

Summary
Many biological functions depend critically upon fine details of tissue molecular architecture that have resisted exploration by existing imaging techniques. This is particularly true for nervous system tissues, where information processing function depends on intricate circuit and synaptic architectures. Here we describe a new imaging method, called array tomography, which combines and extends superlative features of modern optical fluorescence and electron microscopy methods. Based on methods for constructing and repeatedly staining and imaging ordered arrays of ultrathin (50-200 nm), resin-embedded serial sections on glass microscope slides, array tomography allows for quantitative, high-resolution, large-field volumetric imaging of large numbers of antigens, fluorescent proteins, and ultrastructure in individual tissue specimens. Compared to confocal microscopy, array tomography offers the advantage of better spatial resolution, in particular along the z-axis, as well as depth-independent immunofluorescent staining. The application of array tomography can reveal important but previously unseen features of brain molecular architecture.
Introduction
The tools most used today for imaging biological molecular architectures are immunofluorescence microscopy, immunoelectron microscopy and genetically encoded fluorescent protein markers and reporters. These powerful and widely used tools have led to discoveries that cell and tissue functions depend profoundly on the very finest details of molecular architecture, and that molecular architectures can be extremely variable and dynamic. However, each of these imaging methods has significant weaknesses and many important aspects of cell and tissue molecular architecture remain unexplored. While fluorescent protein markers have provided unique opportunities for imaging live cell dynamics, they require the expression of transgenes and are thus impractical for the study of human specimens. They may also perturb the molecular architectures of interest. Both immunofluorescence and immunoelectron microscopy are confined to use on fixed cells and tissues, but since these techniques do not require transgene expression, they are readily applied to human cell and tissues.
Immunofluorescence microscopy provides an ease of use and substantial multiplexing capacity that have made it one of the most widely used tools in cell biology, but it suffers from serious limitations in resolution and quantitative interpretability, especially as applied to tissues where antibody penetration becomes a limitation. Immunoelectron microscopy provides the highest resolution and has been alone in its power to discern the very fine details of molecular architecture, but it has proven difficult to apply to extended three-dimensional architectures and has limited multiplexing capacity. These microscopy limitations have thwarted attempts to describe the tissue architecture of most fundamental interest to neuroscience, that of the neural circuit.
We set out to develop a new imaging technique that would enhance and combine the strengths of presently available methods for imaging tissues in general and neural circuit architectures in particular. We began with an idea for improving the volumetric resolution of immunofluorescence microscopy, by addressing its most severe limitation-resolution along the z (focus) axis. Optical sectioning and improved z-axis resolution can be obtained by confocal as opposed to conventional widefield fluorescence microscopy, but even for the confocal, limiting resolution is far worse along the z-axis (rarely better than 700 nm) than in the x-y plane (∼250 nm) (Pawley, 1995). Furthermore, the optical compromises that typically arise in dealing with actual tissue specimens (e.g., numerical aperture limits, refractive index mismatches, aberrations) cause z-axis resolution to degrade much more rapidly that lateral resolution. Thus, we decided to explore the advantages of physical as opposed to optical sectioning approaches to z-axis resolution improvement. The concept is illustrated schematically in Figure 1, insert. A recent study showed that, in fact, the imaging of immunofluorescent ultrathin cryosections provides much better resolution than conventional, much thicker, cryostat sections imaged either by a fluorescent or confocal microscope (Mori et al., 2006). We chose to use ultrathin sections of tissue embedded in the acrylic resin LR White, because of the ease of handling such sections and well-known advantages of this hydrophilic resin for immunostaining (Newman et al., 1983) and the preservation of GFP fluorescence (Luby-Phelps et al., 2003). While not a routine method, ultrathin or semithin plastic sections have been used before for both preembedding (Burette, Khatri et al. 2001) and postembedding immunofluorescence studies (Albrecht et al., 1990; De Camilli et al., 1983; Fialka et al., 1996; Haraguchi and Yokota, 2002; Herken et al., 1988; Kurth et al., 1996; Ojeda et al., 1989).
Figure 1. Schematic representation of the array tomography method.
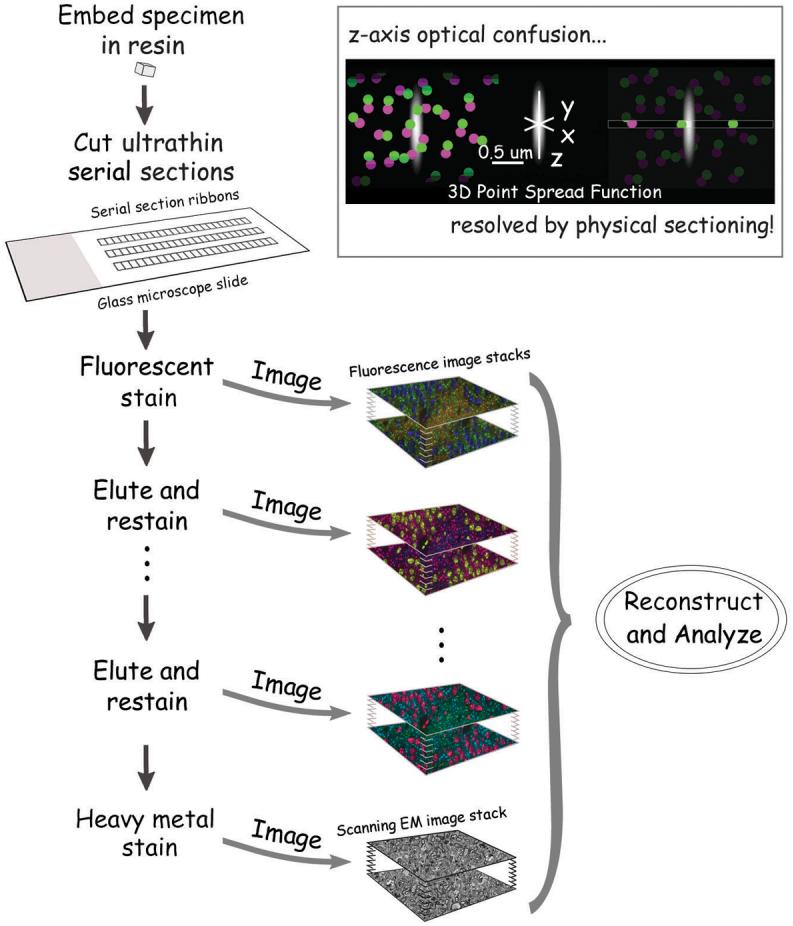
A tissue specimen is embedded in acrylic resin and cut into ribbons of serial ultrathin (50-200 nm) sections, which are then bonded to glass slides. The resulting array is labeled with fluorescent antibodies or other fluorescent stains and imaged to generate ultra-high-resolution volumetric images. The array can be repeatedly eluted, restained and fluorescently imaged, and finally, it can also be stained with heavy metals and imaged under a scanning electron microscope.
Insert illustrates the principle behind the axial resolution enhancement by array tomography. Optical microscopes have their poorest resolution along the optical axis, represented in the figure by z-axis elongation of the 3-D point spread function. Optical sectioning (left) yields an image that is severely degraded by confusion along the z-axis, a problem avoided in array tomography by using ultrathin physical sectioning.
Here we show that ribbons of serial ultrathin sections representing substantial tissue volumes can be cut and collected on a single glass slide and simultaneously processed and imaged for the reconstruction of the three-dimensional distribution of antigens. Staining antibodies can be subsequently eluted and the sections restained a number of times thus allowing the detection of tens of antigens in the same sample. Furthermore, planarization of the three-dimensional specimen completely eliminates problems of both staining and imaging efficiency depending upon depth within a tissue that have previously prevented quantitative interpretation of the resulting volume images. In addition, we show here that array tomography lends itself to the analysis of fluorescent-protein labeled tissues in ways that should strongly complement in vivo dynamic imaging analyses of those same specimens prior to fixation. Finally, tomography arrays can be imaged by backscatter electron microscopy upon completion of the immunofluorescent analysis, and the resulting electron microscopic images can be collated easily in perfect register with the immunofluorescence images. This combination of methods (illustrated schematically in Fig. 1) allows for the quantitative, high-resolution and large-field volumetric imaging of substantial numbers of antigens and fluorescent proteins in essentially perfect correlation with ultrastructural imagery. Because of the use of arrays of serial ultrathin sections, we named this technique Array Tomography. While array tomography is of course limited to use on fixed tissue specimens, we expect that it may prove extremely useful for exploring the currently enigmatic circuit and molecular architectures that underlie the dynamic events of nervous system development and function.
Results
Array Tomography provides improved resolution over Confocal Microscopy
In order to test the hypothesized improvement in resolution by this method, we compared immunofluorescent images from Array Tomography and from confocal microscopy of Vibratome sections. Adult mouse cerebral cortex was fixed using the same procedure in both cases. We chose antigens known to have dense distribution in such tissues: synapsin and α-tubulin (Fig. 2). On single sections in the xy plane, there are more labeled structures visible with the confocal microscope compared to array tomography. This is expected due to the difference in the imaged volume: while the array tomography sections in this case were 200 nm thick, the effective thickness of the volume imaged with the confocal microscope is on the order of 1 μm. Thus, the confocal images are comparable to images obtained by summation of 5 array tomography sections (Fig. 2B, D). In addition, as apparent in the synapsin example (Fig. 2A, inserts), some small immunolabeled structures are lost in the confocal image: thus only bright and relatively big synapsin puncta can be resolved in the confocal image, while in the array tomography image, both big, bright synapsin puncta and small, dimmer puncta can be detected.
Figure 2. Axial resolution and depth-invariance comparisons of array tomography and confocal whole-mount immunofluorescence.
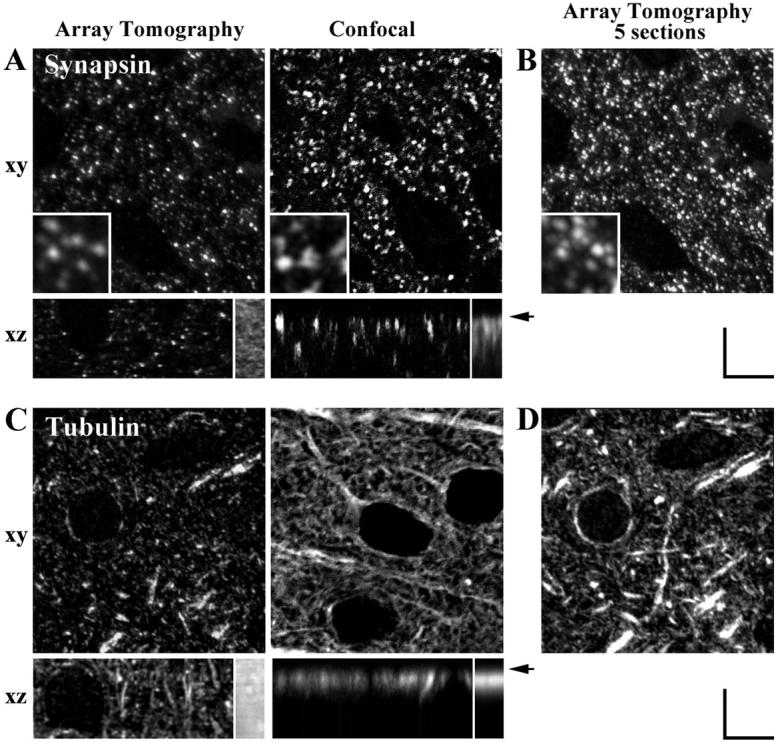
(A) Synapsin I immunostaining for presynaptic boutons in an adult mouse cerebral cortex as imaged with array tomography (40 serial resin sections, each 200 nm thick) and confocal whole-mount immunofluorescence (40 optical sections acquired at 200 nm intervals). Individual xy sections (acquired directly) and xz sections (resampled from stacks of 40 sections). Inserts in the synapsin xy images show a zoomed-in view (4x). Inserts in the xz sections represent the sum of z-sections from 20 μm of tissue and illustrate the depth-distribution of immunofluorescence.
(B) Summation of 5 synapsin-stained array tomography sections (1000 nm total depth) for comparison with individual confocal optical section (also representing approximately 1000 nm total depth).
(C) Tubulin immunostaining for cellular microtubule cytoskeleton. Similar to (A), except for primary antibody.
(D) Similar to (B), except for primary antibody being tubulin. All scale bars, 5 um.
The most obvious difference, however, can be observed when comparing z-sections through image stacks obtained with the methods. In both cases, stacks of 40 images spaced at 200 nm were obtained. For array tomography, this meant that 40 serial sections, each 200 nm thick were imaged, and for confocal microscopy, 40 images where collected through the depth of the specimen by advancing the focal plane 200 nm for each consecutive image. As can be seen from Figure 2A, the spatial resolution in the xy plane and in the z axis is similar for array tomography, while with confocal microscopy, the resolution in the z-axis is much worse and does not allow reliable detection of individual objects. In addition, immunostaining of Vibratome sections encounters the problem of limited depth penetration of antibodies and therefore a nonuniform staining intensity through the depth of the specimen. After an 18 h incubation in the presence of Triton, the synapsin antibody penetrated approximately 6 μm into the tissue, and the tubulin antibody 4 μm. This problem does not exist for array tomography, because each section is subject to the exact same immunostaining conditions.
Thus, in comparison with confocal microscopy, array tomography offers the advantage of better spatial resolution, in particular along the z-axis, as well as depth-independent immunofluorescent staining.
Characteristics of LRWhite postembedding fluorescence: antibodies, photobleaching, GFP fluorescence, antibody penetration
A large portion of the antigens that were explored (Table 1) could be reliably recognized by antibodies in LRWhite sections. The majority of them worked well with the standard paraformaldehyde fixation, but, similarly to their other applications, some required the presence of glutaraldehyde in the fixative (e.g. GABA, PIP2). The secondary fluorescent antibodies tested (FITC, rhodamine red X, CY-5, Alexa 488, Alexa 647) all performed well under these conditions, with the Alexa dyes giving a slightly brighter signal. For example, FITC staining was approximately 85% of the Alexa488 staining at a similar concentration (Fig. 3A).
Table 1.
Primary antibodies tested on ultrathin LRWhite sections from the rodent brain.
| Good immunolabeling | ||
|---|---|---|
| Antibody | Host species | Source |
| Synapsin I | Rabbit | Chemicon |
| Alpha Tubulin | Mouse | Sigma |
| N-cadherin | Goat | Santa Cruz Biotechnology |
| E-cadherein | Rabbit | James Nelson, Stanford University |
| VAMP | Mouse | Synaptic Systems |
| PSD-95 | Mouse | NeuroMabs |
| Beta-actin | Mouse | Sigma |
| Syncam | Rabbit | Sigma |
| GFAP | Rabbit | Sigma |
| GFAP | Mouse | Chemicon |
| SNAP-25 | Rabbit | Sigma |
| Neurofascin | Mouse | NeuroMabs |
| PIP2* | Mouse | Assay Designs |
| Neurofilament 200 | Rabbit | Sigma |
| Bassoon | Mouse | Craig Garner, Stanford University |
| Piccolo | Rabbit | Craig Garner, Stanford University |
| SV2 | Mouse | DSHB, Iowa |
| GABA* | Rabbit | Chemicon |
| Glutamate* | Rabbit | Chemicon |
| GFP | Mouse | Roche |
| GFP | Rabbit | Invitrogen |
| Kv2.1 | Mouse | NeuroMabs |
| SAP-97 | Mouse | NeuroMabs |
| Synaptophysin | Mouse | Abcam |
| No immunolabeling | ||
| MAP2 | Mouse | Chemicon |
| GAD | Rabbit | Chemicon |
| SH3-domain Grb2-like protein | Mouse | NeuroMabs |
| Pan-cadherein | Rabbit | Sigma |
| Tau-1 | Mouse | Sigma |
These antibodies require glutaraldehyde in the fixative solution.
Figure 3. Characterization of fluorescence imaging and immunostaining properties of specimens embedded in LRWhite.
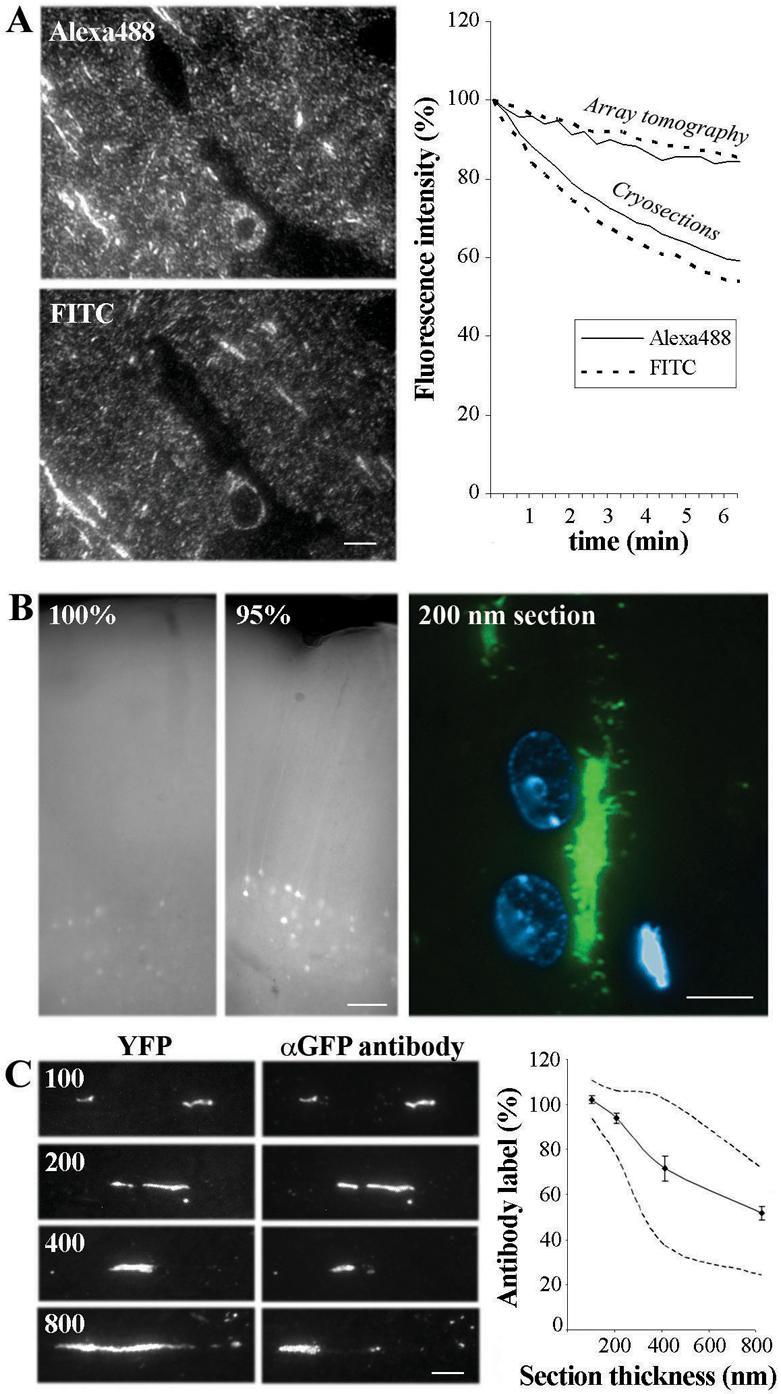
(A) Comparison of Alexa 488 and FITC - conjugated secondary antibodies on adjacent LRWhite ultrathin sections from the mouse cerebral cortex. Tubulin was used as the primary antibody. Graph represents fluorescence intensities of dyes as a function of duration of light exposure. The minimal and very slow photobleaching of both Alexa 488 and FITC as applied to LRWhite ultrathin sections of anti-tubulin stained mouse cerebral cortex is typical of all tags we have tested on such sections. The photobleaching of cryosections stained with the same antibodies and in the same mounting medium is provided for comparison. Scale bar, 5 μm.
(B) Native YFP fluorescence is spared by a partial-dehydration LRWhite embedding method. While some YFP fluorescence can still be detected within the unsectioned block which was completely dehydrated before embedding (100%), partial dehydration up to 95% ethanol (95%) results in bright YFP fluorescence, which can easily be detected within the block and in ultrathin sections (200 nm section). Scale bars, 50 μm (left), 5 μm (right).
(C). Comparison of YFP fluorescence and GFP immunostaining in sections of varying thickness (100, 200, 400 and 800 nm). The graph represents the percentage of the length of YFP containing dendrites labeled with the GFP antibody, which also recognizes YFP, a GFP variant. The dashed lines represent the maximum and minimum values for each thickness. Please note the close correspondence of YFP and GFP antibody label in sections of 100 and 200 nm. Standard errors are shown. Scale bar, 5 μm.
A commonly reported problem for immunofluorescence is the rapid photobleaching of the signal. Under the present conditions very little photobleaching was noticed and the samples could be imaged repeatedly without a noticeable change in fluorescence. To quantify this phenomenon, sections immunolabeled with tubulin followed by either FITC or Alexa488 were imaged for prolonged periods of time. At the end of 6 min of continuous exposure to a 100W mercury lamp, both dyes had retained more than 85% of their initial fluorescence (Fig. 3A, graph). In comparison, 10 μm cryostat sections immunolabeled with the same primary and secondary antibodies, mounted in the same mounting medium (VectaShield with DAPI), and imaged on the same microscope showed significantly more photobleaching. Thus, after 6 min of exposure to a 100 W mercury lamp, the Alexa 488 fluorescence had decreased to 59 % of the initial value and the FITC fluorescence had decreased to 54 %.
In order to expand the applicability of LRWhite sections, we tested different embedding conditions for preserving the fluorescence of GFP and its variants. Brain tissue from a YFP-H transgenic mouse (Feng et al., 2000) was used. Partial dehydration of the tissue (up to 95% ethanol) before LRWhite embedding resulted in the retention of bright YFP fluorescence that could be detected even at low magnification (10x objective) within the resin block of tissue prior to sectioning (Fig. 3B). YFP fluorescence was also very bright in the ultrathin LRWhite sections (70 - 200 nm).
Taking advantage of the preserved YFP fluorescence, we also tested the penetration depth of antibodies in the LR White sections by comparing YFP fluorescence with the GFP antibody immunolabeling on the same structures (Fig. 3C). Because YFP is a GFP variant, it is also recognized by the GFP antibody. There was almost perfect overlap between YFP and the immunolabel in sections of 100 nm and 200 nm thicknesses, while in thicker sections, some YFP structures were not immunolabeled. Based on this experiment, the maximum thickness of LR White sections for immunolabeling was chosen to be 200 nm.
Double immunofluorescence on LRWhite sections allows the resolution of closely apposed structures
The improved resolution capabilities of array tomography make it well suited for determining the spatial relationship of antigens. For example, double immunofluorescence labeling of presynaptic boutons with synapsin and postsynaptic densities with PSD-95 in the rat cerebral cortex (Fig. 4) reveals the complementary distribution of these two structures, which are known to be adjacent to each other with no overlap. A large number of apposed synapsin and PSD-95 puncta can be observed in the 200 nm LRWhite section. Some immunolabeled puncta do not have an adjacent partner, most probably due to the limited thickness of the section, thus leaving the corresponding structure in sections before or after the imaged section. Thus, imaging of a larger number of consecutive sections will be required for precise characterization of the spatial relationship of antigens.
Figure 4. Evidence from double immunofluorescence that array tomography can discriminate individual synapses.
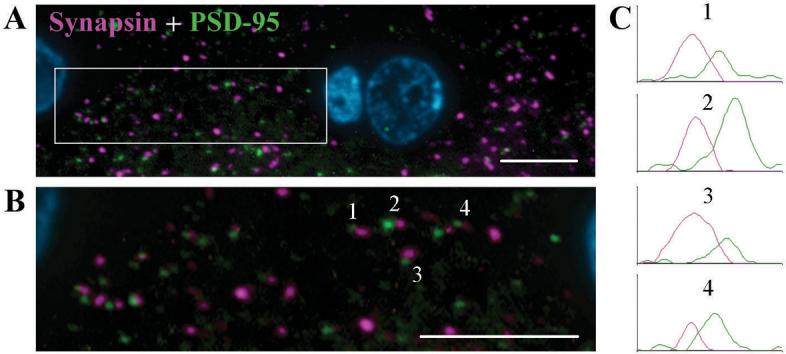
(A) Immunofluorescence image of a single LRWhite section (200 nm) from adult rat cerebral cortex: synapsin (purple), PSD-95 (green) and DAPI (blue).
(B) A zoomed in view of the area within the white rectangle in (A). The presynaptic (synapsin) and postsynaptic (PSD-95) markers can be resolved as being adjacent to one another. Scale bar, 5 μm.
(C) Graphs represent the immunofluorescence intensity in arbitrary units of 4 synapsin - PSD-95 appositions.
Three-dimensional volume imaging
To obtain information about the three-dimensional distribution of antigens, LRWhite embedded tissue was thin sectioned into long series of sections (ribbons), which were then placed onto glass slides forming arrays. After immunostaining of the arrays, fluorescent images from the same region of each section were collected. These images were then aligned and used for 3D reconstructions. On Figure 5A, serial sections (70 nm each) were labeled with antibodies to synapsin and PSD-95. Individual presynaptic boutons labeled with synapsin and the adjacent postsynaptic densities labeled with PSD-95 can be tracked through several ultrathin sections and the data used to obtain a 3D volume image of their spatial relationship.
Figure 5. High-resolution three-dimensional renderings derived from two-dimensional images of serial LRWhite ultrathin sections.
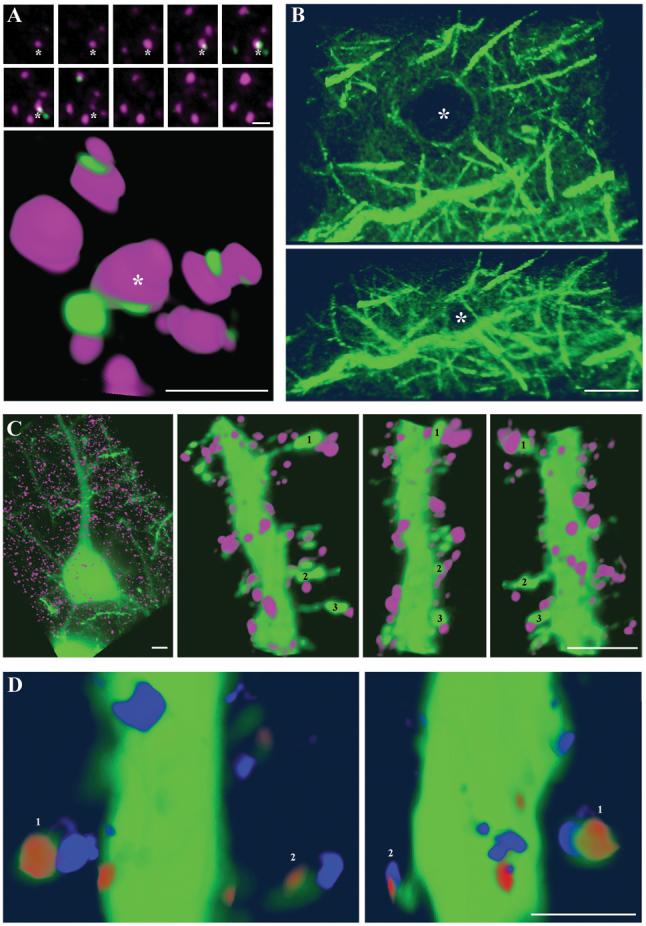
(A) Synapsin (magenta) and PSD-95 (green) immunostaining of ultrathin (70 nm) sections from the mouse cerebral cortex. The 10 consecutive sections (above) used for the 3D reconstruction (below) are shown. One of the synaptic boutons that can be followed through 7 sections is identified with an asterisk. Scale bar, 1 μm.
(B) Two different views from a volume rendering of tubulin immunostaining in the mouse cerebral cortex. Forty sections 200 nm each were used for this reconstruction. Dendritic processes can be seen coming out of a cell body, the nucleus of which is devoid of tubulin staining (marked with asterisk). Scale bar, 5 μm.
(C) 3D reconstruction of synapsin immunofluorescence (purple) on sections from the YFP-H mouse cerebral cortex. A YFP-expressing layer V pyramidal neuron is shown on the left and three different views from a spine-bearing dendrite are shown on the right. Three spines are identified with numbers. Scale bar, 2 μm.
(D) 3D views from a spine-bearing YFP-dendrite immunolabeled with synapsin (blue) and β-actin (red). Two spines are identified with numbers. Note that β-actin is within the spine head and synapsin is adjacent to the spine. Scale bar, 1 μm.
Other abundant antigens, for example tubulin, form a densely entangled mesh within brain tissue which is also difficult to resolve with other immunostaining methods (e.g Fig. 2). Using immunofluorescently labeled serial sections, microtubule bundles can be tracked and used to reconstruct the microtubule cytoskeleton of cells within tissue (Fig. 5B, Suppl. Movie 1). In this example, 40 sections of 200 nm each from the mouse cerebral cortex were labeled with an α-tubulin antibody, fluorescently imaged and volume rendered.
3D volume imaging can also be applied to GFP expressing tissues in combination with immunofluorescence to investigate the distribution of antigens in relation to identified cells (Fig. 5C and D). On Fig. 5C we show the 3D reconstruction of a portion of a dendrite from a series of 200 nm sections labeled with synapsin. Dendritic spines bearing synapses as identified with synapsin labeling can be easily resolved with this method and the synapsin label is seen immediately adjacent to the spines. In comparison, immunolabeling of the same tissue with β-actin reveals the concentration of β-actin within the heads of dendritic spines (Fig. 5D). As can be seen from the above examples, array tomography offers distinct advantages when studying the relative distribution of different antigens by providing high 3D resolution.
Consecutive staining with antibody elution
An additional advantage of ultrathin LRWhite sections for immunofluorescence staining is the possibility of eluting the applied and imaged antibodies and staining with a different combination of antibodies. The majority of antigens that were examined survived the elution procedure and could be detected again with almost identical distribution and very similar intensity of the signal (Suppl. Fig. 1). DAPI staining of cell nuclei, however, was not possible after the elution. Multiple consecutive rounds of staining, imaging and elution could be performed, with no apparent limit. In this way information about the relative distribution of a significant number of antigens (10 or more) can be achieved. In Fig. 6 we performed 9 consecutive double immunolabelings. For alignment purposes, one of the antibodies from the first staining (either synapsin or tubulin) was included in each round. Aligning of the images obtained after each immunostaining from the same region of the section allowed the simultaneous mapping of the spatial distribution of all 10 antigens probed. Comparison of 6 consecutive synapsin immunostainings showed that the same structures exhibit similar immunofluorescence intensity in each round of staining.
Figure 6. Demonstration of a very high immunofluorescence multiplexing capacity.
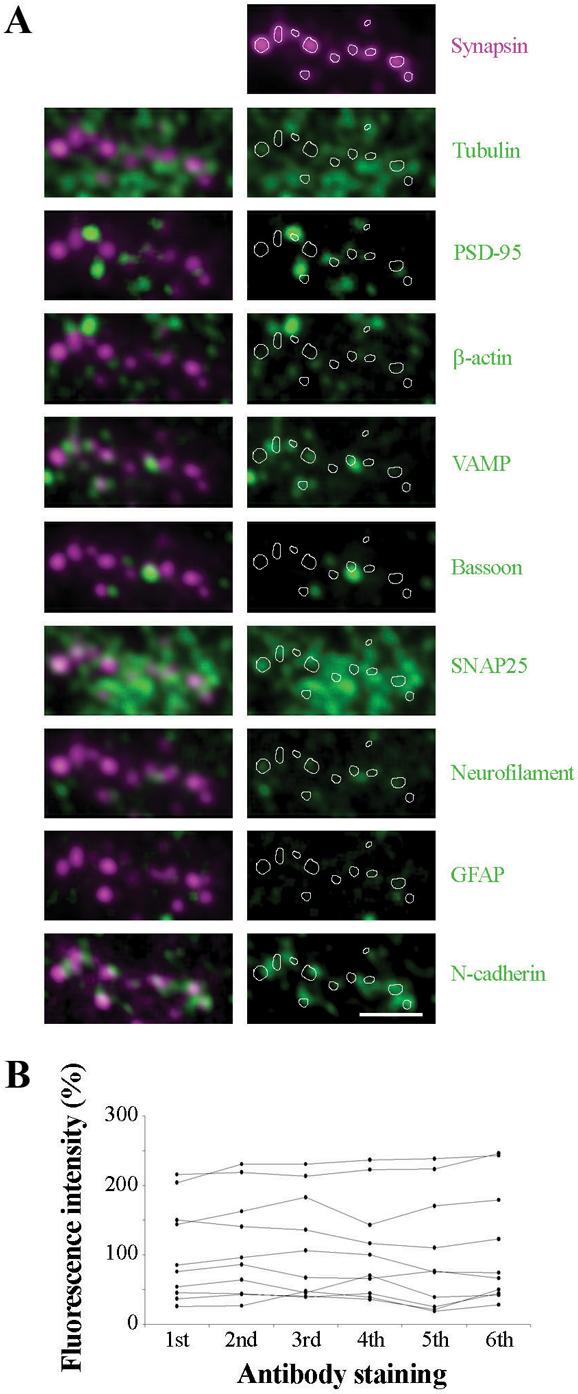
(A) Images from nine cycles of double immunostaining, antibody stripping and restaining of a single array slice from the rat cerebral cortex. For alignment purposes, one of the antibodies from the first staining was always included in consequent rounds. Synapsin was used in all but two cases where tubulin was used instead (co-staining with neurofilament and SNAP25); for simplicity of presentation synapsin is shown in all rounds of immunostaining, even though in two cases the alignment was done using tubulin). Scale bar, 2 μm.
(B) Graph shows the relative level of fluorescence intensity of 10 synapses as identified with synapsin through 6 rounds of immunostaining.
Correlative scanning electron microscopy
Immunofluorescent labeling of arrays of LRWhite sections mounted on glass slides can also be followed by scanning electron microscopy. For this approach to be useful, the tissue has to be prepared with consideration of preserving fine structural details. One possible approach is to process the tissue in a way similar to conventional electron microscopy, i.e. using both paraformaldehyde and glutaraldehyde for fixation and adding postfixation in osmium tetroxide before the dehydration step. While this way of processing the tissue is not optimal for preserving antigenicity and limits the number of antibodies that can be used, it can provide valuable information by bridging the gap between light and electron microscopy. In Fig. 7, immunofluorescent labeling of ultrathin sections with 4 antibodies was combined with scanning electron microscopy. The sections were first labeled with tubulin and GABA antibodies and imaged on the fluorescence microscope. After elution of these antibodies, the sections were restained with β-actin and SNAP-25 and the same region imaged again under the fluorescence microscope. Following poststaining with uranyl acetate and lead citrate, the region was again relocated in the SEM (Fig. 7A). No ultrastructural damage was noted as result of the antibody elution. Higher magnification SEM images were aligned with the immunofluorescent images using landmarks visible in both modes of imaging, such as cell nuclei and blood vessels. (Fig. 7B-D). While it is below the resolution capabilities of traditional TEM, SEM imaging allows the identification of ultrastructural detail, including presynaptic boutons, postsynaptic densities, microtubules, glial processes and correlating it with the immunofluorescent signals. Consistent with the literature (Matsunaga et al., 2002; Matus et al., 1975), tubulin staining was observed on microtubules, which were found mainly in dendrites and cell bodies surrounding the nucleus; glia, smaller presynaptic boutons, dendritic spines were mostly devoid of label. GABA immunostaining was mostly confined to presynaptic boutons and axonal varicosities (Micheva and Beaulieu, 1996), β-actin labeling was prominent in spines and some glial processes (Micheva et al., 1998) and SNAP-25 labeled most presynaptic boutons, but was located outside of synapses as well (Frassoni et al., 2005; Tafoya et al., 2006).
Figure 7. Demonstration of naturally excellent registration of light and electron microscopic imaging of an individual specimen by array tomography.
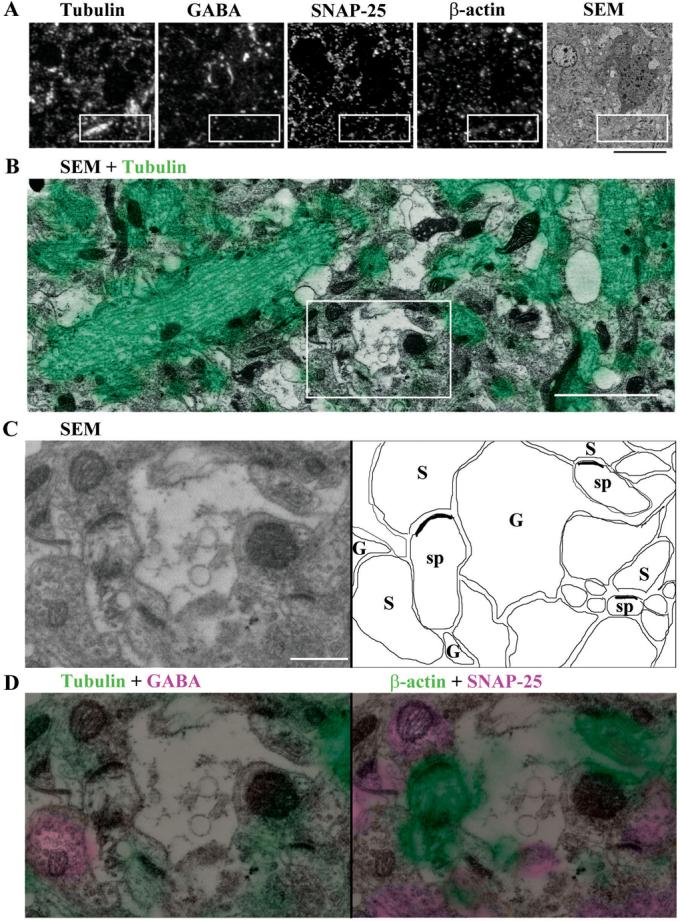
(A) The same region of a 70 nm section from the mouse cerebral cortex is shown as immunostained for tubulin, GABA, SNAP25 and β-actin, and imaged in the SEM. Scale bar, 10 μm.
(B) The boxed region in (A) is imaged at a higher magnification in the SEM and the corresponding immunofluorescent labeling for tubulin (green) is overlayed. Scale bar, 2 μm.
(C) A higher magnification SEM image of the boxed region in (B) and a schematic map of the same region: G - glia, S - presynaptic bouton, sp - spine. Scale bar, 0.5 μm.
(D) Immunofluorescence for tubulin and GABA, and b-actin and SNAP-25 superimposed on the SEM image in c. Scale bar, 0.5 μm.
Automation of array tomography imaging and puncta quantification
Imaging of larger arrays can be very time consuming and thus might limit the application of this technique. For this reason, we developed a procedure for automated imaging of serial section arrays, using commercially available software (AxioVision 4.6, Carl Zeiss Microimaging). Figure 8 and Supplementary Movie 2 show a variety of renderings from a multi-channel volume image collected by imaging an array of 134 sections from layer V of the cerebral cortex of a YFP-H mouse, immunostained with antibodies to synapsin I and to GFP. The collection of this volume image involved the acquisition of 402 separate fluorescence images (3 color channels) under full, unattended automation which included the use of an image-based automatic focus algorithm. These renderings demonstrate that the high axial resolution and depth-independence of array tomography can be obtained over large volumetric fields of view, in this case 120 μm × 110 μm × 27 μm. The intricate detail offered by array tomography can be best appreciated in the monochrome rendering of the anti-GFP fluorescence (Figure 8D). For example, the dendritic spines lining the dendrites, as well as the thin axons crossing the tissue are clearly visible. Also, individual synapsin puncta (Figure 8E, red channel) can be clearly resolved and therefore counted. Using an ImageJ plug-in developed in our laboratory (please see Supplemental Materials) we counted 191,140 synapsin puncta in the tissue volume from Figure 8. This amounts to 0.54 synapsin puncta per μm3 of brain tissue, including cell nuclei and blood vessels.
Figure 8. Results from an automated procedure for the collection of large array tomographic volume images.
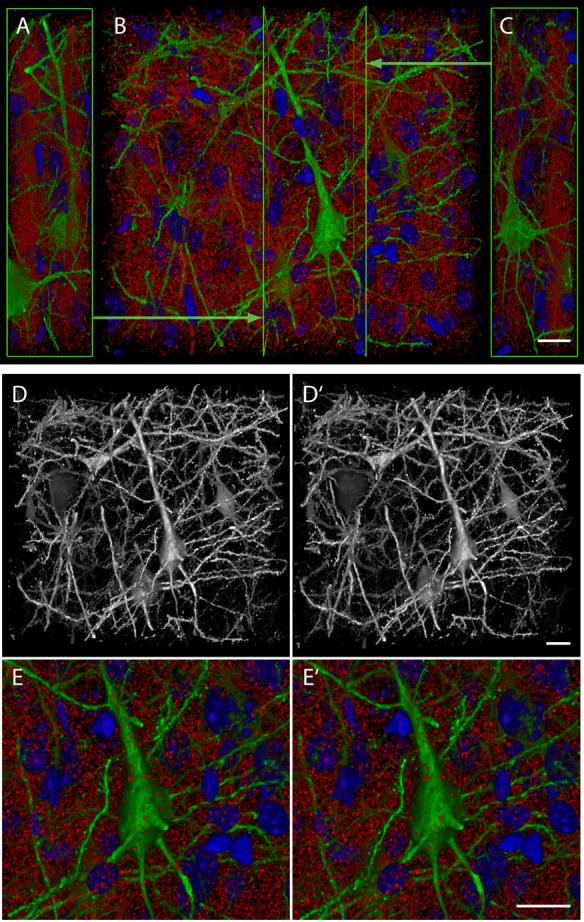
(A -C) Volume renderings from an array tomograph of YFP-H-transgenic mouse cerebral cortex, collected by automated fluorescence microscopy. Tomographic images were acquired in three fluorescence channels (DAPI-DNA, blue; FITC anti-GFP, green; rhodamine anti-synapsin I, red) from 134 sections, each 200 nm thick, using a motorized microscope, a CCD camera and software with image-based automatic focus capability. (B) is a projection along the acquisition Z axis and (A) and (C) are oppositely directed projections along the acquisition X axis, clipped at the Y-Z planes indicated by the bright green lines and arrows in (B). (D) and (D’) are a stereo pair representing solely GFP-channel fluorescence in the same volume as (A-C). (E) and (E’) are a stereo pair of the same volume representing all three fluorescence channels at higher magnification. The very numerous red puncta evident in (A-C) and (E) are consistent with identification as individual presynaptic boutons, while the total density of neural cells within this tissue volume is indicated by the abundant DAPI -stained nuclei evident in these panels. Scale bars, 10 μm.
In order to compare our synapsin counts with the available literature on synapse quantification, we also used our method to count synapsin puncta in the layers of the somatosensory cortex of a control mouse. For ease of comparison, in this case we present our data as numbers per 1μm3 of neuropil (excluding nuclei and blood vessels). After analyzing at least 2520 μm3 per layer the following numbers were obtained: In layer I of the mouse somatosensory cortex there were 0.96 ± 0.04; in layers II/III: 0.72 ± 0.01; layer IV: 0.86 ± 0.07; layer V: 0.64 ± 0.03, and layer VI: 0.37 ± 0.03 synapsin puncta (mean ± st. error), which is very similar to stereological estimations of synaptic bouton numbers in the mouse cortex (e.g. Schüz and Palm, 1989).
Thus, the automation of array tomography allows for a much faster and more precise imaging, as well as quantification of arrays, and should enable the application of array tomography to larger tissue volumes.
Discussion
The imaging method presented in this paper, array tomography, is based on immunostaining and imaging arrays of serial ultrathin (50-200 nm) specimen sections bonded tightly to glass slides. The individual two-dimensional section images are then aligned and collated into volumetric image stacks. Antibody stains can be stripped very efficiently, making it possible to multiplex large numbers of immunostains through repeated cycles of staining, imaging and stripping individual array slides. In addition, with array tomography GFP fluorescence is well-preserved. Fluorescence imaging can be followed by scanning electron microscopy to complement the immunofluorescence data with high resolution structural information.
Array tomography offers a number of advantages to existing imaging methods. For example, array tomography can achieve about an order of magnitude improvement in z-axis resolution over conventional immunofluorescence methods (see Fig. 1 and 2) by relying upon physical ultrathin sectioning (as low as 50 nm) rather than optical sectioning (500 nm in theory (Pawley, 1995), but usually more than 700 nm in practice). This allows for a more precise mapping of antigen distribution, in particular when determining whether antigens are colocalized or confined to adjacent structures. Also, the detection of antigens is uniform throughout the depth of the specimen, allowing quantitative measurements of antigen distribution. For example, due to problems with antibody penetration and the difficulty of resolving small (<1 μm) and densely packed individual structures at the light microscopic level, reliable quantification of synapses has only been possible using electron microscopy, which is very time-consuming and laborious (Calhoun et al., 1996; Geinisman et al., 1996). Array tomography can now provide another reliable means to achieve such quantification faster and within larger tissue volumes, compared to electron microscopy. Another advantage is that tomography arrays also can be imaged at the ultrastructural level using scanning electron microscopy (SEM), as illustrated in Fig. 7. In the present paper, we used conventional methods for chemical fixation and tissue preparation for electron microscopy, which are not optimal for antigen preservation. However, for sensitive antigens, methods of tissue preparation which allow both good ultrastructural and antigenicity preservation are available (e.g. high-pressure freezing, freeze substitution and low temperature, partially hydrated embedding, (Newman and Hobot, 1999) and compatible with array tomography.
While improvement in resolution was a major premise for the development of array tomography, several unexpected advantages were also discovered. For example, conventional fluorescent dyes showed remarkable photostability under the conditions of array tomography. After 6 min of constant illumination with a 100W mercury lamp, 85% of the initial fluorescence intensity remained, compared to a much faster photobleaching of the same dyes on cryostat sections, as observed by us, or in other applications, as reported in the literature (Lee et al., 2004; Panchuk-Voloshina et al., 1999; Sukhanova et al., 2002). The photobleaching mechanisms of organic dyes are not completely understood, but it is known that this process depends on the environment (Song et al., 1995; Zondervan et al., 2004). The relative rigidity of the resin, restricting the mobility of molecules, is likely to contribute to the observed photostability. This property of array tomography allows for the collection of high quality images using longer exposure times.
In addition, in our hands antibodies were found to easily penetrate to a depth of 200 nm in LRWhite embedded material, thus increasing the amount of accessible antigens and allowing for an uninterrupted 3D reconstruction from serial sections as thick as 200 nm. There are conflicting reports about the ability of antibodies to penetrate LRWhite sections (Brorson et al., 1994; Newman and Hobot, 1987) and it is possible that this depends on the conditions of resin polymerization. It has been suggested that the relatively slow thermal polymerization of the resin that we employed limits cross-linking in the plastic and leads to linearity of molecular arrangement, thus facilitating antibody penetration (Newman and Hobot, 1987).
Array tomography offers new opportunities for high-order multiplexing that were not easily available with previous immunoimaging methods. While antibody elution is not a new method (for example, (Kolodziejczyk and Baertschi, 1986; Tramu et al., 1978; Wahlby et al., 2002), it has been limited previously by ultrastructural damage and decrease in antigenicity. Recently, a method using multiple rounds of immunolabeling, fluorescent tag bleaching and restaining of 5 μm cryostat sections, has underlined the importance and power of multiplexing approaches (Schubert et al., 2006). LRWhite ultrathin sections on glass slides proved to be a very favorable substrate for antibody elution, and can undergo a large number of sequential cycles of staining, imaging, stripping and re-staining. We have demonstrated in this paper that quantitative staining can be carried out for nine cycles at least, and the limiting number may be much higher. Thus, where conventional indirect immunostaining methods are used to image four antigens per cycle, for example, 36 distinct antigens might be visualized by nine restaining cycles. It is now well established that most cell and tissue function results from interactions of numerous molecular species at distinct, nanometer-scale focal complexes (e.g., synaptic active zones). Array tomography is well suited for exploring the structure and function of such important focal complexes.
The numerous technical advantages of array tomography combine to offer access to critical aspects of neural circuit molecular architecture that were formerly very difficult to visualize and measure within tissue specimens. For instance, array tomography achieves easy and efficient resolution and quantification of individual synapsin immunolabeled puncta throughout large volumes of mature central nervous system neuropil. While the exact correspondence between synapsin immunolabeling and structurally identified synapses still remains to be demonstrated, all the available data strongly suggest that synapsin is a reliable marker for synapse characterization, and that it can be used as a marker in array tomography for synapse quantification. Traditionally, synapse measurements have required quite slow and circumscribed approaches such electron microscopic stereology. Given that changes in the numbers and volume densities of synapses are believed to be central to many human neurological and cognitive disorders, the potential of measuring such variables with both rigor and high throughput can be expected to have a very large impact. Furthermore, since immunofluorescence and SEM array tomography will lend themselves well to imaging human tissues (unlike methods that strictly require the expression of GFP or other transgenes), array tomography will be useful both for the direct study of human clinical specimens and for critical comparisons of human and animal disease-model tissues. The compatibility of GFP-based imaging methods with array tomography nonetheless offers additional very exciting opportunities to exploit the many special advantages of transgenic animals and to complement and extend modern in vivo imaging techniques with retrospective high-resolution analysis by array tomography. As a powerful technique for the volumetric imaging of brain tissue molecular architecture and ultrastructure, array tomography promises new avenues of attack on many issues that are now coming to the forefront of neuroscience. These include the molecular classification neural cell types, the determination of ion channel and receptor distributions within the tissue context, and circuit connectivity.
Experimental procedures
Tissue preparation
All procedures related to the care and treatment of animals were approved by the Administrative Panel on Laboratory Animal Care at Stanford University. Four adult mice (C57BL/6J), one YFP-H adult mouse (Feng et al., 2000) and one adult Sprague Dawley rat were used for this study. The animals were anesthetized by halothane inhalation and their brains quickly removed and placed in HBSS (4°C). The cerebral cortex was dissected out and fixed by immersion in 4% paraformaldehyde and 2.5% sucrose in phosphate-buffered saline (PBS) using rapid microwave irradiation (PELCO 3451 laboratory microwave system; Ted Pella; one cycle of 1min on - 1 min off - 1 min on at 100 W, and two cycles of 20 s on - 20 s off - 20 s on at 350 W) and ColdSpot (Ted Pella) set at 15°C. The tissue was then left in the fixative for 30 min at room temperature or overnight at 4°C. After rinsing in PBS containing 3.5% sucrose, the tissue was quenched in 50 mM glycine in PBS and then dehydrated in a graded series of ethanol (45 s each at 350W in microwave). For the YFP-H mouse, to preserve YFP fluorescence, the tissue was dehydrated only up to 95% ethanol. The tissue was then infiltrated in LRWhite resin (3 times 45 s each at 350W, and overnight at 4°C), embedded in gelatin capsules and polymerized at 50°C.
For scanning electron microscopy (SEM), the tissue was processed as above except that the fixative contained also 2.5 % glutaraldehyde and before dehydration a poststaining step with osmium tetroxide (1%) and potassium ferricyanide (1.5%), (3 cycles of 1 min on - 1 min off - 1 min on at 100W, followed by 30 min at room temperature) was added.
LRWhite sections
Ultrathin (50 - 200 nm) or semithin (400 - 1000 nm) sections were cut with an ultramicrotome (Ultracut E, Reichert Jung). For cutting ribbons of serial sections, after trimming of the block, glue (Weldwood Contact Cement diluted in xylene) was applied with a paint brush to the top and bottom sides of the block pyramid (modified from (Blumer et al., 2002). After the glue had dried (∼ 2 min), series of 30 or more sections were cut in ribbons using Jumbo Histo Diamond Knife (Diatome). The ribbons were mounted on subbed glass slides (coated with 0.1% gelatin and 0.01% chromium potassium sulphate) and placed on a hot plate (∼ 60°C) for 30 min. The slides were stored at room temperature for up to 3 months. For more details on sectioning and mounting the ribbons, please refer to the Supplementary materials, Array tomography protocol.
Immunofluorescence staining
The sections were encircled with a PAP pen (ImmEdge Pen, Vector Laboratories, Burlingame, CA) and their position was marked with a diamond pen on the opposite side of the slide. Unless stated otherwise in the text, the sections were pretreated with 50 mM glycine in Tris buffer (pH 7.6) for 5 min and the primary antibodies were applied diluted in Tris buffer containing 1% BSA for 2 h. Secondary antibodies in Tris buffer with 1% BSA were applied for 30 min. Between steps and at the end, the sections were extensively washed with Tris buffer. The sections were mounted using Vectashield (with or without DAPI, Vector Laboratories). To elute the applied antibodies, the mounting medium was washed away with dH2O and a solution of 0.15M KMNO4 and 0.01N H2SO4 was applied for 90 s (Tramu et al., 1978). After an extensive wash with dH2O, the slides were placed on a hot plate (60°C) for 30 min.
Microscopy and image processing
Sections were imaged on a Zeiss Axioplan fluorescence microscope using a Zeiss 100x/1.4 NA Plan Apochromat oil objective or 100x/1.3 NA Plan Neofluar objective and a Zeiss AxioCam MRm camera. Using ImageJ, images from serial sections were converted to stacks and were aligned with the StackReg plugin (Thévenaz et al., 1998). The Enhance Contrast and Subtract Background functions of ImageJ were used for image processing. The aligned image stacks were volume rendered with Amira 4.0.
Automation of array tomography imaging
A Zeiss Axiovert 200M fluorescence microscope with motorized stage, AxioCam HRm CCD camera and Zeiss AxioVision 4.6 software was used. A tiled image of the entire ribbon or ribbons of sections on a glass slide was obtained using a 10 x objective and the MosaicX feature of the software. The region of interest was then identified and imaged at a higher magnification with a Zeiss 63x/1.4 NA Plan Apochromat objective, using the image-based automatic focus capability of the software. The obtained stack of images was exported to ImageJ, aligned using the StackReg plugin and imported back into the Axiovision software to generate a 3D view of the stack.
Automation of synapsin puncta counts
Synapsin puncta were counted using an ImageJ plugin, developed in our laboratory by Brad Busse (Supplementary material). Arrays stained with anti-synapsin I antibody were imaged, and the obtained stack of images was aligned using the ImageJ plugin StackReg. Both 200 nm and 70 nm sections were used for synapse quantification and the obtained results were very similar. For synapse quantification in the adult mouse somatosensory cortex, a series of 40 sections, 70 nm each, was used. The sections spanned through all the cortical layers, which were identified using DAPI staining of nuclei and standard morphological criteria. In every cortical layer, stacks of images were taken and synapsin puncta were quantified in at least 9 randomly selected regions of neuropil, each measuring 10 × 10 × 2.8 μm.
Confocal microscopy on Vibratome sections
The brain tissue was obtained following the procedure for Array Tomography as outlined in Tissue preparation, except that after fixation and washing in PBS with glycine, the cortex was cut in 50 μm sections using a Vibratome (Leica VT1000S). For immunostaining, the sections were blocked with 10% BSA and 0.1% Triton in PBS for 30 min at room temperature, then incubated overnight (18 h) on shaker at 4°C with the primary antibody diluted in PBS containing 3% BSA and 0.1% Triton. The next day, the sections were washed in PBS, blocked again with 10% BSA and 0.1% Triton in PBS for 30 min at room temperature and incubated in secondary antibody (FITC) diluted in PBS containing 3% BSA and 0.1% Triton for 1 h on shaker at 4°C. The sections were then transferred on glass slides and without drying were mounted using Vectashield. The sections were imaged on a confocal microscope (Zeiss LSM 510) using the 488 nm laser line with a pinhole of 1 Airy unit and a Zeiss 63x/1.4 NA Plan Apochromat oil objective.
Scanning electron microscope
Following the imaging of the immunostained arrays on the fluorescence microscope, the mounting medium was washed away with dH20 and the sections were placed on a hot plate (60°C) for 30 min. After cooling down they were poststained with 5% uranyl acetate in H20 for 10 min and lead citrate for 90 s. The glass slides were carbon coated using Denton Benchtop Turbo Carbon Evaporator. They were imaged on a Hitachi S4800 scanning electron microscope equipped with field emission gun using the backscattered electron detector.
Supplementary Material
Acknowledgements
This work was supported by grants from the McKnight Endowment Fund for Neuroscience, the Mathers Foundation and National Institutes of Health (NINDS). We would like to thank JoAnn Buchanan for making the first fluorescently immunolabeled LRWhite section in our laboratory which inspired the present study, Brad Busse for his invaluable help with image acquisition and processing software, Dr. Nancy O’Rourke for critical reading of the manuscript, and Dr. Beth Stevens for providing the mouse brain cryosections.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Albrecht U, Seulberger H, Schwarz H, Risau W. Correlation of blood-brain barrier function and HT7 protein distribution in chick brain circumventricular organs. Brain Res. 1990;535:49–61. doi: 10.1016/0006-8993(90)91823-y. [DOI] [PubMed] [Google Scholar]
- Blumer MJ, Gahleitner P, Narzt T, Handl C, Ruthensteiner B. Ribbons of semithin sections: an advanced method with a new type of diamond knife. J. Neurosci. Methods. 2002;120:11–16. doi: 10.1016/s0165-0270(02)00166-8. [DOI] [PubMed] [Google Scholar]
- Brorson S-H, Roos N, Skjcrten F. Antibody penetration into LR-White sections. Micron. 1994;25:453–460. doi: 10.1016/0968-4328(94)00035-2. [DOI] [PubMed] [Google Scholar]
- Calhoun ME, Jucker M, Martin LJ, Thinakaran G, Price DL, Mouton PR. Comparative evaluation of synaptophysin-based methods for quantification of synapses. J. Neurocytol. 1996;25:821–828. doi: 10.1007/BF02284844. [DOI] [PubMed] [Google Scholar]
- De-Camilli P, Cameron R, Greengard P. Synapsin I (protein I), a nerve terminal-specific phosphoprotein. I. Its general distribution in synapses of the central and peripheral nervous system demonstrated by immunofluorescence in frozen and plastic sections. J. Cell Biol. 1983;96:1337–1354. doi: 10.1083/jcb.96.5.1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng G, Mellor RH, Bernstein M, Keller-Peck C, Nguyen QT, Wallace M, Nerbonne JM, Lichtman JW, Sanes JR. Imaging neuronal subsets in transgenic mice expressing multiple spectral variants of GFP. Neuron. 2000;28:41–51. doi: 10.1016/s0896-6273(00)00084-2. [DOI] [PubMed] [Google Scholar]
- Fialka I, Schwarz H, Reichmann E, Oft M, Busslinger M, Beug H. The estrogen-dependent c-JunER protein causes a reversible loss of mammary epithelial cell polarity involving a destabilization of adherens junctions. J. Cell Biol. 1996;132:1115–1132. doi: 10.1083/jcb.132.6.1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frassoni C, Inverardi F, Coco S, Ortino B, Grumelli C, Pozzi D, Verderio C, Matteoli M. Analysis of SNAP-25 immunoreactivity in hippocampal inhibitory neurons during development in culture and in situ. Neuroscience. 2005;131:813–823. doi: 10.1016/j.neuroscience.2004.11.042. [DOI] [PubMed] [Google Scholar]
- Geinisman Y, Gundersen HJ, van der Zee E, West MJ. Unbiased stereological estimation of the total number of synapses in a brain region. J. Neurocytol. 1996;25:805–819. doi: 10.1007/BF02284843. [DOI] [PubMed] [Google Scholar]
- Haraguchi CM, Yokota S. Immunofluorescence technique for 100-nm-thick semithin sections of Epon-embedded tissues. Histochem. Cell. Biol. 2002;117:81–85. doi: 10.1007/s00418-001-0363-1. [DOI] [PubMed] [Google Scholar]
- Herken R, Fussek M, Barth S, Gotz W. LR-White and LR-Gold resins for postembedding immunofluorescence staining of laminin in mouse kidney. Histochem. J. 1988;20:427–432. doi: 10.1007/BF01002428. [DOI] [PubMed] [Google Scholar]
- Kolodziejczyk E, Baertschi AJ. Multiple immunolabeling in histology: a new method using thermo-inactivation of immunoglobulins. J. Histochem. Cytochem. 1986;34:1725–1729. doi: 10.1177/34.12.3023476. [DOI] [PubMed] [Google Scholar]
- Kurth T, Schwarz H, Schneider S, Hausen P. Fine structural immunocytochemistry of catenins in amphibian and mammalian muscle. Cell Tissue Res. 1996;286:1–12. doi: 10.1007/s004410050669. [DOI] [PubMed] [Google Scholar]
- Lee LY, Ong SL, Hu JY, Ng WJ, Feng Y, Tan X, Wong SW. Use of semiconductor quantum dots for photostable immunofluorescence labeling of Cryptosporidium parvum. Appl. Environ. Microbiol. 2004;70:5732–5736. doi: 10.1128/AEM.70.10.5732-5736.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luby-Phelps K, Ning G, Fogerty J, Besharse JC. Visualization of identified GFP-expressing cells by light and electron microscopy. J. Histochem. Cytochem. 2003;51:271–274. doi: 10.1177/002215540305100301. [DOI] [PubMed] [Google Scholar]
- Matsunaga W, Miyata S, Itoh M, Kiyohara T, Maekawa S. Expression of high levels of tubulin and microtubule-associated protein 2d in the neurohypophysial astrocytes of adult rat. Neuroscience. 2002;111:151–162. doi: 10.1016/s0306-4522(01)00607-8. [DOI] [PubMed] [Google Scholar]
- Matus AI, Walters BB, Mughal S. Immunohistochemical demonstration of tubulin associated with microtubules and synaptic junctions in mammalian brain. J. Neurocytol. 1975;4:733–744. doi: 10.1007/BF01181633. [DOI] [PubMed] [Google Scholar]
- Micheva KD, Beaulieu C. Quantitative aspects of synaptogenesis in the rat barrel field cortex with special reference to GABA circuitry. J. Comp. Neurol. 1996;373:340–354. doi: 10.1002/(SICI)1096-9861(19960923)373:3<340::AID-CNE3>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- Micheva KD, Vallee A, Beaulieu C, Herman IM, Leclerc N. beta-Actin is confined to structures having high capacity of remodelling in developing and adult rat cerebellum. Eur. J. Neurosci. 1998;10:3785–3798. doi: 10.1046/j.1460-9568.1998.00391.x. [DOI] [PubMed] [Google Scholar]
- Mori M, Ishikawa G, Takeshita T, Goto T, Robinson JM, Takizawa T. Ultrahigh-resolution immunofluorescence microscopy using ultrathin cryosections: subcellular distribution of caveolin-1alpha and CD31 in human placental endothelial cells. J. Electron Microsc. (Tokyo) 2006;55:107–112. doi: 10.1093/jmicro/dfl011. [DOI] [PubMed] [Google Scholar]
- Newman GR, Hobot JA. Modern acrylics for post-embedding immunostaining techniques. J. Histochem. Cytochem. 1987;35:971–981. doi: 10.1177/35.9.3302021. [DOI] [PubMed] [Google Scholar]
- Newman GR, Hobot JA. Resins for combined light and electron microscopy: a half century of development. Histochem. J. 1999;31:495–505. doi: 10.1023/a:1003850921869. [DOI] [PubMed] [Google Scholar]
- Newman GR, Jasani B, Williams ED. A simple post-embedding system for the rapid demonstration of tissue antigens under the electron microscope. Histochem. J. 1983;15:543–555. doi: 10.1007/BF01954145. [DOI] [PubMed] [Google Scholar]
- Ojeda JL, Ros MA, Icardo JM. A technique for fluorescence microscopy in semithin sections. Stain Technol. 1989;64:243–248. doi: 10.3109/10520298909107009. [DOI] [PubMed] [Google Scholar]
- Panchuk-Voloshina N, Haugland RP, Bishop-Stewart J, Bhalgat MK, Millard PJ, Mao F, Leung WY, Haugland RP. Alexa dyes, a series of new fluorescent dyes that yield exceptionally bright, photostable conjugates. J. Histochem. Cytochem. 1999;47:1179–1188. doi: 10.1177/002215549904700910. [DOI] [PubMed] [Google Scholar]
- Pawley JB. Handbook of biological confocal microscopy. Plenum Press; New York: 1995. [Google Scholar]
- Schubert W, Bonnekoh B, Pommer AJ, Philipsen L, Bockelmann R, Malykh Y, Gollnick H, Friedenberger M, Bode M, Dress AW. Analyzing proteome topology and function by automated multidimensional fluorescence microscopy. Nat. Biotechnol. 2006;24:1270–1278. doi: 10.1038/nbt1250. [DOI] [PubMed] [Google Scholar]
- Schüz A, Palm G. Density of neurons and synapses in the cerebral cortex of the mouse. J. Comp. Neurol. 1989;286:442–455. doi: 10.1002/cne.902860404. [DOI] [PubMed] [Google Scholar]
- Song L, Hennink EJ, Young IT, Tanke HJ. Photobleaching kinetics of fluorescein in quantitative fluorescence microscopy. Biophys. J. 1995;68:2588–2600. doi: 10.1016/S0006-3495(95)80442-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sukhanova A, Venteo L, Devy J, Artemyev M, Oleinikov V, Pluot M, Nabiev I. Highly stable fluorescent nanocrystals as a novel class of labels for immunohistochemical analysis of paraffin-embedded tissue sections. Lab. Invest. 2002;82:1259–1261. doi: 10.1097/01.lab.0000027837.13582.e8. [DOI] [PubMed] [Google Scholar]
- Tafoya LCR, Mameli M, Miyashita T, Guzowski JF, Valenzuela CF, Wilson MC. Expression and Function of SNAP-25 as a Universal SNARE Component in GABAergic Neurons. J. Neurosci. 2006;26:7826–7838. doi: 10.1523/JNEUROSCI.1866-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thévenaz P, Ruttimann UE, Unser M. A pyramid approach to subpixel registration based on intensity. IEEE Transactions on Image Processing. 1998;7:27–41. doi: 10.1109/83.650848. [DOI] [PubMed] [Google Scholar]
- Tramu G, Pillez A, Leonardelli J. An efficient method of antibody elution for the successive or simultaneous localization of two antigens by immunocytochemistry. J. Histochem. Cytochem. 1978;26:322–324. doi: 10.1177/26.4.207771. [DOI] [PubMed] [Google Scholar]
- Wahlby C, Erlandsson F, Bengtsson E, Zetterberg A. Sequential immunofluorescence staining and image analysis for detection of large numbers of antigens in individual cell nuclei. Cytometry. 2002;47:32–41. [PubMed] [Google Scholar]
- Zondervan R, Kulzer F, Kol’chenko MA, Orrit M. Photobleaching of rhodamine 6G in Poly(vinyl alcohol) at the ensemble and single-molecule levels. J. Phys. Chem. 2004;108:1657–1665. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.