

Abstract
Introduction
Deep venous thrombosis (DVT) resolution involves the plasmin and the matrix metalloproteinase (MMP) system. This study tested the hypothesis that pharmacological inhibition of the plasmin system would impair DVT resolution and worsen vein wall damage.
Methods
A rat model of stasis DVT by inferior vena cava (IVC) ligation was performed with intravenous control saline or aprotinin (AP; 2.8 mg/kg at operation), and harvest of thrombosed IVC at 7 days. After laser Doppler imaging, DVT were separated, weighed, and vein wall stiffness was assessed by tensiometry. Thrombus and vein wall tissue analysis included total collagen by colorimetric assay, cytokines, chemokines, and d-dimer by ELISA, urokinase-plasminogen activator (uPA) and plasminogen activator inhibitor-1 (PAI-1) by immuno-blotting, MMP-2 and -9 by zymography, and neutrophil (PMN) and monocyte (ED-1) leukocytes by immunohistochemistry.
Results
DVT weights were 2 fold greater in the AP treated rats (P<.05), but no significant differences in thrombus perfusion, collagen, or d-dimer levels were found. Vein wall stiffness was reduced 50% (P<.05), suggesting less biomechanical injury. The total vein wall MMP-9 was increased (P<.05) 5 fold in the AP group compared with controls, while MMP-2 was elevated but had not reached significance. No difference was found in vein wall TNFα, TGFβ, vein wall or thrombus monocytes, PMN, or uPA/PAI-1 ratio between groups.
Discussion
AP inhibition of the plasmin system was associated with larger thrombi but less vein wall injury, but no difference in other measures of resolution, possibly because of increased vein wall MMP-9 activity. These data suggest an important redundant mechanism for DVT resolution.
Keywords: inflammation, thrombosis, venous, plasmin, matrix metalloproteinase, fibrin
INTRODUCTION
Post thrombotic syndrome (PTS) is a common clinical problem after deep venous thrombosis (DVT), of which the incidence has not decreased over the last 25 years, with treatment costs in the hundreds of millions of dollars.1 The end result of DVT resolution is valvular dysfunction and stiff vein walls that clinically manifest as venous hypertension with pain, swelling, and sometimes recalcitrant ulceration.2, 3 Anticoagulants, such as low molecular weight heparin (LMWH), are effective when used for prophylaxis and treatment of DVT, but no evidence exists that these agents decrease vein wall scarring2, 4 Clinically, larger and increased time to thrombus resolution may increase the risk of PTS.5
The formation of plasmin from plasminogen by the action of tissue-type (tPA) and urokinase-type (uPA) plasminogen activators is thought to be the main pathway by which fibrin deposition is regulated in the vascular tree and by which pericellular fibrinolysis, required for cell migration in tissues and thrombi, is activated.6-9 Aprotinin (AP; Trasylol® West Haven, CT), is a naturally occurring polypeptide that reversibly inhibits serine proteases including the plasminogen activators such as urokinase-type plasminogen activation (uPA). Serine proteases mediate a variety of reactions in the body, including activation of the coagulation cascade, the fibrinolytic system, and inflammatory responses. At low plasma concentrations, AP attenuates fibrinolysis via plasmin inhibition.10, 11
Matrix metalloproteinases (MMP) also play an important role in the degradation of extracellular matrix and basement membrane components to allow the migration of vascular smooth muscle cells, as well as contribute to acute and chronic fibrotic diseases such as interstitial lung disease, interstitial nephritis, and vascular disease.12-15 MMP’s are secreted in a latent proform, and physiological activation of proMMP’s may involve plasmin.16, 17 Plasmin directly activates proMMP-1, -3, -9, -10, and -13,14, 16, 18 although other stimuli may cause MMP activation.19 Furthermore, several active MMP’s can activate other proMMP’s, thus representing positive feedback mechanisms.20-24 MMP-2 and -9 are both collagenolytic and elastolytic and are involved with vessel remodeling,25,26 although, unlike other MMP’s, these are not known to be directly fibrinolytic. 27 However, recent work has suggested reciprocal activation in vivo of the MMP and plasmin systems in a mouse model of DVT resolution; namely, that uPA upregulation may compensate partially for MMP inhibition.28
This study tested the hypotheses that early inhibition of plasmin via AP would result in larger thrombi, greater vein wall damage, and a reciprocal increase in MMP activity in a rat model of DVT.
METHODS
Animal model
Male Sprague-Dawley rats (250-450 g) were used for all studies and all protocols were approved by the University of Michigan Animal Care Protocol. For all surgical procedures, the rats underwent general anesthesia with isoflurane/O2 with full physiological monitoring. Stasis thrombosis was induced by IVC ligation as described.29, 30 Briefly, a laparotomy with ligation of the IVC below the renal veins and all visible side branches was performed. Rats were injected with Trasylol® (2.8 mg/kg AP; Bayer Pharmaceuticals Corporation, West Haven, CT) or control saline via tail vein at time of operation. At 7 days, the thrombosed IVC was carefully dissected and removed for histological analysis while tissue studies were done after thrombus-vein wall separation.
Laser Doppler
The Lisca laser Doppler (Lisca, Inc., Lingköping, Sweden) was used to assess in vivo micro-vascular IVC blood flow, based on the aforementioned techniques.29-31 Flow through the exposed IVC region of interest was assessed before ligation and at harvest. Depth was constant for all rodents to ensure consistent estimation of the mid-coronal IVC section. These scans were saved and accompanying image software was used to estimate the mean flow by using a standardized area of analysis. Flow intensities were reported as % baseline flow.
Tensiometric vein wall analysis
Each harvested IVC was divided longitudinally and force extension curves were generated for each segment using an Instron Tensiometer (model 5542, Instron Corporation, Canton, MA) with data analysis as described.30, 32
Collagen Assay
Thrombus collagen (Types I-V) content was estimated using a commercially available kit according to manufacturer’s instructions (Biocolor LTD, Belfast, North Ireland). Total collagen content was corrected to mg thrombus weight, with an estimated sensitivity of 0.5 mcg/mL.30, 33
D-dimer ELISA
Thrombus d-dimer content was estimated using a commercially available kit for human d-dimer with cross reactivity to rat according manufacturer’s instructions (Diagnostica Stago, Germany).34 D-dimer content was corrected to mg thrombus weight.
Chemokine/cytokine ELISA
Tissue homogenate of the IVC’s for rat TNFα, βFGF, TGFβ, and RANTES were performed with species specific primary antibodies quantified using a double ligand technique as has been described for similar chemokines.30, 35, 36 Quantification of peptide mediators was normalized to total protein in the sample.
SDS-PAGE Gelatin Zymography
As described28, 30 homogenized IVC tissue and thrombus were subjected to substrate zymography for MMP-2 and -9 using pre-cast 10% SDS-polyacrylamide gels containing 1 mg/mL of gelatin (unless otherwise stated, all zymography supplies were from Novex, San Diego, CA). Densitometry analysis was performed using a FOTO/Analyst CCD CAMERA (Fotodyne, Hartland, WI) and GEL-Pro Analyzer software version 3.1 (Media Cybernetics, Silver Springs, MD). Pro and active MMP-2 and -9 activity optical densities were summed and normalized to IVC or thrombus protein.
Western immunoblotting for uPA and PAI-1
Protein was isolated from IVC tissue and thrombus using TRIzol Reagent (Life Technologies, Carlsbad, CA) and dissolved in 1% SDS. Rabbit anti-rat uPA (1:2500; Santa Cruz Biotechnology, Santa Cruz, CA) and mouse anti-rat PAI-1 (1:2500; BD Biosciences, Franklin Lakes, NJ) monoclonal antibodies were used. Blotting was performed and immunoreactive bands visualized as described.30, 33
Immunohistochemical staining
Immunohistochemical staining was performed on the paraffin embedded tissue sections (10 μm) as described.28, 37 Anti-ED-1 (1:100; Serotec, Oxford, UK) and anti-PMN (1:500, Accurate Chemical, Westbury, CT) antibodies were used. In a blinded fashion, cellular positive staining quantification was determined by direct counting of cells in 5 hpf (1000X) radially around the IVC wall or thrombus and totaled.
Aprotinin assay
Blood was drawn from rats 1 hour after Trasylol® injection. AP plasma concentration was determined using a commercially available kit according to manufacturer’s instructions for use with rat serum (Unitest Protenin assay, Technoclone Ltd, Dorling, UK).
Statistical analysis
All data are represented as mean ± SE. Two-tailed unpaired Student’s T-test was used for comparison between groups (Sigma Plot, SPSS, Inc. Chicago, IL). P ≤.05 was assigned significance.
RESULTS
Aprotinin causes larger DVT but a similar cellular and cytokine composition
Thrombus weights are a simple and reliable measure of thrombus resolution.28, 29,37 The AP assay 38 confirmed significant plasma AP concentrations at one hour after injection (P < .001). Thrombus size was increased nearly 2-fold in AP animals compared with controls (P = .048) (Figure 1a). Qualitatively, thrombi in the AP group were less solid and more gel-like than controls. There were no differences in thrombus fibrin, measured indirectly by d-dimer34(Figure 1b), or thrombus collagen (Figure 1c) content between groups. In addition, there was no difference in thrombus perfusion measured by laser Doppler between groups (56±7% to 51±8% arbitrary units of baseline non-thrombosed blood flow, n=6-7) and values were consistent with 7 day re-established flow.30 Evaluation of thrombus concentrations of TNFα, TGFβ, βFGF and cellular counts of ED-1 and PMN showed similar values (data not shown).
Figure 1.
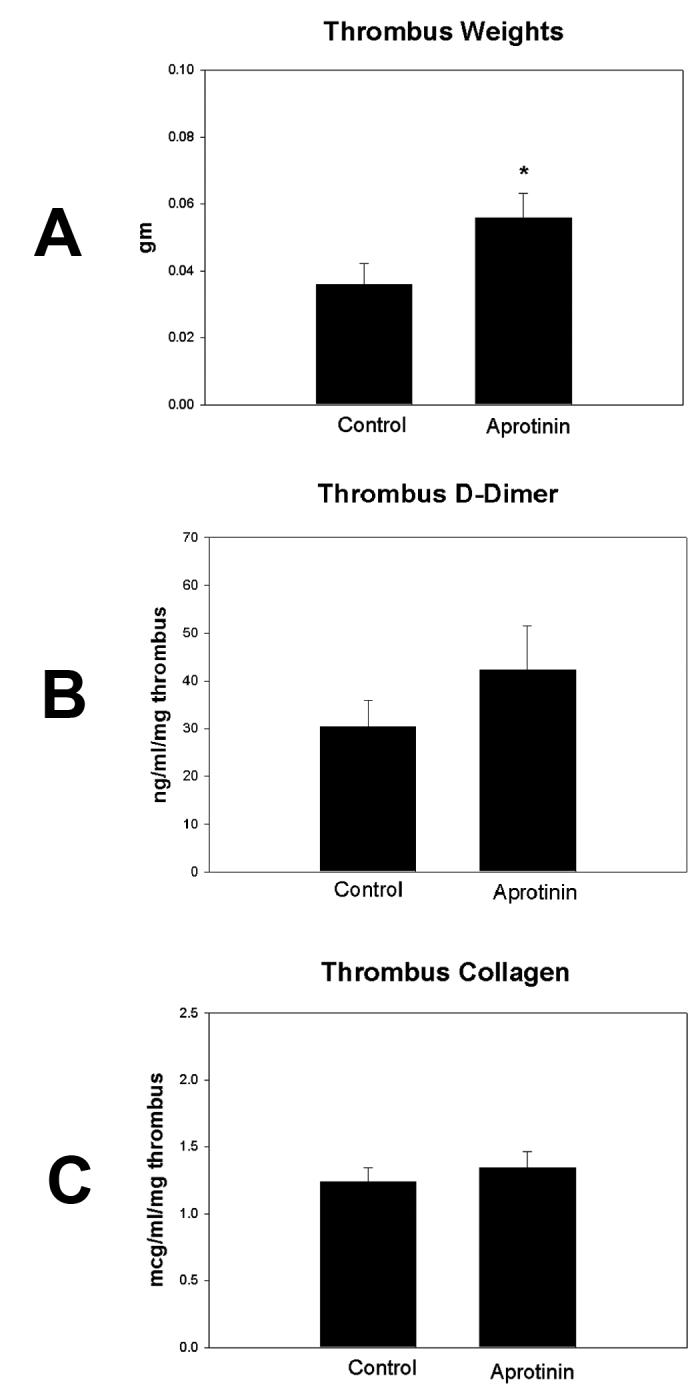
a) Comparison of thrombus weights showed increased thrombus weight in the aprotinin group compared with control, suggesting that DVT resolution was impaired in the aprotinin group (*=P ≤ .05, n=12-14. b). Comparison of thrombus d-dimer concentrations by colorimetric assay showed no significant difference between groups, suggesting that fibrin concentration was not different between groups (n=3-4). c) Comparison of thrombus total collagen concentration by colorimetric assay showed no significant difference between groups (n=9-11).
Aprotinin reduces vein wall damage
Vein wall stiffness is the approximate inverse of vein wall compliance, and is a biochemical measure of vein wall injury during DVT resolution.30, 32 Vein wall stiffness was ∼2 fold less in the AP group compared with controls (P = .05) (Figure 2). Vein wall inflammation is associated with elevation of cytokines and chemokines.30, 35 RANTES is a chemokine that plays an active role in recruiting leukocytes into inflammatory sites as well as being released by platelets. Vein wall RANTES concentration was increased 2-fold in AP treated rats compared with controls (P = .035) (Figure 3). Similarly, there was also no difference in vein wall concentrations of inflammatory cytokines TNFα, βFGF, or TGFβ between groups (data not shown). There was no difference between groups in the number of PMN or ED-1 positive cells in vein wall (data not shown).
Figure 2.
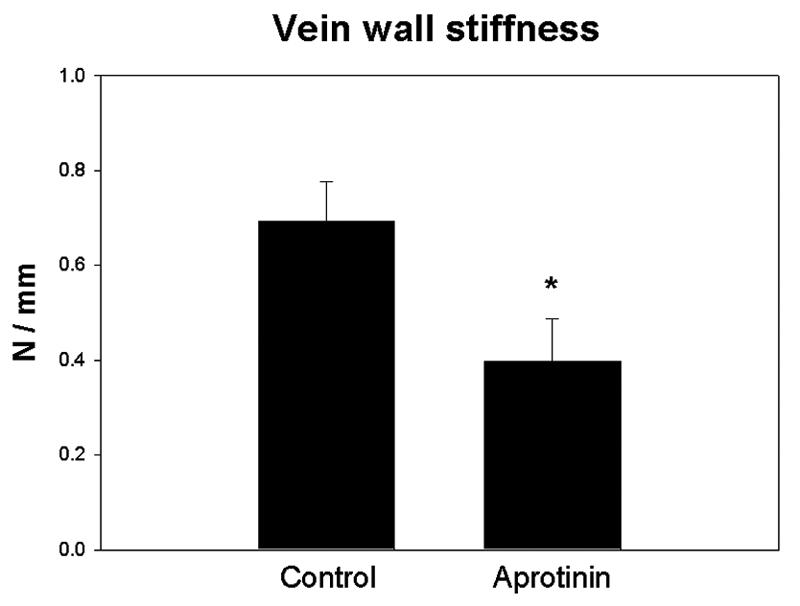
Vein wall stiffness is an approximate converse of elasticity. Normal veins are elastic and compliant to accommodate blood volume changes. Comparison of vein wall stiffness by tensiometric analysis showed decreased stiffness in the aprotinin group compared to control, suggesting that vein wall damage was reduced in the aprotinin group (*= P ≤ .05, n=4).
Figure 3.
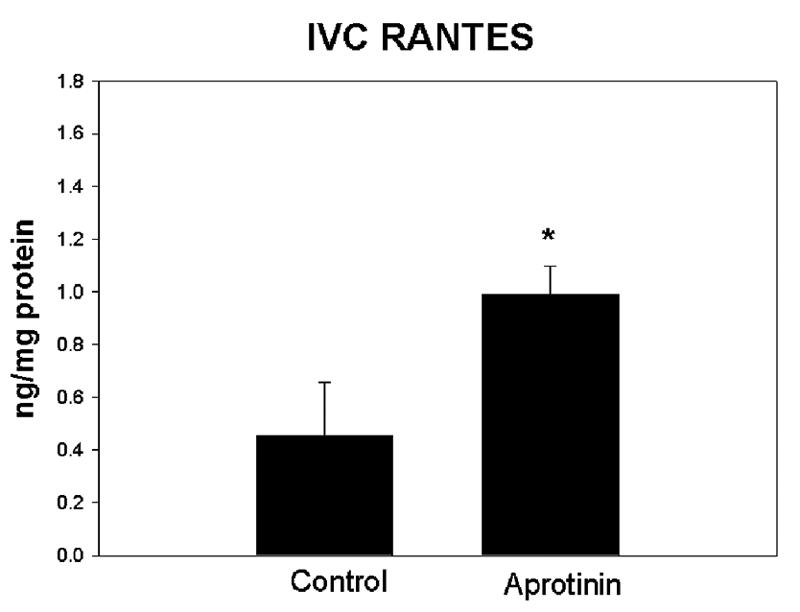
Comparison of RANTES in vein wall showed increased concentration in the aprotenin group as compared with control (*= P ≤ .05, n=6-7). Other pro-inflammatory mediators were not significantly altered by aprotinin treatment (TNFα, IL1β, TGFβ, bFGF) in the thrombus or vein wall.
Vein wall and thrombus matrix reorganization
Total uPA antigen in the thrombi was increased 3-fold in controls compared to the AP group (P=.11), suggesting a trend in increased total uPA activity in control thrombi (Figure 4a). There was no significant difference in uPA concentration in vein walls between groups (.011±.002 vs .010±.002 OD/mg protein; n=5-6; P=.80). To better evaluate whether uPA balance was altered, PAI-1 Ag was also evaluated by WB as uPA is inhibited by PAI-1. Thus, the uPA/PAI-1 ratio is indicative of the overall balance of uPA activity per molecule.33 There were no differences between groups in uPA/PAI-1 ratios measured in thrombus (AP=.67±.19 vs. control= .94±.17; n=3-4; P=.34) or vein wall (AP=.52±.03 vs. control=.57±.20; n=3-4; P=.81), suggesting that there was no difference in overall uPA activity per molecule between groups.
Figure 4.

a) Comparison of thrombus uPA between groups showed a trend of increased concentration in controls (n=6, P =.11). b) Total MMP-9 activity in thrombus and vein wall. The vein wall MMP-9 activity was significantly increased in the aprotinin group, and trended so in the thrombus, as compared with control (n=4-6).
Conversely, total MMP-9 activity of the AP group was increased 5-fold in the vein wall (P=.048) and increased 2-fold in the thrombus (P=.19) (Figure 4b). Similar increases were found analyzing solely pro and active forms of MMP-9 (data not shown). Total MMP-2 activity in the vein wall was increased 2-fold in the AP group (P=.10), but less so in the thrombus as compared with control (P=.18).
DISCUSSION
Plasmin is the primary fibrinolytic protease that directly degrades fibrin and the pericellular matrix, as well as activating other matrix-degrading enzymes, such as proMMP’s.39, 40 The purpose of this study was to use AP as a selective tool to inhibit plasmin activation, and determine its role on vein wall injury as well as MMP activation. We also used this strategy to determine if greater thrombus size was associated with increased vein wall damage. Four main conclusions can be drawn from this study; at 7 days after thrombosis, AP: 1) increases thrombus size, 2) increases total vein wall MMP-9 activity, 3) reduces vein wall stiffness, and 4) does not affect the cellular or inflammatory cytokine profile of the thrombus. This is counterintuitive in many respects, given the proximal position of plasmin mediated fibrinolysis, and is counter to what has been shown in the arterial system with plasmin dependent MMP-9 activation.25 However, multiple pathways provide for plasmin activation and subsequent MMP-activation,41 and prior work has suggested that the thrombus composition is more important than the size.30, 32
Thrombus weights are a simple and reliable measure of thrombus resolution.37 Not surprisingly, AP increased thrombus size at 7 days, suggesting reduced thrombus resolution. This was expected because AP inhibits plasmin, which is largely responsible for fibrinolysis in DVT resolution.6-9 Similarly, it has been shown that uPA, rather than tPA, is most important in thrombus resolution in the venous system.9 Although thrombi were larger in the AP group, other measures of DVT resolution (thrombus perfusion, collagen, and d-dimer content)28-30 did not show a significant difference between groups. Thus, although the thrombi were larger with AP, the resolution was similar. For example, recannalization with interim thrombus blood flow (LD imaging) and collagenolysis were likely similar. We speculate that the fibrin or platelet content may make up the size difference.
MMP-2 and -9 have collagenolytic and elastolytic activity, and are elevated in the vein wall during DVT resolution.15, 28, 33 Previous studies in mice DVT model have suggested a reciprocal activation of the plasmin and MMP systems.28 Consistently, studies have shown that thrombus resolution is impaired in veins of uPA-/- mice,9 although MMP activity was not evaluated in that study. Pharmacological inhibition of the plasmin system was associated with increased vein wall MMP-9 activity, but not a significant increase of MMP-2. It is likely that the MMP-9 is derived from influxed monocytes, as few PMN are present.30 Alternative mechanisms of MMP-9 activation accounted for this observed increase in MMP-9,15, 41 or the single peri operative dose of AP may have only briefly impaired plasmin production. We found that an increased dose of AP (5 fold) showed a similar increase in thrombus size and similar decrease in vein wall stiffness (data not shown).
Although the thrombi were larger in the AP group, the vein wall damage was less, as reflected by the biomechanical measure of stiffness. Clinical and experimental studies suggest that the longer a DVT is in contact with the vein wall, the greater the resulting damage42-44(Henke, unpublished). In this study, AP impaired DVT resolution and paradoxically reduced vein wall stiffness. As this is a full stasis model of DVT, persistent thrombus - vein wall contact occurs, and this mechanism of DVT genesis was the same between both the AP and control groups. This suggests that the resulting damage in the vein wall may be caused by direct activity of the plasmin - activated proteases rather than simply duration of thrombus - vein wall contact. This study indirectly suggests that MMP’s are not the main agents responsible for vein wall fibrotic injury, but are important for DVT resolution. Interestingly, this has been observed with PMN depletion in experimental stasis DVT whereby early loss of thrombus MMP-9 was associated with increased vein wall stiffness.30 Similarly, over expression of uPA has been associated with cardiac injury in a mouse model.45 Another possible explanation is that attenuation of the inflammatory response by AP caused reduced vein wall injury, or AP inhibited other matrix enzymes such as cathepsins or serum elastase.46 The latter contribution may be most important, and is a subject of future study.
Cellular and inflammatory cytokine content was also similar between the groups, corrected to total protein in the sample. In contrast to uPA genetic deleted mice, we found no difference in thrombus monocytes. However, the leukocyte deficiency noted in uPA-/- mice in Singh’s study was most striking at later time points. Although there was no difference in vein wall PMN or ED-1 cell counts between groups, impaired DVT resolution may explain the significant increase in chemokine RANTES as a compensatory response to increase leukocyte influx in the AP group. However, there was no difference in pro-inflammatory cytokine TNFα concentrations between groups. AP is also known to be able to attenuate inflammatory responses,11 which may have influenced RANTES secretion. Alternatively, the increased RANTES may also reflect a greater platelet mass as RANTES is released from platelets after stimulation.
The current data suggest a complex role for plasmin activators and the adjacent vein wall response. We are currently investigating the mediators involved with uPA -/- mice as well of some of the non-MMP mechanisms involved with DVT resolution. While native plasmin is essential for fibrinolysis, this may paradoxically worsen vein wall injury. The ultimate clinical goal is to provide rapid fibrinolysis and decrease the vein wall injury while conferring minimal bleeding risk.
Acknowledgments
Supported in part by NIH HL675317, HL083918 (PKH).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Heit JA, Silverstein MD, Mohr DN, et al. The epidemiology of venous thromboembolism in the community. Thromb Haemost. 2001;86:452–63. [PubMed] [Google Scholar]
- 2.Prandoni P, Lensing AW, Cogo A, et al. The long-term clinical course of acute deep venous thrombosis. Ann Intern Med. 1996;125:1–7. doi: 10.7326/0003-4819-125-1-199607010-00001. [DOI] [PubMed] [Google Scholar]
- 3.Delis KT, Bountouroglou D, Mansfield AO. Venous Claudication in Iliofemoral Thrombosis: Long-term Effects on Venous Hemodynamics, Clinical Status, and Quality of Life. Ann Surg. 2004;239:118–126. doi: 10.1097/01.sla.0000103067.10695.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Goldhaber SZ, Tapson VF. A prospective registry of 5,451 patients with ultrasound-confirmed deep vein thrombosis. Am J Cardiol. 2004;93:259–262. doi: 10.1016/j.amjcard.2003.09.057. [DOI] [PubMed] [Google Scholar]
- 5.Johnson BF, Manzo RA, Bergelin RO, Strandness DEJ. Relationship between changes in the deep venous system and the development of the postthrombotic syndrome after an acute episode of lower limb deep vein thrombosis: A one- to six-year follow-up. J Vasc Surg. 1995;21:307–13. doi: 10.1016/s0741-5214(95)70271-7. [DOI] [PubMed] [Google Scholar]
- 6.Tabrizi P, Wang L, Seeds N, et al. Tissue plasminogen activator (tPA) deficiency exacerbates cerebrovascular fibrin deposition and brain injury in a murine stroke model: studies in tPA-deficient mice and wild-type mice on a matched genetic background. Arterioscler Thromb Vasc Biol. 1999;19:2801–6. doi: 10.1161/01.atv.19.11.2801. [DOI] [PubMed] [Google Scholar]
- 7.Bugge TH, Flick MJ, Danton MJ, et al. Urokinase-type plasminogen activator is effective in fibrin clearance in the absence of its receptor or tissue-type plasminogen activator. Proc Natl Acad Sci U S A. 1996;93:5899–904. doi: 10.1073/pnas.93.12.5899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ploplis VA, French EL, Carmeliet P, Collen D, Plow EF. Plasminogen deficiency differentially affects recruitment of inflammatory cell populations in mice. Blood. 1998;91:2005–9. [PubMed] [Google Scholar]
- 9.Singh I, Burnand KG, Collins M, et al. Failure of thrombus to resolve in urokinase-type plasminogen activator gene-knockout mice: rescue by normal bone marrow-derived cells. Circulation. 2003;107:869–75. doi: 10.1161/01.cir.0000050149.22928.39. [DOI] [PubMed] [Google Scholar]
- 10.van Oeveren W, Harder MP, Roozendaal KJ, Eijsman L, Wildevuur CR.Aprotinin protects platelets against the initial effect of cardiopulmonary bypass J Thorac Cardiovasc Surg 199099788–96. discussion 796-7 [PubMed] [Google Scholar]
- 11.Wachtfogel YT, Kucich U, Hack CE, et al. Aprotinin inhibits the contact, neutrophil, and platelet activation systems during simulated extracorporeal perfusion J Thorac Cardiovasc Surg 19931061–9. discussion 9-10 [PubMed] [Google Scholar]
- 12.Dollery CM, McEwan JR, Henney AM. Matrix metalloproteinases and cardiovascular disease. Circ Res. 1995;77:863–8. doi: 10.1161/01.res.77.5.863. [DOI] [PubMed] [Google Scholar]
- 13.Bendeck MP, Zempo N, Clowes AW, Galardy RE, Reidy MA. Smooth muscle cell migration and matrix metalloproteinase expression after arterial injury in the rat. Circ Res. 1994;75:539–45. doi: 10.1161/01.res.75.3.539. [DOI] [PubMed] [Google Scholar]
- 14.Woessner JF., Jr. Matrix metalloproteinases and their inhibitors in connective tissue remodeling. Faseb J. 1991;5:2145–54. [PubMed] [Google Scholar]
- 15.Galis ZS, Khatri JJ. Matrix metalloproteinases in vascular remodeling and atherogenesis: the good, the bad, and the ugly. Circ Res. 2002;90:251–62. [PubMed] [Google Scholar]
- 16.Eeckhout Y, Vaes G. Further studies on the activation of procollagenase, the latent precursor of bone collagenase. Effects of lysosomal cathepsin B, plasmin and kallikrein, and spontaneous activation. Biochem J. 1977;166:21–31. doi: 10.1042/bj1660021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Birkedal-Hansen H. Proteolytic remodeling of extracellular matrix. Curr Opin Cell Biol. 1995;7:728–35. doi: 10.1016/0955-0674(95)80116-2. [DOI] [PubMed] [Google Scholar]
- 18.Matrisian LM. The matrix-degrading metalloproteinases. Bioessays. 1992;14:455–63. doi: 10.1002/bies.950140705. [DOI] [PubMed] [Google Scholar]
- 19.Keski-Oja J, Lohi J, Tuuttila A, Tryggvason K, Vartio T. Proteolytic processing of the 72,000-Da type IV collagenase by urokinase plasminogen activator. Exp Cell Res. 1992;202:471–6. doi: 10.1016/0014-4827(92)90101-d. [DOI] [PubMed] [Google Scholar]
- 20.Suzuki K, Enghild JJ, Morodomi T, Salvesen G, Nagase H. Mechanisms of activation of tissue procollagenase by matrix metalloproteinase 3 (stromelysin) Biochemistry. 1990;29:10261–70. doi: 10.1021/bi00496a016. [DOI] [PubMed] [Google Scholar]
- 21.Ogata Y, Enghild JJ, Nagase H. Matrix metalloproteinase 3 (stromelysin) activates the precursor for the human matrix metalloproteinase 9. J Biol Chem. 1992;267:3581–4. [PubMed] [Google Scholar]
- 22.Okada Y, Gonoji Y, Naka K, et al. Matrix metalloproteinase 9 (92-kDa gelatinase/type IV collagenase) from HT 1080 human fibrosarcoma cells. Purification and activation of the precursor and enzymic properties. J Biol Chem. 1992;267:21712–9. [PubMed] [Google Scholar]
- 23.Imai K, Yokohama Y, Nakanishi I, et al. Matrix metalloproteinase 7 (matrilysin) from human rectal carcinoma cells. Activation of the precursor, interaction with other matrix metalloproteinases and enzymic properties. J Biol Chem. 1995;270:6691–7. doi: 10.1074/jbc.270.12.6691. [DOI] [PubMed] [Google Scholar]
- 24.Sato H, Takino T, Okada Y, et al. A matrix metalloproteinase expressed on the surface of invasive tumour cells. Nature. 1994;370:61–5. doi: 10.1038/370061a0. [DOI] [PubMed] [Google Scholar]
- 25.Visse R, Nagase H. Matrix metalloproteinases and tissue inhibitors of metalloproteinases: structure, function, and biochemistry. Circ Res. 2003;92:827–39. doi: 10.1161/01.RES.0000070112.80711.3D. [DOI] [PubMed] [Google Scholar]
- 26.Coats WD, Jr., Whittaker P, Cheung DT, Currier JW, Han B, Faxon DP. Collagen content is significantly lower in restenotic versus nonrestenotic vessels after balloon angioplasty in the atherosclerotic rabbit model. Circulation. 1997;95:1293–300. doi: 10.1161/01.cir.95.5.1293. [DOI] [PubMed] [Google Scholar]
- 27.Lijnen HR. Matrix metalloproteinases and cellular fibrinolytic activity. Biochemistry (Mosc) 2002;67:92–8. doi: 10.1023/a:1013908332232. [DOI] [PubMed] [Google Scholar]
- 28.Henke PK, Pearce CG, Moaveni DM, et al. Targeted Deletion of CCR2 Impairs Deep Vein Thombosis Resolution in a Mouse Model. J Immunol. 2006;177:3388–97. doi: 10.4049/jimmunol.177.5.3388. [DOI] [PubMed] [Google Scholar]
- 29.Varma MR, Moaveni DM, Dewyer NA, et al. Deep vein thrombosis resolution is not accelerated with increased neovascularization. J Vasc Surg. 2004;40:536–42. doi: 10.1016/j.jvs.2004.05.023. [DOI] [PubMed] [Google Scholar]
- 30.Henke PK, Varma MR, Deatrick KB, et al. Neutrophils modulate post-thrombotic vein wall remodeling but not thrombus neovascularization. Thromb Haemost. 2006;95:272–81. doi: 10.1160/TH05-02-0099. [DOI] [PubMed] [Google Scholar]
- 31.Couffinhal T, Silver M, Kearney M, et al. Impaired collateral vessel development associated with reduced expression of vascular endothelial growth factor in ApoE-/- mice. Circulation. 1999;99:3188–98. doi: 10.1161/01.cir.99.24.3188. [DOI] [PubMed] [Google Scholar]
- 32.Myers DD, Jr., Henke PK, Bedard PW, et al. Treatment with an oral small molecule inhibitor of P selectin (PSI-697) decreases vein wall injury in a rat stenosis model of venous thrombosis. J Vasc Surg. 2006;44:625–32. doi: 10.1016/j.jvs.2006.05.021. [DOI] [PubMed] [Google Scholar]
- 33.Deatrick KB, Eliason JL, Lynch EM, et al. Vein wall remodeling after deep vein thrombosis involves matrix metalloproteinases and late fibrosis in a mouse model. J Vasc Surg. 2005;42:140–8. doi: 10.1016/j.jvs.2005.04.014. [DOI] [PubMed] [Google Scholar]
- 34.Gossage JA, Humphries J, Modarai B, Burnand KG, Smith A. Adenoviral urokinase-type plasminogen activator (uPA) gene transfer enhances venous thrombus resolution. J Vasc Surg. 2006;44:1085–90. doi: 10.1016/j.jvs.2006.07.020. [DOI] [PubMed] [Google Scholar]
- 35.Wakefield TW, Strieter RM, Wilke CA, et al. Venous thrombosis-associated inflammation and attenuation with neutralizing antibodies to cytokines and adhesion molecules. Arterioscler Thromb Vasc Biol. 1995;15:258–68. doi: 10.1161/01.atv.15.2.258. [DOI] [PubMed] [Google Scholar]
- 36.Rectenwald JE, Deatrick KB, Sukheepod P, et al. Experimental pulmonary embolism: effects of the thrombus and attenuation of pulmonary artery injury by low-molecular-weight heparin. J Vasc Surg. 2006;43:800–8. doi: 10.1016/j.jvs.2005.12.010. [DOI] [PubMed] [Google Scholar]
- 37.Henke PK, Varga A, De S, et al. Deep vein thrombosis resolution is modulated by monocyte CXCR2-mediated activity in a mouse model. Arterioscler Thromb Vasc Biol. 2004;24:1130–7. doi: 10.1161/01.ATV.0000129537.72553.73. [DOI] [PubMed] [Google Scholar]
- 38.Cardigan RA, Mackie IJ, Gippner-Steppert C, Jochum M, Royston D, Gallimore MJ. Determination of plasma aprotinin levels by functional and immunologic assays. Blood Coagul Fibrinolysis. 2001;12:37–42. doi: 10.1097/00001721-200101000-00006. [DOI] [PubMed] [Google Scholar]
- 39.Lijnen HR, Van Hoef B, Lupu F, Moons L, Carmeliet P, Collen D. Function of the plasminogen/plasmin and matrix metalloproteinase systems after vascular injury in mice with targeted inactivation of fibrinolytic system genes. Arterioscler Thromb Vasc Biol. 1998;18:1035–45. doi: 10.1161/01.atv.18.7.1035. [DOI] [PubMed] [Google Scholar]
- 40.Saksela O, Rifkin DB. Cell-associated plasminogen activation: regulation and physiological functions. Annu Rev Cell Biol. 1988;4:93–126. doi: 10.1146/annurev.cb.04.110188.000521. [DOI] [PubMed] [Google Scholar]
- 41.Lijnen HR, Collen D. Matrix metalloproteinase system deficiencies and matrix degradation. Thromb Haemost. 1999;82:837–45. [PubMed] [Google Scholar]
- 42.Meissner MH, Manzo RA, Bergelin RO, Markel A, Strandness DE., Jr.Deep venous insufficiency: the relationship between lysis and subsequent reflux J Vasc Surg 199318596–605. discussion 606-8 [PubMed] [Google Scholar]
- 43.Mewissen MW. Catheter-directed thrombolysis for lower extremity deep vein thrombosis. Tech Vasc Interv Radiol. 2001;4:111–4. doi: 10.1016/s1089-2516(01)90005-8. [DOI] [PubMed] [Google Scholar]
- 44.See-Tho K, Harris EJ., Jr.Thrombosis with outflow obstruction delays thrombolysis and results in chronic wall thickening of rat veins J Vasc Surg 199828115–22. discussion 123 [DOI] [PubMed] [Google Scholar]
- 45.Moriwaki H, Stempien-Otero A, Kremen M, Cozen AE, Dichek DA. Overexpression of urokinase by macrophages or deficiency of plasminogen activator inhibitor type 1 causes cardiac fibrosis in mice. Circ Res. 2004;95:637–44. doi: 10.1161/01.RES.0000141427.61023.f4. [DOI] [PubMed] [Google Scholar]
- 46.Cheng XW, Kuzuya M, Sasaki T, et al. Increased expression of elastolytic cysteine proteases, cathepsins S and K, in the neointima of balloon-injured rat carotid arteries. Am J Pathol. 2004;164:243–51. doi: 10.1016/S0002-9440(10)63114-8. [DOI] [PMC free article] [PubMed] [Google Scholar]