a main energy source via fermentative metabolism under aerobic and anaerobic conditions. We found the parasite to use a variety of amino acids as energy substrates (Fig. 2) (10), with arginine dihydrolase metabolism a major pathway for energy production (fig. S7) (11). We confirmed a central role for aminotransferases (Fig. 2 and table S9) and glutamate dehydrogenase as indicated previously (12, 13); these pathways are likely catabolic but may be reversible to allow the parasite to synthesize glutamate, aspartate, alanine, glutamine, and glycine. Genes required for synthesis of proline from arginine (fig. S7) and for threonine metabolism (fig. S8) were identified. We also identified a de novo biosynthesis pathway for cysteine via cysteine synthase, an LGT candidate (fig. S8) (14), and genes encoding enzymes involved in methionine metabolism, including its possible regeneration (fig. S9).
Earlier studies indicated that de novo lipid biosynthesis in T. vaginalis is confined to the major phospholipid phosphatidylethanolamine (PE) (15), whereas other lipids, including cholesterol, are likely acquired from exogenous sources. We found an absence of several essential enzyme-encoding genes in the synthesis and degradation pathways of nearly all lipids (4), in contrast to the PE synthetic pathway, which appears complete; however, experimental verification of these results is required.
T. vaginalis is microaerophilic with a primarily anaerobic life style and thus requires redox and antioxidant systems to counter the detrimental effects of oxygen. Genes encoding a range of defense molecules, such as superoxide dismutases, thioredoxin reductases, peroxiredoxins, and rubrerythrins, were identified (table S10).
T. vaginalis demonstrates a broad range of transport capabilities, facilitated by expansion of particular transporter families, such as those for sugar and amino acids (table S11). The parasite also possesses more members of the cation-chloride cotransporter (CCC) family than any other sequenced eukaryote, likely reflecting osmotic changes faced by the parasite in a mucosal environment.
None of the proteins required for glycosyl-phosphatidylinositol (GPI)-anchor synthesis were identified in the genome sequence, making T. vaginalis the first eukaryote known to lack an apparent GPI-anchor biosynthetic pathway. Whether T. vaginalis has evolved an unusual biosynthetic pathway for synthesis of its nonprotein lipid anchors, such as the inositol-phosphoceramide of surface lipophosphoglycans (16), remains to be determined.
Massively expanded gene families
Many gene families in the T. vaginalis genome have undergone expansion on a scale unprecedented in unicellular eukaryotes (Table 3). Such “conservative” gene family expansions are likely to improve an organism’s adaptation to its environment (17). Notably, the selective expansion of subsets of the membrane trafficking machinery, critical for secretion of pathogenic proteins, endocytosis of host proteins, and phagocytosis of bacteria and host cells (table S12), correlates well with the parasite’s active endocytic and phagocytic life-style.
Table 3.
Large gene families in T. vaginalis. Only gene families, or aggregates of gene families mediating a given process, for which there are more than 30 members and that have been assigned a putative function are listed. (See SOM text for subfamily organization.) The small guano-sine triphosphatases (GTPases) are the sum of Rab and ARF small GTPases only; all other GTPases are shown in table S12. ABC, ATP-binding cassette; MFS, major facilitator superfamily; MOP, multi-drug/oligosaccharidyl-lipid/polysaccharide; AAAP, amino acid/auxin permease.
| Gene family (functional unit) | Members |
|---|---|
| Protein kinases | 927 |
| BspA-like gene family | 658 |
| Membrane trafficking: small GTPases | 328 |
| rDNA gene cluster | ~250 |
| Cysteine peptidase (clan CA, family C19) | 117 |
| Membrane trafficking: vesicle formation | 113 |
| ABC transporter superfamily | 88 |
| GP63-like (leishmanolysin) | 77 |
| MFS transporter family | 57 |
| MOP flippase transporter family | 47 |
| Cysteine peptidase (clan CA, family C1) | 48 |
| AAAP transporter family | 40 |
| Dynein heavy chain | 35 |
| P-ATPase transporter family | 33 |
| Serine peptidase (clan SB, family S8) | 33 |
| Membrane trafficking: vesicle fusion | 31 |
Massively amplified gene families also occur in the parasite’s kinome, which comprises ~880 genes (SOM text) encoding distinct eukaryotic protein kinases (ePKs) and ~40 atypical protein kinases, making it one of the largest eukaryotic kinomes known. The parasite has heterotrimeric guanine nucleotide–binding proteins and components of the mitogen-activated protein kinase (MAPK) pathway, suggesting yeast-like signal transduction mechanisms. Unusually, the T. vaginalis kinome contains 124 cytosolic tyrosine kinase–like (TKL) genes, yet completely lacks receptor serine/threonine ePKs of the TKL family. Inactive kinases were found to make up 17% of the T. vaginalis kinome (table S13); these may act as substrates and scaffolds for assembly of signaling complexes (18). ePK accessory domains are important for regulating signaling pathways, but just nine accessory domain types were identified in 8% (72/883) of the T. vaginalis ePKs (table S14), whereas ~50% of human ePKs contain at least 1 of 83 accessory domain types. This suggests that regulation of protein kinase function and cell signaling in T. vaginalis is less complex than that in higher eukaryotes, a possible explanation for the abundance of T. vaginalis ePKs.
T. vaginalis possesses several unusual cyto-skeletal structures: the axostyle, the pelta, and the costa (19). Most actin- and tubulin-related components of the cytoskeleton are present (table S15), with the exception of homologs of the actin motor myosin. In contrast, homologs of the microtubular motors kinesin and dynein are unusually abundant (Table 3). Thus, T. vaginalis intracellular transport mechanisms are mediated primarily by kinesin and cytoplasmic dynein, as described for Dictyo-stelium and filamentous fungi, raising the possibility that the loss of myosin-driven cytoplasmic transport is not uncommon in unicellular eukaryotes (20). Whether the structural remodeling of amoeboid T. vaginalis during host colonization (see below) is actin-based, as described for other eukaryotes, or driven by novel cytoskeletal rearrangements remains an open question.
We identified homologs of proteins involved in DNA damage response and repair, chromatin restructuring, and meiosis, the latter a process not thought to occur in the parasite (table S16). Of the 29 core meiotic genes found, several are general repair proteins required for meiotic progression in other organisms (21), and eight are meiosis-specific proteins. Thus, T. vaginalis contains either recent evolutionary relics of meiotic machinery or genes functional in meiotic recombination in an as-yet undescribed sexual cycle.
Molecular mechanisms of pathogenesis
T. vaginalis must adhere to host cells to establish and maintain an infection. A dense glycocalyx composed of lipophosphoglycan (LPG) (Fig. 3) and surface proteins has been implicated in adherence (22), but little is known about this critical pathogenic process. We identified genes encoding enzymes predicted to be required for LPG synthesis (table S17). Of particular interest are the genes required for synthesis of an unusual nucleotide sugar found in T. vaginalis LPG, the monosaccharide rhamnose, which is absent in the human host, making it a potential drug target. Genes (some of which are LGT candidates) were identified that are involved in sialic acid biosynthesis, consistent with the reported presence of this sugar on the parasite surface (23).
Fig. 3.
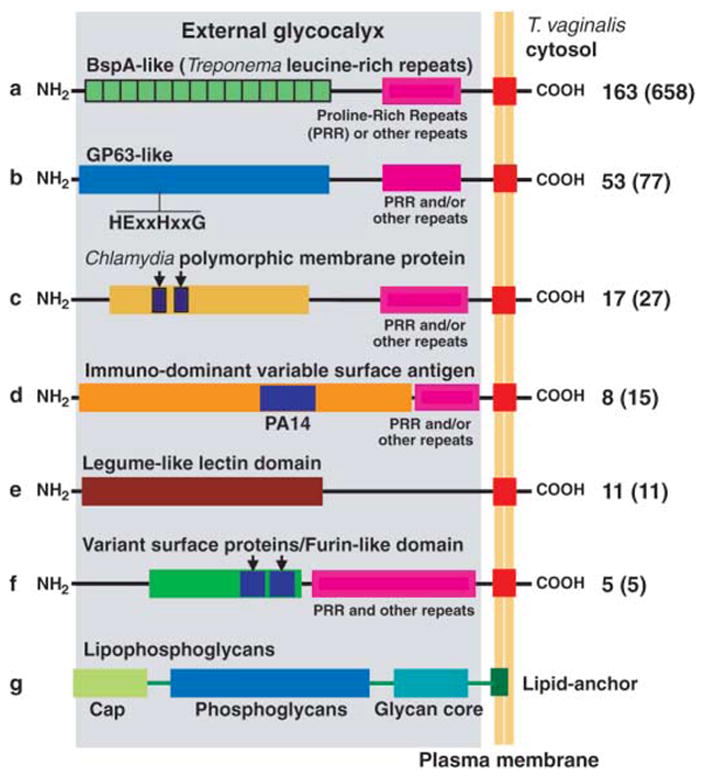
Structural organization of putative T. vaginalis surface molecules involved in host cell adherence and cyto-toxicity (28). Candidate surface protein families (a to f) are depicted as part of the glycocalyx (gray shading), known to be composed of LPG (g). The number of proteins with an inferred transmembrane domain (red box) is indicated at right, with the size of the entire family shown in parentheses. Substantial length variation exists between and within families (proteins are not drawn to scale). Additional information can be found in table S18.
We identified eight families containing ~800 proteins (4) that represent candidate surface molecules (Fig. 3 and table S18), including ~650 highly diverse BspA-like proteins characterized by the Treponema pallidum leucine-rich repeat, TpLLR. BspA-like proteins are expressed on the surface of certain pathogenic bacteria and mediate cell adherence and aggregation (24). The only other eukaryote known to encode BspA-like proteins, the mucosal pathogen Entamoeba histolytica (25), contains 91 such proteins, one of which was recently localized to the parasite surface (26).
There are >75 T. vaginalis GP63-like proteins, homologs of the most abundant surface proteins of Leishmania major, the leishmanolysins, which contribute to virulence and pathogenicity through diverse functions in both the insect vector and the mammalian host (27). Most T. vaginalis GP63-like genes possess the domains predicted to be required for a catalytically active metallopeptidase, including a short HEXXH motif (28) (Fig. 3). Unlike trypanosomatid GP63 proteins, which are predicted to be GPI-anchored, most T. vaginalis GP63-like proteins have a predicted C-terminal transmembrane domain as a putative cell surface anchor, consistent with the apparent absence of GPI-anchor biosynthetic enzymes. Other T. vaginalis protein families share domains with Chlamydia polymorphic membrane proteins, Giardia lamblia variant surface proteins, and E. histolytica immunodominant variable surface antigens (Fig. 3 and table S18).
After cytoadherence, the parasite becomes amoeboid, increasing cell-to-cell surface contacts and forming cytoplasmic projections that interdigitate with target cells (19). We have identified genes encoding cytolytic effectors, which may be released upon host-parasite contact. T. vaginalis lyses host red blood cells, presumably as a means of acquiring lipids and iron and possibly explaining the exacerbation of symptoms observed during menstruation (29). This hemolysis is dependent on contact, temperature, pH, and Ca2+, suggesting the involvement of pore-forming proteins (30) that insert into the lipid bilayer of target cells, mediating osmotic lysis. Consistent with this, we have identified 12 genes (TvSaplip1 to TvSaplip12) containing saposin-like (SAPLIP) pore-forming domains (fig. S10). These domains show a predicted six-cysteine pattern and abundant hydrophobic residues in conserved positions while displaying high sequence variability (fig. S11). The TvSaplips are similar to amoebapore proteins secreted by E. histolytica and are candidate trichopores that mediate a cytolytic effect.
The degradome
Peptidases perform many critical biological processes and are potential virulence factors, vaccine candidates, and drug targets (31). T. vaginalis contains an expanded degradome of more than 400 peptidases (SOM text), making it one of the most complex degradomes described (table S19). Of the three families of aspartic peptidases (table S20), T. vaginalis contains a single member of the HIV-1 retropepsin family that might serve as a putative candidate for anti-HIV peptidase inhibitors. Many studies have implicated papain family cysteine peptidases as virulence factors in trichomonads; we identified >40 of them, highlighting the diversity of this family. Cysteine peptidases that contribute to the 20S proteosome (ubiquitin C-terminal hydrolases) are abundant (117 members, ~25% of the degradome), emphasizing the importance of cytosolic protein degradation in the parasite. T. vaginalis has nine NlpC/P60-like members (table S20; several of which are LGT candidates), which play a role in bacterial cell wall degradation and the destruction of healthy vaginal microflora, making the vaginal mucosa more sensitive to other infections.
We also identified many subtilisin-like and several rhomboid-like serine peptidases, candidates for processing T. vaginalis surface proteins. In addition to the first asparaginase-type of threonine peptidase found in a protist, 13 families of metallopeptidases were also identified, as well as three cystatin-like proteins, natural peptidase inhibitors, which may regulate the activity of the abundant papain-like cysteine peptidases (table S20).
The hydrogenosome
Several microaerophilic protists and fungi, including trichomonads and ciliates, lack typical mitochondria and possess double-membrane hydrogenosomes, which produce adenosine triphophate (ATP) and molecular hydrogen through fermentation of metabolic intermediates produced in the cytosol. Although the origin of these organelles has been controversial, most evidence now supports a common origin with mitochondria (3). Few genes encoding homologs of mitochondrial transporters, translocons, and soluble proteins were identified in the T. vaginalis genome (fig. S12), suggesting that its hydrogenosome has undergone reductive evolution comparable to other protists whose mitochondrial proteomes are reduced (e.g., Plasmodium). Because nuclear-encoded hydrogenosomal matrix proteins are targeted to the organelle by N-terminal presequences that are proteolytically cleaved upon import (32) similar to mitochondrial precursor proteins, we screened the genome for consensus 5-to 20-residue presequences containing ML(S/T/A) X(1...15)R (N/F/E/XF), MSLX(1...15)R(N/F/XF), or MLR(S/N)F (28) motifs. A total of 138 genes containing putative presequences were identified, 67% of which are similar to known proteins, primarily ones involved in energy metabolism and electron-transport pathways (fig. S13 and table S21).
The production of molecular hydrogen, the hallmark of the hydrogenosome, is catalyzed by an unusually diverse group of iron-only [Fe]-hydrogenases that possess, in addition to a conserved H cluster, four different sets of functional domains (fig. S14), indicating that hydrogen production may be more complex than originally proposed. The pathway that generates electrons for hydrogen (fig. S12) is composed of many proteins encoded by multiple genes (tables S22 to S24). Our analyses extend the evidence that T. vaginalis hydrogenosomes contain the complete machinery required for mitochondria-like intraorganellar FeS cluster formation (33) and also reveal the presence of two putative cytosolic auxiliary proteins, indicating that hydrogenosomes may be involved in biogenesis of cytosolic FeS proteins. Some components of FeS cluster assembly machinery have also been found in mitosomes (mitochondrial remnants) of G. lamblia, supporting a common evolutionary origin of mitochondria, hydrogenosomes, and mitosomes (3).
A new predicted function of hydrogenosomes revealed by the genome sequence is amino acid metabolism. We identified two components of the glycine-cleavage complex (GCV), L protein and H protein. Another component of this pathway is serine hydroxymethyl transferase (SHMT), which in eukaryotes exists as both cytosolic and mitochondrial isoforms. A single gene coding for SHMT of the mitochondrial type with a putative N-terminal hydrogenosomal presequence was identified. Because both GCV and SHMT require folate (fig. S12), which T. vaginalis apparently lacks, the functionality of these proteins remains unclear.
The 5-nitroimidazole drugs metronidazole (Mz) and tinidazole are the only approved drugs for treatment of trichomoniasis. These prodrugs are converted within the hydrogenosome to toxic nitroradicals via reduction by ferredoxin (Fdx) (fig. S12). Clinical resistance to Mz (MzR) is estimated at 2.5 to 5% of reported cases and rising (34) and is associated with decreases in or loss of Fdx (35). We identified seven Fdx genes with hydrogenosomal targeting signals (tables S21 and S25), the redundancy of which provides explanations for the low frequency of MzR and for why knockout of a single Fdx gene does not lead to MzR (35). Our analyses also provide clues to the potential mechanisms that clinically resistant parasites may use, such as the presence of nitroreductase (NimA-like), reduced nicotinamide adenine dinucleotide phosphate (NADPH)–nitroreductase, and NADH-flavin oxidoreductase genes (tables S23 and S26), which have been implicated in MzR in bacteria (36, 37).
Summary and concluding remarks
Our investigation of the T. vaginalis genome sequence provides a new perspective for studying the biology of an organism that continues to be ignored as a public health issue despite the high number of trichomoniasis cases worldwide. The discovery of previously unknown metabolic pathways, the elucidation of pathogenic mechanisms, and the identification of candidate surface proteins likely involved in facilitating invasion of human mucosal surfaces provide potential leads for the development of new therapies and novel methods for diagnosis.
The analysis presented here of one of the most repetitive genomes known has undoubtedly been hampered by the sheer number of highly similar repeats and transposable elements. Why did this genome expand so dramatically in size? We hypothesize that the most recent common ancestor of T. vaginalis underwent a population bottleneck during its transition from an enteric environment (the habitat of most trichomonads) to the urogenital tract. During this time, the decreased effectiveness of selection resulted in repeat accumulation and differential gene family expansion. Genome size and cell volume are positively correlated (38, 39); hence, the increased genome size of T. vaginalis achieved through rapid fixation of repeat copies could have ultimately resulted in a larger cell size. T. vaginalis cell volume is greater than that of T. tenax and related intestinal species Pentratrichomonas hominis (40) and T. gallinae (41), and it generally conforms to the relationship of genome size to cell volume reported for protists (41). T. vaginalis is also a highly predatory parasite that phagocytoses bacteria, vaginal epithelial cells, and host erythrocytes (42) and is itself ingested by macrophages. Given these interactions, it is tempting to speculate that an increase in cell size could have been selected for in order to augment the parasite’s phagocytosis of bacteria, to reduce its own phagocytosis by host cells, and to increase the surface area for colonization of vaginal mucosa.
Supplementary Material
Footnotes
Supporting Online Material
www.sciencemag.org/cgi/content/full/315/5809/207/DC1
Materials and Methods
SOM Text
References
Data
References and Notes
- 1.Global Prevalence and Incidence of Selected Curable Sexually Transmitted Infections. World Health Organization; Geneva: 2001. www.who.int/docstore/hiv/GRSTI/006.htm. [Google Scholar]
- 2.Adl SM, et al. J Eukaryot Microbiol. 2005;52:399. doi: 10.1111/j.1550-7408.2005.00053.x. [DOI] [PubMed] [Google Scholar]
- 3.Embley TM, Martin W. Nature. 2006;440:623. doi: 10.1038/nature04546. [DOI] [PubMed] [Google Scholar]
- 4.Materials and methods are available as supporting material on. Science. Online. [Google Scholar]
- 5.Vanacova S, Yan W, Carlton JM, Johnson PJ. Proc Natl Acad Sci USA. 2005;102:4430. doi: 10.1073/pnas.0407500102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schumacher MA, Lau AO, Johnson PJ. Cell. 2003;115:413. doi: 10.1016/s0092-8674(03)00887-0. [DOI] [PubMed] [Google Scholar]
- 7.Muller M. In: Evolutionary Relationship Among Protozoa. Coombs GH, Vickerman V, Sleigh MA, Warren A, editors. Chapman & Hall; London: 1998. pp. 109–132. [Google Scholar]
- 8.Silva JC, Bastida F, Bidwell SL, Johnson PJ, Carlton JM. Mol Biol Evol. 2005;22:126. doi: 10.1093/molbev/msh260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kutisova K, et al. Parasitology. 2005;131:309. doi: 10.1017/s0031182005008000. [DOI] [PubMed] [Google Scholar]
- 10.Zuo X, Lockwood BC, Coombs GH. Microbiology. 1995;141:2637. doi: 10.1099/13500872-141-10-2637. [DOI] [PubMed] [Google Scholar]
- 11.Kleydman Y, Yarlett N, Gorrell TE. Microbiology. 2004;150:1139. doi: 10.1099/mic.0.26939-0. [DOI] [PubMed] [Google Scholar]
- 12.Lowe PN, Rowe AF. Mol Biochem Parasitol. 1986;21:65. doi: 10.1016/0166-6851(86)90080-0. [DOI] [PubMed] [Google Scholar]
- 13.Turner AC, Lushbaugh WB. Exp Parasitol. 1988;67:47. doi: 10.1016/0014-4894(88)90007-0. [DOI] [PubMed] [Google Scholar]
- 14.Westrop GD, Goodall G, Mottram JC, Coombs GH. J Biol Chem. 2006;281:25062. doi: 10.1074/jbc.M600688200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Beach DH, Holz GG, Jr, Singh BN, Lindmark DG. Mol Biochem Parasitol. 1991;44:97. doi: 10.1016/0166-6851(91)90225-u. [DOI] [PubMed] [Google Scholar]
- 16.Singh BN, Beach DH, Lindmark DG, Costello CE. Arch Biochem Biophys. 1994;309:273. doi: 10.1006/abbi.1994.1113. [DOI] [PubMed] [Google Scholar]
- 17.Vogel C, Chothia C. PLoS Comput Biol. 2006;2:e48. doi: 10.1371/journal.pcbi.0020048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Parsons M, Worthey EA, Ward PN, Mottram JC. BMC Genomics. 2005;6:127. doi: 10.1186/1471-2164-6-127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Benchimol M. Microsc Microanal. 2004;10:528. doi: 10.1017/S1431927604040905. [DOI] [PubMed] [Google Scholar]
- 20.Richards TA, Cavalier-Smith T. Nature. 2005;436:1113. doi: 10.1038/nature03949. [DOI] [PubMed] [Google Scholar]
- 21.Ramesh MA, Malik SB, Logsdon JM., Jr Curr Biol. 2005;15:185. doi: 10.1016/j.cub.2005.01.003. [DOI] [PubMed] [Google Scholar]
- 22.Bastida-Corcuera FD, Okumura CY, Colocoussi A, Johnson PJ. Eukaryot Cell. 2005;4:1951. doi: 10.1128/EC.4.11.1951-1958.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dias Filho BP, Andrade AF, de Souza W, Esteves MJ, Angluster J. Microbios. 1992;71:55. [PubMed] [Google Scholar]
- 24.Ikegami A, Honma K, Sharma A, Kuramitsu HK. Infect Immun. 2004;72:4619. doi: 10.1128/IAI.72.8.4619-4627.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Loftus B, et al. Nature. 2005;433:865. doi: 10.1038/nature03291. [DOI] [PubMed] [Google Scholar]
- 26.Davis PH, et al. Mol Biochem Parasitol. 2006;145:111. doi: 10.1016/j.molbiopara.2005.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yao C, Donelson JE, Wilson ME. Mol Biochem Parasitol. 2003;132:1. doi: 10.1016/s0166-6851(03)00211-1. [DOI] [PubMed] [Google Scholar]
- 28.Single-letter abbreviations for the amino acid residues are as follows: A, Ala; C, Cys; D, Asp; E, Glu; F, Phe; G, Gly; H, His; I, Ile; K, Lys; L, Leu; M, Met; N, Asn; P, Pro; Q, Gln; R, Arg; S, Ser; T, Thr; V, Val; W, Trp; X, any amino acid; and Y, Tyr.
- 29.Lehker MW, Chang TH, Dailey DC, Alderete JF. J Exp Med. 1990;171:2165. doi: 10.1084/jem.171.6.2165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fiori PL, Rappelli P, Rocchigiani AM, Cappuccinelli P. FEMS Microbiol Lett. 1993;109:13. doi: 10.1111/j.1574-6968.1993.tb06136.x. [DOI] [PubMed] [Google Scholar]
- 31.Klemba M, Goldberg DE. Annu Rev Biochem. 2002;71:275. doi: 10.1146/annurev.biochem.71.090501.145453. [DOI] [PubMed] [Google Scholar]
- 32.Bradley PJ, Lahti CJ, Plumper E, Johnson PJ. EMBO J. 1997;16:3484. doi: 10.1093/emboj/16.12.3484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sutak R, et al. Proc Natl Acad Sci USA. 2004;101:10368. doi: 10.1073/pnas.0401319101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schmid G, et al. J Reprod Med. 2001;46:545. [PubMed] [Google Scholar]
- 35.Land KM, et al. Mol Microbiol. 2004;51:115. doi: 10.1046/j.1365-2958.2003.03791.x. [DOI] [PubMed] [Google Scholar]
- 36.Kwon DH, et al. Antimicrob Agents Chemother. 2001;45:2609. doi: 10.1128/AAC.45.9.2609-2615.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Leiros HK, et al. J Biol Chem. 2004;279:55840. doi: 10.1074/jbc.M408044200. [DOI] [PubMed] [Google Scholar]
- 38.Cavalier-Smith T. Ann Bot (London) 2005;95:147. doi: 10.1093/aob/mci010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gregory TR. Biol Rev Camb Philos Soc. 2001;76:65. doi: 10.1017/s1464793100005595. [DOI] [PubMed] [Google Scholar]
- 40.Honigberg BM, Brugerolle G. In: Trichomonads Parasitic in Humans. Honigberg BM, editor. Springer-Verlag; New York: 1990. pp. 5–35. [Google Scholar]
- 41.Shuter BJ. Am Nat. 1983;122:26. [Google Scholar]
- 42.Rendon-Maldonado JG, Espinosa-Cantellano M, Gonzalez-Robles A, Martinez-Palomo A. Exp Parasitol. 1998;89:241. doi: 10.1006/expr.1998.4297. [DOI] [PubMed] [Google Scholar]
- 43.We are grateful to the Trichomonas research community and M. Gottlieb for support and guidance during the duration of this project. We thank M. Delgadillo-Correa, J. Shetty, S. van Aken, and S. Smith for technical support; T. Creasy, S. Angiuoli, H. Koo, J. Miller, J. Orvis, P. Amedeo, and E. Lee for engineering support; W. Majoros and G. Pertea for data manipulation; and S. Sullivan and E. F. Merino for editing. Funding for this project was provided by the National Institute of Allergy and Infectious Diseases (grants UO1 AI50913-01 and NIH-N01-AI-30071). The Burroughs Wellcome Fund and the Ellison Medical Foundation provided funds for Trichomonas community meetings during the genome project. The Chang Gung T. vaginalis Systems Biology Project was supported by grants (SMRPD33002 and SMRPG33115) from Chang Gung Memorial Hospital and Taiwan Biotech Company Limited. This whole genome shotgun project has been deposited at DNA Data Bank of Japan/European Molecular Biology Laboratory/GenBank under the project accession AAHC00000000. The version described here is the first version, AAHC01000000.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.