

Summary
Background
Carbamazepine is widely accepted as a drug of first choice for patients with partial onset seizures. Several newer drugs possess efficacy against these seizure types but previous randomised controlled trials have failed to inform a choice between these drugs. We aimed to assess efficacy with regards to longer-term outcomes, quality of life, and health economic outcomes.
Methods
SANAD was an unblinded randomised controlled trial in hospital-based outpatient clinics in the UK. Arm A recruited 1721 patients for whom carbamazepine was deemed to be standard treatment, and they were randomly assigned to receive carbamazepine, gabapentin, lamotrigine, oxcarbazepine, or topiramate. Primary outcomes were time to treatment failure, and time to 12-months remission, and assessment was by both intention to treat and per protocol. This study is registered as an International Standard Randomised Controlled Trial, number ISRCTN38354748.
Findings
For time to treatment failure, lamotrigine was significantly better than carbamazepine (hazard ratio [HR] 0·78 [95% CI 0·63–0·97]), gabapentin (0·65 [0·52–0·80]), and topiramate (0·64 [0·52–0·79]), and had a non-significant advantage compared with oxcarbazepine (1·15 [0·86–1·54]). For time to 12-month remission carbamazepine was significantly better than gabapentin (0·75 [0·63–0·90]), and estimates suggest a non-significant advantage for carbamazepine against lamotrigine (0·91 [0·77–1·09]), topiramate (0·86 [0·72–1·03]), and oxcarbazepine (0·92 [0·73–1·18]). In a per-protocol analysis, at 2 and 4 years the difference (95% CI) in the proportion achieving a 12-month remission (lamotrigine-carbamazepine) is 0 (−8 to 7) and 5 (−3 to 12), suggesting non-inferiority of lamotrigine compared with carbamazepine.
Interpretation
Lamotrigine is clinically better than carbamazepine, the standard drug treatment, for time to treatment failure outcomes and is therefore a cost-effective alternative for patients diagnosed with partial onset seizures.
Introduction
Epilepsy is a common disorder worldwide (50 per 100 000 people; 0·5–1%).1 Studies of the natural history of the condition indicate that as many as 70% of patients enter long-term remission shortly after starting drug therapy.2,3 Carbamazepine is currently recommended as the first-line antiepileptic drug treatment for patients with partial onset seizures.4,5 This recommendation is based largely on the results of randomised controlled trials comparing carbamazepine and valproate, a meta-analysis of which provided evidence that carbamazepine was the better treatment for the outcomes of time to first seizure and time to 12-month remission.6
The past decade and a half has seen the licensing and introduction of several new antiepileptic drugs. These have all been licensed initially on the basis of add-on randomised controlled trials in patients with refractory partial epilepsy. Aggregate data meta-analyses of these trials7,8 indicate by indirect comparisons that some agents could be more effective than others. Some comparative randomised trials of the new drugs compared with carbamazepine9-19 or other standard drugs20-23 have appeared. Such trials comparing carbamazepine with tiagabine,16 vigabatrin,13 and remacemide17 indicate that these drugs are not as effective as carbamazepine and should not be investigated further as possible first-line treatments. For gabapentin, lamotrigine, oxcarbazepine, and topiramate, randomised controlled trials suggest efficacy as monotherapy, but for several reasons these trials fail to inform clinical practice or policy, an issue that has been highlighted in NICE appraisals.24,25 Firstly, the trials were too short to measure important clinical outcomes of longer-term seizure control.26 Secondly, existing randomised controlled trials have not systematically addressed quality of life outcomes, and have not been structured to assess health economic issues. Despite these limitations, there has been a steady rise in the prescribing of new antiepileptic drugs from 0·1% of total antiepileptic prescriptions in 1991 to 20% in 2002, where new drugs accounted for 69% of the total costs of such drugs to the UK National Health Service (NHS; GBP£99 million of £142 million).24
Because most patients who develop epilepsy are treated with one type of drug and might continue to take them for many years, standard and new drugs need to be compared so as to establish which should, in the future, be first choice for appropriate groups of patients. We have therefore undertaken two concurrent unblinded randomised controlled trials comparing Standard and New Antiepileptic Drugs (SANAD), which examine seizure control, tolerability, quality of life, and health economic outcomes. Arm A of SANAD is reported here and compares carbamazepine, gabapentin, lamotrigine, oxcarbazepine, and topiramate.
SANAD was commissioned by the Health Technology Assessment Programme of NHS Research and Development. It received appropriate multicentre and local ethics and research committee approvals, and was managed according to the Medical Research Council's Good Clinical Practice Guidelines.27 Patients gave informed written consent to inclusion and to long-term follow-up.
Methods
Patients and procedures
Patients were included in arm A of SANAD if they had a history of two or more clinically definite unprovoked epileptic seizures in the previous year and if carbamazepine was deemed the better standard treatment option, compared with valproate, by the recruiting clinician. This allocation allowed inclusion of patients with newly diagnosed epilepsy, patients who had failed treatment with previous monotherapy (as long as the drug failure did not include one of the drugs present in the randomisation), and patients who had entered a period of remission from seizures but had relapsed after withdrawal of treatment. Patients were excluded if the clinician or patient felt that treatment was contraindicated, if all their seizures had been acute symptomatic seizures (including febrile seizures), they were aged 4 years or younger, or if there was a history of progressive neurological disease.
Information recorded at entry to the study included patient demographics, a history of learning disability or developmental delay, neurological history including head injury, stroke, intra-cerebral infection and acute symptomatic seizures, and a history of epilepsy in a first degree family member. Clinicians were asked to classify seizures and epilepsy syndromes by International League Against Epilepsy classifications28,29 as far as was possible, at least to differentiate between partial onset (focal) or generalised onset seizures. However, where there was uncertainty, patients were recorded as having unclassified convulsive or other unclassified seizures. Results of any electroencephalography or brain imaging around the time of randomisation were documented.
Participants were randomly allocated to treatment in a ratio of 1:1:1:1:1. However, fewer patients were randomised to oxcarbazepine, since it was only included in this randomisation after June 1, 2001. To randomise a patient, the clinician telephoned a central randomisation service, and provided patient identifying information as well as the clinical factors used for stratification of randomisation, which were centre, sex, and treatment history (newly diagnosed and untreated, treated with ineffective monotherapy, relapse after remission of epilepsy). The central randomisation centre then allocated patients using a computer programme that used a minimisation procedure. Although choice of drug was randomised, drug dose and preparation was that used by the clinician in their everyday practice. The rate of titration, initial maintenance dose, and any subsequent increments or decrements were decided by the clinician aided by guidelines (webtable 1). The aim of treatment was to control seizures with a minimum effective dose of drug. This treatment necessitated dosage increments if further seizures happened as is usual clinical practice.
Patients were to be seen for follow-up at 3 months, 6 months, 1 year, and at successive yearly intervals from the date of randomisation. If clinically indicated, more frequent follow-up was undertaken. At every visit, details of the occurrence of seizures, adverse events, hospital admissions, and antiepileptic drug treatment were documented. For adverse effects, clinicians were asked to indicate whether they were clinically important. Where patients ceased attending hospital clinics, follow-up was obtained from general practitioners, or directly from the patient via a telephone interview.
There were two primary outcome measures: the time from randomisation to treatment failure (stopping the randomised drug due either to inadequate seizure control or to intolerable side-effects, or both, or the addition of other antiepileptic drugs, whichever was the earliest); and the time from randomisation to the achievement of a 1 year period of remission of seizures. Secondary clinical outcomes were: the time from randomisation to a first seizure (an efficacy outcome that is to some degree dependent on choice of the initial drug dose); time to achieve a 2-year remission; the incidence of clinically important adverse events and side-effects emerging after randomisation. Additionally, quality of life outcomes and cost-effectiveness of the different drugs were assessed.
The calculations of sample size were based on the two primary outcomes and informed by a meta-analysis of individual patient data comparing valproate and carbamazepine.30 We wished to establish that the lower 95% confidence limit for the treatment comparisons between old and new drugs exceeded −10% (non-inferiority), rather than establishing equivalence within 10%. With α=0·05 and β=10%, giving a 95% CL of 10% around an overall 1 year remission rate of 70% and a retention rate of 70% (ie, treatment failure rate of 30%) at a median of 2·5 years follow-up with power 90% (β=0·10), needed 445 patients per treatment group.
Details of methods used in assessing quality of life and health economic outcomes are detailed in the webappendix, webfigure 1, and webtables 2 and 3. For both adults and children, the quality of life assessment involved use of several previously validated generic and epilepsy-specific measures. For adults, we used the Newly Diagnosed Epilepsy Quality of Life battery, for which the included measures have been previously validated. For health economic assessment, patients' use of resources was categorised under three general headings: consumption of antiepileptic drugs; resource use associated with the management of adverse events requiring hospitalisation; and other health care and social services resource use.
Because oxcarbazepine was added to arm A only after the trial had been running for some time, two separate analyses are presented: (1) a comparison of carbamazepine with gabapentin, lamotrigine, and topiramate using patient data from the entire trial period; and (2) a comparison of oxcarbazepine with gabapentin, lamotrigine, carbamazepine, and topiramate using data from patients recruited after June 1, 2001.
Statistical analysis
Initial analyses of time-to-event data used log-rank tests and Cox proportional hazard models. For analysis of the primary clinical outcomes, we planned to undertake both an intention-to-treat analysis and a per-protocol analysis. Intention-to-treat analysis would be most conservative for tests of differences between drugs, but per-protocol analysis would be most conservative when considering issues of equivalence. The populations for these two analysis approaches are described in figure 1. For the per-protocol analyses, the clinical and statistical issues of informative censoring have been identified. The problem arises for the remission outcomes as follows: patients who have a treatment failure before achieving a period of remission will be censored at the date of treatment failure, and the log-rank analysis assumes that time to achieve a remission for an individual is independent of the reason for censoring. However, patients with a poor prognosis of remission would more likely be withdrawn from a drug for inadequate seizure control, leading to selection bias in a log-rank analysis. For this reason, the log-rank analysis is not considered appropriate here and the cumulative incidence analysis is preferred; however, the p values from the log-rank analysis are presented for consistency. For time to treatment failure, further analyses were undertaken to assess the two main reasons for treatment failure—inadequate seizure control or unacceptable adverse effects. To allow for possible dependence between the different withdrawal risks, cumulative incidence analyses are presented, the analysis which does not assume that censoring is non-informative.
Figure 1.

Study flow diagram for SANAD arm A
*Three patients did not take drug but did not have seizure data.
This study is registered as an International Standard Randomised Controlled Trial, number ISRCTN38354748.
Role of the funding source
SANAD was funded by the Health Technology Assessment Programme, with an additional 20% of resources coming from companies with products assessed. The funding sources had no role in study design, data collection, analysis, interpretation of data, or in writing this report. All authors had full access to the data. The corresponding author had full access to the data and had final responsibility for the decision to submit for publication.
Results
The first patient was randomised into the study on Dec 1, 1999, and randomisation continued up to Aug 31, 2004. Attempts were made to follow-up all patients to, at the latest, a point in time between May 1, 2005, and Aug 31, 2005, although some follow-up data was obtained up to Jan 13, 2006. 1721 patients were randomised: 378 to carbamazepine; 377 to gabapentin; 378 to lamotrigine; 210 to oxcarbazepine; and 378 to topiramate. The treatment groups were well balanced for demographic and clinical factors (table 1), with 88% of patients classified as having a cryptogenic or symptomatic partial epilepsy and 10% classified as having an unclassified epilepsy. 49 patients were excluded from all analyses (figure 1), 44 because the diagnosis of epilepsy was subsequently changed, and five for other reasons. During the course of the study 38 patients declined further follow-up and one patient left the country; these 39 patients contributed to the analysis up to the time of their last follow-up.
Table 1.
Baseline demographic and clinical characteristics for arm A
| Carbamazepine (n=378) | Gabapentin (n=377) | Lamotrigine (n=378) | Oxcarbazepine (n=210) | Topiramate (n=378) | Total (n=1721) | |
|---|---|---|---|---|---|---|
| Sex, n (%) | ||||||
| Men | 208 (55·0) | 207 (54·9) | 208 (55·0) | 111 (52·9) | 208 (55·0) | 942 (54·7) |
| Women | 170 (45·0) | 170 (45·1) | 170 (45·0) | 99 (47·1) | 170 (45·0) | 779 (45·3) |
| Treatment history, n (%) | ||||||
| Untreated | 309 (81·8) | 306 (81·2) | 308 (81·5) | 181 (86·2) | 308 (81·5) | 1412 (82·1) |
| Monotherapy (not optimum treatment) | 60 (15·9) | 60 (15·9) | 61 (16·1) | 25 (11·9) | 60 (15·9) | 266 (15·5) |
| Recent seizures after remission | 9 (2·4) | 11 (2·9) | 9 (2·4) | 4 (1·9) | 10 (2·7) | 43 (2·5) |
| History, n (%) | ||||||
| Learning disability | 20 (5·3) | 17 (4·5) | 23 (6·1) | 4 (1·9) | 21 (5·6) | 85 (4·9) |
| Neurological deficit | 34 (9·0) | 28 (7·4) | 32 (8·5) | 15 (7·1) | 30 (7·9) | 139 (8·1) |
| Neurological disorder, n (%) | ||||||
| Stroke/cerebrovascular | 32 (8·5) | 27 (7·2) | 20 (5·3) | 10 (4·8) | 19 (5·0) | 108 (6·3) |
| Intracranial surgery | 13 (3·4) | 17 (4·5) | 15 (4·0) | 2 (1·0) | 24 (6·4) | 71 (4·1) |
| Head injury | 12 (3·2) | 17 (4·5) | 18 (4·8) | 10 (4·8) | 26 (6·9) | 83 (4·8) |
| Meningitis/encephalitis | 4 (1·1) | 7 (1·9) | 12 (3·2) | 3 (1·4) | 8 (2·1) | 34 (2·0) |
| Other | 28 (7·4) | 24 (6·4) | 29 (7·7) | 11 (5·2) | 32 (8·5) | 124 (7·2) |
| History of seizures, n (%) | ||||||
| Febrile convulsions | 27 (7·1) | 16 (4·2) | 25 (6·6) | 7 (3·3) | 17 (4·5) | 92 (5·4) |
| Any other acute symptomatic seizures | 6 (1·6) | 15 (4·0) | 18 (4·8) | 8 (3·8) | 13 (3·4) | 60 (3·5) |
| Epilepsy in first degree relatives | 39 (10·3) | 44 (11·7) | 38 (10·1) | 24 (11·4) | 34 (9·0) | 179 (10·0) |
| Epilepsy syndrome, n (%) * | ||||||
| Idiopathic partial | 4 (1·1) | 5 (1·3) | 6 (1·6) | 3 (1·4) | 6 (1·6) | 24 (1·4) |
| Symptomatic or cryptogenic partial | 338 (89·4) | 333 (88·6) | 330 (88·0) | 180 (85·7) | 322 (85·4) | 1503 (87·6) |
| Idiopathic generalised | 3 (0·8) | 3 (0·8) | 4 (1·1) | 5 (2·4) | 7 (1·9) | 22 (1·3) |
| Other syndrome | 2 (0·5) | 0 (0) | 0 (0) | 1 (0·5) | 1 (0·3) | 4 (0·2) |
| Unclassified | 31 (8·2) | 35 (9·3) | 35 (9·3) | 21 (10·0) | 41 (10·9) | 163 (9·5) |
| Median interval between first and most recent seizure (25th–75th centile), days† | 465 (162–1720) | 446 (156–2195) | 492 (165–1765) | 463 (155–1470) | 488 (153–1949) | 467 (156–1889) |
| Median interval between most recent seizure and randomisation (25th–75th centile), days‡ | 13 (4–37) | 13 (3–37) | 14 (3–38) | 14 (4–41) | 12 (3–33) | 13 (3–37) |
| Median number of seizures (25th–75th centile)§ | 12 (4–65) | 12 (4–70) | 12 (4–60) | 11 (4–51) | 12 (4–80) | 12 (4–63) |
| Mean age (SD), years | 39·2 (18·3) | 37·8 (17·9) | 36·8 (18·3) | 40·1 (18·0) | 38·4 (18·6) | 38·3 (18·3) |
Missing data for epilepsy syndrome for one individual on gabapentin, three individuals on lamotrigine, and one individual on topiramate.
Missing dates for seizures for one individual on topiramate.
Missing data for dates of seizures for one individual on topiramate.
Missing number of seizures for one individual on topiramate.
Follow-up in arm A was 94% complete, with 5406 patient years of follow-up, compared with 5762 years that could have been expected had follow-up been complete. Of the 71 deaths, ten were judged as related to epilepsy (including accidental deaths caused by seizures, status epilepticus, and all sudden deaths), one to carbamazepine, two to gabapentin, four to lamotrigine, three to oxcarbazepine, and none to topiramate. The remaining 61 deaths were from other causes. Because of the pragmatic nature of the trial design and the absence of blinding, we needed to assess the doses of drugs used and consider the degree to which the full dose ranges were explored before treatment failure events (table 2).
Table 2.
Dose taken by adults at withdrawal or last follow-up
| Carbamazepine | Gabapentin | Lamotrigine | Topiramate | Oxcarbazepine * | |
|---|---|---|---|---|---|
| Inadequate seizure control | n=33 | n=83 | n=42 | n=40 | n=15 |
| 991 (347); 400–1800 | 2414 (899); 300–4800 | 355 (175); 85–800 | 291 (168); 50–600 | 1480 (525); 300–2100 | |
| Unacceptable adverse events | n=50 | n=35 | n=30 | n=68 | n=29 |
| 546 (189); 200–1000 | 1366 (636); 400–3000 | 178 (113); 25–550 | 137 (77); 25–400 | 895 (351); 300–2100 | |
| Inadequate seizure control and unacceptable adverse events |
n=18 | n=23 | n=9 | n=16 | n=9 |
| 711 (323); 200–1400 | 1878 (875); 600–3600 | 219 (178); 50–550 | 218 (110); 75–400 | 1150 (525); 450–1950 | |
| Other reason for withdrawal | n=13 | n=14 | n=19 | n=14 | n=7 |
| 569 (317); 200–1200 | 1314 (466); 300–2100 | 184 (62); 50–300 | 189 (103); 75–500 | 814 (285); 600–1200 | |
| Remission of seizures | n=14 | n=12 | n=9 | n=13 | n=7 |
| 614 (337); 200–1400 | 1475 (663); 300–2700 | 158 (92); 50–300 | 133 (57); 50–200 | 771 (293); 300–1200 | |
| Still on randomised drug | n=140 | n=120 | n=168 | n=126 | n=87 |
| 662 (311); 100–2000 | 1496 (669); 300–3600 | 249 (136); 20–800 | 181 (108); 25–600 | 1019 (467); 300–2850 |
Data are mean (SD); range.
Figures in this column use data only for patients randomised after June 1, 2001.
There is satisfactory evidence that clinicians did explore a full dosing range before accepting treatment failure because of inadequate seizure control. As might be expected, doses associated with unacceptable adverse events were consistently lower than those associated with inadequate seizure control.
Treatment failure for unacceptable side-effects was largely limited to the early post-randomisation period, whereas the timing of withdrawal for inadequate seizure control (with or without unacceptable adverse events) was later because upward titration of dose is needed before withdrawal for inadequate seizure control could have taken place; the median number of days to failure (25th–75th centile) for unacceptable adverse events was 84 (26–215) and for inadequate seizure control was 313 (152–642).
For time to treatment failure for any reason (inadequate seizure control or unacceptable adverse events; table 3) there are significant overall differences, although inevitably there is some reduction in power in the analysis using data from June 1, 2001, onwards. When comparisons are made across the whole duration of the trial, lamotrig ine is better than all other drugs for pairwise comparisons (figure 2). Carbamazepine and oxcarbazepine are inter mediate between these options and appear broadly similar, although the CI is wide and should not be taken to imply equivalence between the two drugs. Gabapentin and topiramate are the poorest performing drugs. Sensitivity analyses (not shown) indicate that including only patients with definite partial seizures or including patients subsequently withdrawn as “not epilepsy” does not affect the results.
Table 3.
Treatment failure (all events)
| Year | ||||||
|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | |
| Any reason | ||||||
| Number at risk | ||||||
| Carbamazepine | 225 | 160 | 106 | 65 | 26 | 7 |
| Gabapentin | 195 | 133 | 87 | 51 | 24 | 8 |
| Lamotrigine | 266 | 178 | 121 | 84 | 42 | 11 |
| Topiramate | 207 | 136 | 81 | 54 | 24 | 7 |
| Oxcarbazepine | 118 | 73 | 29 | 3 | ||
| Percentage still on drug (95% CI) | ||||||
| Carbamazepine | 65 (61 to 70) | 57 (52 to 62) | 53 (47 to 58) | 50 (44 to 56) | 45 (38 to 52) | 40 (31 to 49) |
| Difference in percentage without failure compared with carbamazepine (95% CI) | ||||||
| Gabapentin | −8 (−15 to −1) | −8 (−16 to −1) | −10 (−18 to −2) | −11 (−19 to −3) | −7 (−16 to 2) | −4 (−15 to 7) |
| Lamotrigine | 12 (6 to 19) | 8 (1 to 15) | 6 (−2 to 13) | 4 (−4 to 12) | 6 (−4 to 15) | 11 (1 to 20) |
| Topiramate | −5 (−12 to 2) | −9 (−16 to −2) | −8 (−16 to −1) | −8 (−16 to 0) | −5 (−14 to 4) | −4 (−16 to 7) |
| Oxcarbazepine | −2 (−11 to 8) | −1 (−11 to 9) | −1 (−13 to 10) | −1 (−14 to 12) | ||
| For unacceptable adverse events | ||||||
| Percentage without failure (95% CI) | ||||||
| Carbamazepine | 74% (70 to 79) | 70% (65 to 75) | 69% (64 to 74) | 69% (64 to 74) | 68% (63 to 73) | 68% (63 to 73) |
| Difference in percentage without failure compared with carbamazepine (95% CI) | ||||||
| Gabapentin | 9 (3 to 15) | 11 (4 to 17) | 11 (4 to 17) | 11 (4 to 17) | 12 (5 to 18) | 12 (5 to 18) |
| Lamotrigine | 10 (4 to 16) | 11 (4 to 17) | 10 (4 to 17) | 11 (4 to 17) | 10 (3 to 17) | 10 (3 to 17) |
| Topiramate | 0 (−6 to 6) | 0 (−7 to 7) | −1 (−8 to 6) | 0 (−7 to 7) | 1 (−7 to 8) | −2 (−10, 7) |
| Oxcarbazepine* | 3 (−6 to 11) | 4 (−5 to 13) | 4 (−6 to 13) | 5 (−5 to 15) | ||
| For inadequate seizure control | ||||||
| Percentage without failure (95% CI) | ||||||
| Carbamazepine | 92 (55 to 95) | 87 (41 to 91) | 84 (33 to 88) | 82 (28 to 86) | 77 (20 to 83) | 73 (15 to 81) |
| Difference in percentage without failure compared with carbamazepine (95% CI) | ||||||
| Gabapentin | −17 (−22 to −11) | −19(−25 to −13) | −21 (−27 to −14) | −21 (−29 to −14) | −18 (−27 to −10) | −15 (−26 to −4) |
| Lamotrigine | 0 (−4 to 4) | −2 (−8 to 3) | −4 (−10 to 2) | −6 (−12 to 1) | −3 (−12 to 5) | 1 (−9 to 12) |
| Topiramate | −4 (−9 to 0) | −8 (−14 to −3) | −7 (−13 to 0) | −7 (−14 to 0) | −5 (−13 to 4) | −2 (−13 to 9) |
| Oxcarbazepine* | −4 (−9 to 2) | −4 (−11 to 4) | −4 (−14 to 5) | −6 (−17 to 5) | ||
Figures in this row use data for oxcarbazepine and carbamazepine only from patients randomised after June 1, 2001.
Figure 2.
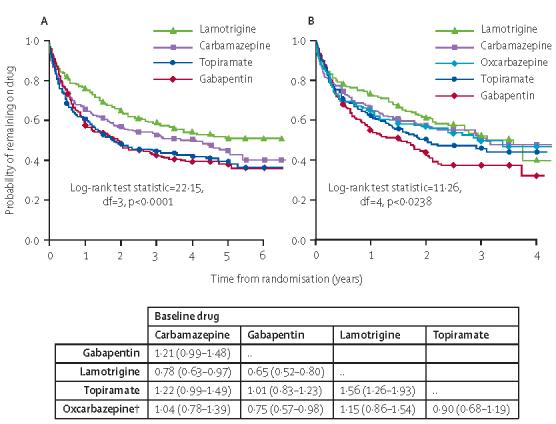
Time to treatment failure for whole recruitment period (A) and recruitment after June, 2001 (B)
*HR greater than 1 indicates that failure takes place more rapidly on drug compared with baseline.†Analysis for oxcarbazepine use data only from patients randomised after June 1, 2001.
The reasons for treatment failure vary according to drug (webtable 4). The cumulative incidence analysis shows that carbamazepine (lamotrigine:carbamazepine 0·62 [0·46–0·83]) and topiramate (lamotrigine: topiramate 0·62 [0·46, 0·84]) are most frequently associated with treatment failure for unacceptable adverse events, and lamotrigine and gabapentin (gabapentin:carbamazepine 0·60 [0·44–0·81]) are least likely to produce this treatment failure (webfigure 2). By contrast, gabapentin is most likely to be associated with treatment failure due to inadequate seizure control and carbamazepine the least likely (gabapentin:carbamazepine 2·45 (1·81–3·32). Carbamazepine was also better than topiramate (topiramate: carbamazepine 1·43 [1·03–1·98]), with no significant difference between lamotrigine and carbamazepine (lamotrigine:carbamazepine 1·17 [0·84–1·64]; webfigure 3). When examining estimates for the proportion of patients with treatment failure events (table 4), compared with carbamazepine, lamotrigine is 10–11% better for treatment withdrawal for adverse events and statistically different at all time points between 1 and 6 years. Lamotrigine is similar to carbamazepine for incidence of treatment failure due to inadequate seizure control, with point estimates varying between 1% superiority at 6 years and 6% inferiority at 4 years. For this efficacy outcome, examination of the lower 95% CI for differences in withdrawal rates indicates that we have excluded any inferiority of lamotrigine greater than 12% (years 4 and 5). At other time points, non-inferiority limits (according to the lower 95% CI) were 4% at 1 year, 8% at 2 years, and 9% at 6 years. This result lends support to the non-inferiority for efficacy of lamotrigine compared with carbamazepine.
Table 4.
Seizure outcomes by drug
| Events/total | Year 1 | Year 2 | Year 3 | Year 4 | Year 5 | Year 6 | |
|---|---|---|---|---|---|---|---|
| Time to 12-month remission—intention to treat | |||||||
| Number at risk | |||||||
| Carbamazepine | 254/362 | 347 | 120 | 73 | 41 | 16 | 6 |
| Gabapentin | 215/359 | 337 | 141 | 76 | 45 | 25 | 7 |
| Lamotrigine | 245/365 | 356 | 126 | 59 | 36 | 19 | 4 |
| Topiramate | 225/358 | 338 | 126 | 74 | 50 | 24 | 5 |
| Oxcarbazepine | 128/200 | 189 | 58 | 21 | 3 | ||
| Percentage of 12-month remission (95% CI) | |||||||
| Carbamazepine | 254/362 | 36 (31 to 41) | 60 (55 to 66) | 69 (63 to 74) | 77 (72 to 82) | 82 (77 to 87) | 85 (79 to 91) |
| Difference in percentage of 12-month remission compared with carbamazepine (95% CI) | |||||||
| Gabapentin | 215/359 | −12 (−19 to −5) | −9 (−16 to −1) | −6 (−13 to 2) | −6 (−14 to 1) | −9 (−17 to −1) | −9 (−18 to 0) |
| Lamotrigine | 245/365 | −7 (−13 to 0) | −3 (−11 to 4) | 3 (−4 to 11) | −1 (−8 to 7) | −3 (−10 to 5) | −2 (−11 to 7) |
| Topiramate | 225/358 | −3 (−10 to 4) | −5 (−13 to 2) | −2 (−10 to 5) | −6 (−14 to 1) | −7 (−14 to 1) | −4 (−14 to 6) |
| Oxcarbazepine | 128/200 | −1 (−11 to 9) | −3 (−13 to 7) | −1 (−12 to 9) | −2 (−15 to 10) | ||
| Time to 12-month remission—per protocol | |||||||
| Number at risk | |||||||
| Carbamazepine | 164/352 | 222 | 47 | 26 | 13 | 5 | |
| Gabapentin | 126/351 | 194 | 42 | 16 | 8 | 5 | |
| Lamotrigine | 170/358 | 264 | 66 | 25 | 16 | 6 | |
| Topiramate | 138/353 | 203 | 40 | 15 | 5 | 3 | |
| Oxcarbazepine | 80/195 | 117 | 25 | 6 | |||
| Percentage of 12-month remission (95% CI) | |||||||
| Carbamazepine | 164/352 | 29 (24 to 34) | 44 (39 to 50) | 48 (42 to 53) | 50 (44 to 55) | 53 (47 to 58) | |
| Difference in percentage of 12-month remission compared with carbamazepine (95% CI) | |||||||
| Gabapentin | 126/351 | −9 (−16 to −3) | −9 (−17 to −2) | −9 (−17 to −1) | −9 (−17 to −1) | −12 (−20 to −4) | |
| Lamotrigine | 170/358 | −4 (−11 to 3) | 0 (−8 to 7) | 4 (−4 to 12) | 5 (−3 to 12) | 3 (−5 to 11) | |
| Topiramate | 138/353 | −4 (−11 to 3) | −6(−13 to 2) | −6 (−14 to 1) | −6 (−14 to 2) | −8 (−16 to 0) | |
| Oxcarbazepine | 80/195 | −2 (−12 to 7) | −7(−17 to 4) | −5 (−16 to 6) | |||
| Time to 24 months remission—intention to treat | |||||||
| Number at risk | |||||||
| Carbamazepine | 168/362 | 295 | 116 | 71 | 28 | 8 | |
| Gabapentin | 132/359 | 283 | 125 | 74 | 36 | 8 | |
| Lamotrigine | 155/365 | 296 | 120 | 67 | 30 | 9 | |
| Topiramate | 140/358 | 284 | 124 | 80 | 41 | 12 | |
| Oxcarbazepine | 68/200 | 138 | 39 | 5 | |||
| Percentage of 24 month remission (95% CI) | |||||||
| Carbamazepine | 168/362 | 32 (27 to 37) | 50 (44 to 56) | 57 (51 to 63) | 65 (58 to 72) | 69 (61 to 78) | |
| Difference in percentage of 24 month remission compared with carbamazepine (95% CI) | |||||||
| Gabapentin | 132/359 | −13 (−20 to −6) | −12 (−21 to −4) | −7 (−16 to 2) | −7 (−17 to 4) | −8 (−20 to 4) | |
| Lamotrigine | 155/365 | −6 (−13 to 1) | −7 (−15 to 1) | −1 (−9 to 8) | −3 (−13 to 6) | −5 (−17 to 6) | |
| Topiramate | 140/358 | −5 (−12 to 3) | −7 (−16 to 1) | −6 (−15 to 3) | −11 (−21 to −2) | −8 (−20 to 4) | |
| Oxcarbazepine | 68/200 | −4 (−15 to 7) | −6 (−19 to 6) | 0 (−18 to 17) | |||
| Time to first seizure—intention to treat | |||||||
| Number at risk | |||||||
| Carbamazepine | 259/362 | 126 | 92 | 53 | 34 | 17 | 4 |
| Gabapentin | 288/359 | 83 | 53 | 39 | 18 | 7 | 1 |
| Lamotrigine | 290/365 | 106 | 76 | 48 | 29 | 10 | 5 |
| Topiramate | 268/358 | 112 | 76 | 50 | 33 | 13 | 4 |
| Oxcarbazepine* | 144/200 | 67 | 42 | 11 | |||
| Percentage of first seizure (95% CI) | |||||||
| Carbamazepine | 259/362 | 63 (58 to 68) | 69 (64 to 73) | 73 (68 to 78) | 73 (68 to 78) | 74 (69 to 79) | 78 (69 to 87) |
| Difference in percentage of first seizure compared with carbamazepine (95% CI) | |||||||
| Gabapentin | 288/359 | 12 (5 to 19) | 11 (4 to 17) | 8 (1 to 14) | 9 (3 to 16) | 11 (3 to 18) | 6 (−4 to 17) |
| Lamotrigine | 290/365 | 7 (1 to 14) | 6 (0 to 13) | 7 (0 to 13) | 8 (2 to 15) | 10 (3 to 17) | 5 (−5 to 15) |
| Topiramate | 268/358 | 4 (−3 to 11) | 5 (−2 to 11) | 3 (−4 to 9) | 5 (−2 to 12) | 4 (−3 to 11) | 0 (−10 to 10) |
| Oxcarbazepine* | 144/200 | 1 (−8 to 10) | 1 (−8 to 10) | 5 (−4 to 15) | |||
Figures in this row for use data for oxcarbazepine and carbamazepine only from patients randomised after June 1, 2001.
For oxcarbazepine, analyses including patients randomised after June 1, 2001, indicate that overall treatment failure rates for oxcarbazepine and carbamazepine are similar (oxcarbazepine:carbamaze-pine 1·04 [0·78–1·39]), but oxcarbazepine might be less likely than carbamazepine to fail because of adverse effects (oxcarbazepine: carbamazepine 0·85 [0·59–1·24]), but more likely to fail because of inadequate seizure control (oxcarbazepine: carbamazepine 1·33 [0·82–2·15]). Point estimates for differences in the proportion of failures due to inadequate seizure control vary between 4% inferiority for oxcarbazepine versus carbamazepine (years 1–3) and 6% at 4 years, but examination of the CIs indicates that we cannot exclude oxcarbazepine being between 9% inferior at 1 year and 17% inferior at 4 years. This finding would not seem to support a claim for non-inferiority of oxcarbazepine compared with carbamazepine for this efficacy outcome, although this could be due to reduced power because of the fewer patients available for this analysis. Results for time to 12-month remission of seizures are presented in table 4, figure 3, and webfigure 4.
Figure 3.

Time to 12-month remission for whole recruitment period (A) and recruitment after June, 2001 (B)
*HR greater than 1 indicates that failure takes place more rapidly on drug compared with baseline.†Analysis for oxcarbazepine use data only from patients randomised after June 1, 2001.
The intention-to-treat analysis shows that times to achieve a 12-month remission are statistically different across drugs and identify gabapentin and topiramate as the least favoured options (figure 3). For gabapentin the differences from carbamazepine (and indeed oxcarbazepine and lamotrigine) are significant and of likely clinical importance, though the differences for topiramate from these drugs are less and are not significant. The standard drug, carbamazepine, seems to be the preferred treatment for this outcome in all pair-wise comparisons, although differences between lamotrigine and oxcarbazepine are smaller (figure 3). Restricting analyses to patients with partial onset seizures produces no substantive differences.
In intention-to-treat analyses, follow-up data after a treatment failure have been included. Thus some patients achieve a 1-year remission on drug regimens other than the one to which they were randomised, and still contribute to outcome for the drug to which patients were originally randomised. In examining data for intention to treat for time to 12-month remission, understanding how clinicians chose to switch treatment after treatment failure events becomes important. After failure on one of the newer drugs (except oxcarbazepine), remission would probably happen after switching to carbamazepine, but failure on carbamazepine then switching to lamotrigine was most commonly associated with achieving remission (webtable 5).
For these reasons, a per-protocol analysis is also presented (table 4, webfigure 4), in which observations were censored at the point of treatment failure (a 1-year remission is only counted as an event for patients achieving remission on the drug to which they were randomised). Overall, the best performing drugs result in around 50% of patients achieving a 1-year remission on the drug to which they were randomised, compared with around 75% remission rates for the best performing drugs in the intention-to-treat analyses (at 5 years after randomisation). For the comparisons across the whole treatment period, differences between carbamazepine and lamotrigine seem small. Although the point estimates suggest a 4% inferiority for lamotrigine at 1 year, there is no difference at 2 years, and lamotrigine is better at 4–5 years. Examination of the lower 95% CI around point estimates indicates that there is sufficient power in these comparisons to exclude lamotrigine being any more than 11% inferior to carbamazepine at 1 year, 8% at 2 years, 4% at 3 years, 3% at 4 years, and 5% at 5 years after randomisation. These estimates might be sufficient to support non-inferiority of lamotrigine compared with carbamazepine for this primary efficacy outcome. This per-protocol analysis is the most conservative when assessing issues of non-inferiority. When the per-protocol analysis for the period after inclusion of oxcarbazepine is considered, oxcarbazepine produces similar 12-month remission rates but the CIs do not exclude the drug varying between 12% (at 1 year) and 17% (at 2 years) inferior to carbamazepine. There is insufficient evidence of non-inferiority.
Intention-to-treat analyses for time to 2-year remission are shown in table 4. Overall, carbamazepine is better than all other drugs and statistically so compared with gabapentin (HR gabapentin:carbamazepine 0·72 [0·58–0·91]) and topiramate (topiramate:carbamazepine 0·80 [0·64–1·00]). For the period after the addition of oxcarbazepine, this drug seems statistically better than gabapentin (1·51 [1·05–2·18]). For this period, carbamazepine remains the preferred option but the differences in pair-wise comparisons between oxcarbazepine and carbamazepine, lamotrigine and topiramate were not significant.
Data for time to first seizure outcomes are shown in table 4. Gabapentin seems least effective in preventing first seizures, with carbamazepine being most effective. Topiramate and lamotrigine are intermediate. Pair wise comparisons for the entire period show carbamazepine to be statistically better than both gabapentin (1·35 [1·14, 1·60]) and lamotrigine (1·23 [1·04, 1·45]), but not topiramate (1·05 [0·89, 1·25]). Results for the period after addition of oxcarbazepine suggest carbamazepine and oxcarbazepine are similar, but the CI is wide (1·06 [0·84, 1·33]).
For the primary outcomes, regression models were used to assess the effect of age and to investigate an age-treatment interaction. For time to treatment failure, children (younger than 16 years) were significantly more likely to have treatment failure (1·25 [1·00–1·56]), whereas the older age-group (65 years or older) were significantly less likely to have treatment failure (0·72 [0·55–0·94]) than the middle age-group (16–65 years). For time to 12-month remission, children and the older age-group had a significantly better chance of remission than the middle age-group (1·22 [1·01–1·48] and 1·70 [1·39–2·09], respectively). There was no evidence of an age-treatment interaction for either outcome, hence we have no evidence that the relative treatment effects differ across these age-groups.
During follow-up, clinicians recorded adverse events described by patients, and indicated whether they judged the events to be clinically important. Table 5 summarises intention-to-treat rates of adverse events judged as clinically important. An intention-to-treat approach summarises adverse events associated with the randomised policy, but since patients might have had their treatment changed during follow up, this approach does not clearly present adverse events attributable to specific drugs. We therefore present both intention-to-treat and per-protocol rates for adverse events (table 5).
Table 5.
Frequency of clinically important adverse events
| Carbamazepine | Gabapentin | Lamotrigine | Oxcarbazepine | Topiramate | Total | |
|---|---|---|---|---|---|---|
| Number randomised | 378 | 377 | 378 | 210 | 378 | 1721 |
| Total number (%) of patients with at least one adverse event | 183 (48%) | 178 (47%) | 169 (45%) | 100 (48%) | 200 (53%) | 830 (48%) |
| Tiredness/drowsiness/fatigue/lethargy | 48 [36] | 46 [34] | 31 [17] | 22 [16] | 43 [33] | 190 [136] |
| Depression | 14 [8] | 18 [10] | 20 [13] | 7 [5] | 29 [24] | 88 [60] |
| Headache | 21 [9] | 20 [15] | 21 [13] | 9 [6] | 17 [11] | 88 [54] |
| Allergic rash | 38 [32] | 13 [4] | 17 [15] | 20 [16] | 17 [8] | 105 [75] |
| Memory problems | 20 [12] | 22 [19] | 13 [10] | 13 [8] | 26 [19] | 94 [68] |
| Dizziness/vertigo | 14 [10] | 23 [15] | 15 [9] | 13 [12] | 15 [8] | 80 [54] |
| Other psychiatric | 16 [7] | 17 [9] | 11 [7] | 7 [5] | 37 [31] | 88 [59] |
| Worsening of seizures | 17 [5] | 22 [13] | 17 [12] | 3 [1] | 17 [8] | 76 [39] |
| Other neurological | 9 [6] | 21 [14] | 15 [9] | 8 [5] | 18 [12] | 71 [46] |
| Other general | 13 [6] | 19 [11] | 19 [13] | 9 [6] | 16 [12] | 76 [48] |
| Behaviour/personality change/aggression | 12 [4] | 9 [6] | 12 [7] | 2 [1] | 24 [19] | 59 [37] |
| Ataxia | 9 [6] | 24 [12] | 14 [9] | 8 [6] | 9 [3] | 64 [36] |
| Confusion/difficulty thinking/disoriented | 9 [9] | 16 [15] | 8 [4] | 8 [6] | 22 [19] | 63 [53] |
| Anxiety/agitation/nervousness | 7 [7] | 15 [11] | 8 [5] | 7 [6] | 15 [12] | 52 [41] |
| Weight loss | 2 [1] | 4 [2] | 4 [2] | 3 [1] | 29 [27] | 42 [33] |
| Diplopia | 5 [2] | 11 [4] | 4 [2] | 8 [6] | 6 [3] | 34 [17] |
| Nausea | 9 [6] | 7 [3] | 9 [6] | 15 [13] | 4 [4] | 44 [32] |
| Weight gain | 9 [7] | 15 [12] | 4 [1] | 1 [0] | 5 [4] | 34 [24] |
| Accidental injury | 7 [2] | 11 [6] | 12 [8] | 3 [1] | 8 [3] | 41 [20] |
| Pins and needles/dysaesthesia | 4 [1] | 5 [1] | 3 [1] | 0 [0] | 26 [24] | 38 [27] |
| Sleep disturbance | 5 [2] | 4 [4] | 9 [8] | 4 [2] | 9 [8] | 31 [24] |
| Other events* | 108 [71] | 113 [73] | 110 [70] | 46 [38] | 103 [64] | 480 [316] |
For adverse events, data outside brackets are intention to treat and inside brackets are per protocol.
Sorted by descending total frequency: other cardiac or vascular; other skin and appendages; abdominal pain, dyspepsia; other gastrointestinal; other visual disturbance; other renal tract or genital; diarrhoea; tremor; aches and pains; constipation; infection; mouth or gum problem; other respiratory or pulmonary; ischaemic heart disease or myocardial infarct; other haematological; other musculoskeletal; vomiting; impotence or libido problems; alopecia; word finding difficulty; status epilepticus; stroke—infarction; diabetes mellitus; hearing problem or tinnitus; hypertension; anorexia; bruising; flu like symptoms; haemorrhage; malignancy; short of breath; vaginal bleeding; arthritis; eczema; peptic ulceration; asthma; other hepatobiliary; urinary retention; abnormal liver function tests; anaemia; child birth; myalgia; other endochrine; psoriasis; upper respiratory tract infection, catarrh, sinusitis, rhinorrhoea; urinary tract infection; faints; hallucinations; hepatitis; pancreatitis; psychosis; transient ischaemic attack; tachycardia; thyroid disease; venous thrombosis.
Notably, around 50% of patients reported adverse events at some point in the study and that the differences between drugs were not great. For the intention-to-treat population, lamotrigine was the drug with the least number of patients reporting adverse events (45% intention to treat, 37% per protocol) with topiramate the most (53% intention to treat, 49% per protocol).
For the individual symptoms reported, tiredness, headache, and fatigue were the most common symptoms, though these did not seem specific to any individual drug. Depression, memory disturbance, and various psychiatric symptoms were common and particularly associated with topiramate. Rash was a common non-CNS symptom, especially with carbamazepine and oxcarbazepine. Rates of rash were lower with lamotrigine. Gabapentin's particular adverse event profile was characterised by a high rate of dizziness, ataxia, and weight gain, topiramate by psychiatric symptoms, including anxiety, weight loss, and paraesthesiae. The profiles of lamotrigine and oxcarbazepine were non-specific. These profiles were consistent across intention-to-treat and per-protocol summaries. Overall, the most common adverse effect associated with treatment failure was rash (7% of patients allocated carbamazepine), which accounted for 21% of carbamazepine treatment failures. Similarly 6% of patients had a rash on oxcarbazepine which accounted for 19% of the oxcarbazepine treatment failures. The overall rash rate on lamotrigine was 3% of patients and rash accounted for 14% of treatment failures. We should note that in the study neither patients nor clinicians were masked to drug treatment, which might have affected the symptoms reported to the clinicians and their assessment of the clinical importance.
The quality of life analysis was confined to adult patients only (although quality of life data were obtained for children younger than 16 years, the numbers eligible were small and the measures used were different from those for adults). 1345 randomised adults were sent baseline quality-of-life questionnaires. The overall response rate in this group of the trial was 84% at baseline and 75% at 2-year follow-up (webtable 6) and there were no significant differences between the drugs. Comparison of responders and non-responders to the baseline and 2-year follow-up assessments revealed that there were important differences between them, with potential to create bias in the interpretation of the results of the quality of life study. Women were more likely to respond to the questionnaires than men, and the median age for responders was higher than for non-responders.
Of adult participants who returned a questionnaire at baseline, there were differences between those who subsequently returned a 2-year follow-up questionnaire and those who did not for the quality of life measures defined as primary: anxiety, depression, neurotoxicity, other adverse effects of antiepileptic drugs, EQ-5D scores, and self-rated global quality of life. Non-responders at 2 years reported worse baseline levels of anxiety and depression, higher neurotoxicity and adverse events, poorer EQ-5D scores, and poorer global quality of life. 2-year non-responders were also less likely to have achieved a 12-month remission of seizures before the 2-year follow up; 21% of those who achieved a 12-month remission were non-responders at 2 years compared with 32% of those who had not (p<0·0001). Similarly, non-responders were more likely to have had a treatment failure on the randomised drug; 23% of those without treatment failure were non-responders compared with 31% of those who had treatment failure (p<0·0002).
Few significant differences in quality of life between treatment groups at 2 years were identified (webtable 7), although some trends in the data with regard to direction of treatment effects were evident. Thus, based both on mean scores and caseness (ie, whether a patient was classified as clinically anxious), the likelihood of anxiety was reduced for topiramate (despite this symptom being commonly reported to clinicians by patients taking the drug), compared with carbamazepine and gabapentin (though the size of the reduction was small and the CIs relatively wide); and there was a non-significant reduction in risk for topiramate compared with lamotrigine or oxcarbazepine. Likewise, based both on mean scores and caseness (ie, whether the patient was classified as clinically depressed) there was a trend for reduced risk of depression for lamotrigine compared with the other antiepileptic drugs; and the difference was significant for lamotrigine compared with gabapentin (though again the difference was small and the CIs relatively wide). There were no important differences or trends for scores on the Adverse Events Profile, the Neurotoxicity Scale, the EQ-5D, or for global quality of life. The lack of differences between treatment groups might indicate that those with the poorest quality of life outcomes failed to return a 2-year questionnaire, so that important effects were diluted or missed.
By contrast, there were several significant differences both for achieving a positive (ie, remission of seizures) and a negative (ie, treatment failure of the original randomised drug) clinical outcome (webtable 8). Thus, achieving a 12-month remission by 2-year follow-up was associated with a decreased risk of anxiety and depression (measured both by mean scores and caseness), a decreased risk of cognitive (neurotoxicity) and other antiepileptic drug adverse effects, and a reduced likelihood of scoring negatively for global quality of life. There was also a small but significant improvement for quality of life as measured by scores on the EQ-5D. Treatment failure on the randomised drug by 2-year follow-up was associated with increased risk of anxiety and depression, increased risk of cognitive and other antiepileptic drug adverse effects, poorer quality of life as measured by EQ-5D score, and an increased likelihood of scoring negatively for global quality of life.
The cost per quality adjusted life year (QALY) analysis was restricted to adults, because the EQ-5D (from which QALYs were derived) had not been validated for completion by children or by proxy. Since the estimation of QALYs and resource use were dependent on patients returning completed questionnaires, results could have been subject to response bias as outlined above. Two analyses are presented, the first compared carbamazepine, gabapentin, lamotrigine, and topiramate and included all 636 patients who were randomised to one of these drugs and provided complete EQ-5D responses. Tables 6 and 7 show the point estimates of the incremental cost-effectiveness ratios, which were estimated using the lowest costs for carbamazepine and lamotrigine. Disaggregated costs are presented in webtable 9.
Table 6.
Incremental cost-effectiveness ratios—cost per QALY
| Cost (£) | QALYs | Incremental cost (£) |
Incremental QALYs |
Incremental cost-effectiveness ratio (£/QALY) |
|
|---|---|---|---|---|---|
| Comparison excluding oxcarbazepine | |||||
| Carbamazepine | 1226 | 1·477 | .. | .. | .. |
| Topiramate | 2009 | 1·501 | 783 | 0·024 | Extended dominance |
| Lamotrigine | 2257 | 1·564 | 248 | 0·063 | 11851 |
| Gabapentin | 2561 | 1·491 | 304 | −0·073 | Dominated |
| Comparison including oxcarbazepine | |||||
| Carbamazepine | 1095 | 1·491 | .. | .. | .. |
| Oxcarbazepine | 1839 | 1·611 | 744 | 0·12 | 6200 |
| Topiramate | 1930 | 1·541 | 91 | −0·07 | Dominated |
| Lamotrigine | 2078 | 1·563 | 148 | 0·022 | Extended dominance |
| Gabapentin | 2573 | 1·480 | 495 | −0·083 | Dominated |
Table 7.
Incremental cost-effectiveness ratios—cost per seizure avoided
| Cost (£) | Seizures | Incremental cost (£) |
Incremental seizures avoided |
Incremental cost-effectiveness ratio (£/seizure avoided) |
|
|---|---|---|---|---|---|
| Comparison excluding oxcarbazepine | |||||
| Carbamazepine | 1266 | 52·6 | .. | .. | .. |
| Topiramate | 2008 | 63·1 | 742 | −10·5 | Dominated |
| Lamotrigine | 2134 | 41·7 | 126 | 21·4 | 80 |
| Gabapentin | 2494 | 69·8 | 360 | −28·1 | Dominated |
| Comparison including oxcarbazepine | |||||
| Carbamazepine | 1151 | 50·9 | .. | .. | .. |
| Oxcarbazepine | 1815 | 32·0 | 664 | 18·9 | 35 |
| Lamotrigine | 1946 | 50·9 | 131 | −18·9 | Dominated |
| Topiramate | 2059 | 59·4 | 113 | −8·5 | Dominated |
| Gabapentin | 2594 | 85·3 | 535 | −25·9 | Dominated |
Gabapentin has a positive incremental cost and a negative incremental QALY gain and is therefore dominated by lamotrigine. Because lamotrigine has a lower incremental cost-effectiveness ratio than topiramate, topiramate is ruled out on the grounds of extended dominance. The incremental cost-effectiveness ratio for lamotrigine relative to carbamazepine is £11 851. Bootstrapping methods (webappendix) were used to generate cost effectiveness acceptability curves, and table 8 summarises the probabilities that each of the new antiepileptic drugs is cost-effective at ceiling ratios of £10 000, £30 000, and £50 000 per QALY.
Table 8.
Probabilities that the new antiepileptic drugs are cost effective relative to carbamazepine across a range of ceiling ratios (λ)
| Gabapentin | Lamotrigine | Topiramate | Oxcarbazepine | |
|---|---|---|---|---|
| Cost per QALY excluding oxcarbazepine | ||||
| £10 000 | 0·04 | 0·42 | 0·20 | |
| £30 000 | 0·31 | 0·82 | 0·47 | |
| £50 000 | 0·41 | 0·89 | 0·54 | |
| Cost per QALY including oxcarbazepine | ||||
| £10 000 | 0·04 | 0·36 | 0·39 | 0·69 |
| £30 000 | 0·21 | 0·66 | 0·63 | 0·86 |
| £50 000 | 0·30 | 0·73 | 0·67 | 0·89 |
| Cost per seizure avoided excluding oxcarbazepine | ||||
| £160 | 0·08 | 0·70 | 0·17 | |
| £400 | 0·13 | 0·79 | 0·22 | |
| £800 | 0·15 | 0·82 | 0·24 | |
| £1600 | 0·16 | 0·84 | 0·25 | |
| Cost per seizure avoided including oxcarbazepine | ||||
| £160 | 0·05 | 0·41 | 0·27 | 0·85 |
| £400 | 0·08 | 0·48 | 0·33 | 0·90 |
| £800 | 0·10 | 0·50 | 0·35 | 0·90 |
| £1600 | 0·10 | 0·52 | 0·37 | 0·91 |
The second analysis compared carbamazepine, gabapentin, lamotrigine, oxcarbazepine, topiramate and included the 414 adults who provided complete EQ-5D responses. Both topiramate and gabapentin have positive incremental costs and negative incremental QALY gains and are therefore dominated by oxcarbazepine and lamotrigine, respectively (table 6).
For cost per seizure avoided analysis in adults and children, two analyses were undertaken, the first comparing carbamazepine, gabapentin, lamotrigine, and topiramate, and included 823 children and adults for whom we had data for both numbers of seizures and resource use. Topiramate and gabapentin have positive incremental costs and a negative incremental number of seizures avoided, and are therefore dominated by carbamazepine and lamotrigine, respectively. Topiramate is dominated by carbamazepine, hence the incremental cost-effectiveness ratio for lamotrigine presented in table 7 (£80) has been recalculated relative to carbamazepine.
The second analysis compared carbamazepine, gabapentin, lamotrigine, oxcarbazepine, and topiramate, and was based on 547 adults and children (table 6). Lamotrigine, topiramate, and gabapentin have positive incremental costs and negative incremental seizures avoided and are therefore dominated by oxcarbazepine. The incremental cost-effectiveness ratio for oxcarbazepine relative to carbamazepine is £35 (table 7).
Discussion
For patients with partial onset seizures that need monotherapy, we have found lamotrigine to be significantly better for time to treatment failure than the current standard treatment, carbamazepine, and the newer drugs gabapentin and topiramate. For time to 12-month remission from seizures, lamotrigine was non-inferior to carbamazepine.
SANAD was designed as a pragmatic trial to assess whether any of the newly licensed antiepileptic drugs should become first-choice treatment and replace the existing first-line agents, carbamazepine or valproate. If there were clinical or quality of life benefits from these new drugs, we wished to assess the incremental costs associated with such benefits.
Because epilepsy is a chronic disorder, we wished to assess treatments over a relevant amount of time. Therefore, several decisions were made about the methods used that should be considered when assessing the results. We wished the trial to have strong external validity so that results could be applied to everyday clinical practice. Entry criteria were therefore as inclusive as possible and clinicians were encouraged to use their everyday clinical practice in the management of patients. We provided some guidelines for initial target drug dosing, but allowed clinicians to vary the dose on clinical grounds as they saw fit throughout the course of the study to ensure as far as possible that patients received optimum doses for seizure control on the one hand and avoidance of adverse effects on the other.
The study was unmasked because this situation is closer to clinical practice and because it greatly reduced the cost of the study, while increasing practicability. For a five-way comparison of drugs (in arm A), we would have been unable to provide a single matching tablet for all treatment options, and patients would have had to take an active treatment as well as one or more placebo tablets to match the remaining treatment options. This restriction, in addition to the central provision of drug supplies that would be needed to deliver medications formulated specifically for the trial and for protracted periods of follow-up, would have presented unfeasible logistical problems and have been prohibitively expensive. For a long study, there would have been practical difficulties in maintaining masking for drugs that have differing interactions with important treatments such as the oral contraceptive or warfarin. Similarly, management of women in the child-bearing years would have been greatly complicated. All these decisions, especially the lack of blinding, could be seen as compromising the internal validity of the study, and we cannot exclude the possibility that the use of some of the drugs might not have been optimum as far as dosing and the use of modified-release preparations are concerned. To compensate for these concerns, we have been able to randomise more than 1721 patients and achieve a high level of follow-up, something that would have been impossible with a more explanatory shorter clinical trial.
Previous comparative drug studies in similar patients had shown differences in tolerability, but had failed to show differences in efficacy. We therefore felt that the possibility of equivalence, or at least non-inferiority for efficacy outcomes, should be addressed. Thus, if a new antiepileptic drug showed better tolerability to its standard comparator, we wished to have power to exclude clinically important differences in efficacy, before accepting the drug as being first choice according to clinical outcomes. This decision resulted in power calculations needing 445 patients per treatment group. Although we were unable to recruit this many patients, we were able to extend the length of the study, with an increase in the number of outcome events and corresponding protection of power. We have presented two analyses of seizure-outcome results. In the intention-to-treat analyses, clinical data after a treatment failure on the randomised drug are included. Thus these analyses are of a policy of initial treatment with the randomised drug followed by, where necessary, switching to an alternative regimen, which was usually monotherapy with the standard drug or, if the standard drug failed, with lamotrigine. By contrast, the per-protocol analyses censored observations at the time of a treatment failure, so that only information while on the randomised drug is included. The intention-to-treat analyses should be regarded as most conservative, but when assessing possible equivalence or non-inferiority then per-protocol analyses are more conservative and should be given greatest weight.31
SANAD is the only large comparative drug study to our knowledge that includes quality of life and health economic assessments alongside clinical assessments. Our findings of differences between responders and non-responders for baseline quality of life profile and trial clinical outcomes accord with previous research showing that responders to surveys are likely to make favourable reports and to be more successful in their current status than non-responders.32,33 The implications of this responder bias for interpretation of the quality of life data and calculation of QALYs must therefore be considered when interpreting results. We can, however, be confident that the measures used are sensitive in view of the fact that there are clear quality of life benefits from achieving 12-month remission and harms from treatment failure.
The health economics analysis was done with two distinct incremental cost-effectiveness ratios, namely cost per QALY gained and cost per seizure avoided. Although number of seizures is an important clinical outcome, it constitutes a narrow measure of benefit in an economic assessment because it focuses on only one aspect of patient outcome. By contrast, the QALY is a much broader measure of benefit because it measures health-related quality of life, which is affected by not only the various clinical outcomes, but also other factors, such as the consequences of drug side-effects on patients' health.
Analysis of the clinical primary outcomes provides results with some precision. Lamotrigine has the lowest incidence of treatment failure and is better for this outcome than all drugs except oxcarbazepine (when this comparison is restricted to patients randomised after June 1, 2001). The differences are clinically important, with 12% and 8% fewer patients having treatment failure on lamotrigine than carbamazepine at 1 year and 2 years after randomisation, respectively. Competing risks analysis shows that lamotrigine is better because of its tolerability advantage over carbamazepine, since carbamazepine has fewest contributions to treatment failure from inadequate seizure control and is better than lamotrigine for the secondary efficacy outcome of time to first seizure. The difference between lamotrigine and carbamazepine for time to 12-month remission is small compared with the difference in tolerability and is not significant. Indeed, the CIs around comparisons between carbamazepine and lamotrigine for treatment failure due to inadequate seizure control and time to 12-month remission are sufficiently small to infer non-inferiority of lamotrigine for these outcomes (varying between 5–12% for both per protocol analyses). Carbamazepine is better than lamotrigine for time to first seizure, but this efficacy outcome might be dependent on initial dosing and could indicate that initial lamotrigine dosing in the trial was conservative, which would favour better tolerability outcomes, but detract from its efficacy early in the study. This inference is supported by the way in which per-protocol analysis of time to 12-month remission shows lamotrigine catching up with and eventually overtaking carbamazepine. There is also a lower rate of rash in patients randomised to lamotrigine in this arm of the study than might have been expected, a further potential consequence of conservative initial dosing.34,35
One further issue that might affect the lamotrigine-carbamazepine comparison is the choice of prescribing carbamazepine as either a standard preparation or as modified release. We did not obtain information systematically on this prescribing, but most collaborating clinicians indicated that they routinely prescribe the modified rather than the standard release versions of the drug. We feel that prescribing of ordinary release carbamazepine is unlikely to have adversely affected carbamazepine assessment in the study.
Our study recruited across a wide range of ages, which allowed us to assess whether the results from arm A were as applicable to children and to people older than 65 years as they were to those between the extremes of age. Although age itself does affect outcomes, there is no evidence of an interaction between age and drug treatment groups, which indicates that individual drug results are applicable through life. These findings do not accord with those of Rowan and colleagues,36 who studied older patients with epilepsy and reported that both lamotrigine and gabapentin were preferred to carbamazepine in this age-group.
Although there might be circumstances where other drugs are preferred (consideration of teratogenicity, bone health, drug interactions), the better tolerability seen in lamotrigine than carbamazepine, with non-inferiority of longer-term efficacy outcomes, lends support to lamotrigine as first choice treatment for most patients with partial epilepsy. Although the improved clinical outcomes are not indicated by improvements for individual core domains of quality of life with the exception of depression, there is no evidence from such data that would detract from the clinical conclusion. The economic analysis lends support to lamotrigine being preferred to carbamazepine in terms of both cost per seizure avoided and cost per QALY gained. There seems to be a high probability that lamotrigine is a cost-effective alternative to carbamazepine at what might be considered affordable (to the NHS) values of the ceiling ratio (λ). The National Institute for Health and Clinical Excellence should now reconsider its guidance about the first-line antiepileptic drug for patients with partial onset seizures.
We see no reasons to prefer gabapentin or topiramate to the standard drug carbamazepine, except where there might be individual mitigating factors. Both are associated with a higher risk of treatment failure that are not significant, gabapentin because of poor efficacy and topiramate because of poor tolerability and lesser efficacy than carbamazepine. The health economic assessment supports this view. For all clinical outcomes, there is some similarity between carbamazepine and oxcarbazepine, but the smaller numbers of patients available to the comparison reduce the statistical power and we could not conclude that they are equivalent. The economic analysis that included oxcarbazepine provides some evidence that it is preferred to carbamazepine. The point estimates of the incremental cost per seizure avoided are low, ranging between £31 and £35. In the cost per QALY analysis, the probability that oxcarbazepine is a cost-effective alternative to carbamazepine is high across the range of ceiling ratio values (λ). Indeed, data from this period of the study suggest than oxcarbazepine is the most cost-effective of the drugs assessed. However, in the absence of firm conclusions about oxcarbazepine's clinical effectiveness, further data for both clinical and health economic outcomes are needed before the drug can be accepted or rejected as a first-line treatment.
Other studies have compared lamotrigine with carbamazepine in similar populations, though over much shorter periods, and a meta-analysis of individual patient data has been undertaken.26 This meta-analysis showed that lamotrigine was better tolerated, and less likely to be associated with treatment failure, in agreement with our results. Time to first seizure also agreed with SANAD in indicating that time tended to be longer for carbamazepine, but SANAD is the first study to our knowledge that has allowed examination of the more clinically important time to 12-month remission efficacy outcome, where the difference between the two drugs is much smaller. Two studies have compared lamotrigine and gabapentin in elderly and adult patients, respectively, and recorded little difference.36,37 They were, however, too short to allow meaningful comparison of efficacy outcomes, so that the treatment failure outcomes reported in the studies were dominated by the drugs' similar and good tolerability.
The SANAD study has successfully shown the feasibility of doing large pragmatic epilepsy studies in the NHS in a way that would be difficult in many other health-care systems. Since its design, three further new antiepileptic drugs have been licensed in the UK: levetiracetam, pregabalin, and zonisamide. The same questions that applied to gabapentin, lamotrigine, oxcarbazepine, and topiramate, now apply to these drugs, though for partial epilepsies they will now need to be compared with lamotrigine and possibly oxcarbazepine, rather than carbamazepine. SANAD has shown that we have a robust method to answer these questions.
Acknowledgments
The study was supported by a grant from the Health Technology Assessment Programme of the NHS. There were further contributions from GlaxoSmithKline, Janssen-Cilag, Novartis Pfizer, Sanofi-Synthelabo, and the Wellcome Trust that supported related studies.
Footnotes
Conflict of interest statement
AGM has received fees and reimbursement for attending conferences from Jannsen Cilag, GlaxoSmithKline, Novartis, Pfizer, and Sanofi Synthelabo, and research funding from Pfizer; AMA-K has received speaker fees and reimbursement for attending conferences from Jannsen Cilag, GlaxoSmithKline, Novartis, Pfizer, and Sanofi Synthelabo; RA has received consultancy fees, speaker fees, and reimbursement for attending conferences from Jannsen Cilag, GlaxoSmithKline, and Sanofi Synthelabo; GAB has received research funding, speaker fees, and reimbursement for attending conferences from Jannsen Cilag, Glaxo SmithKline, Novartis, Pfizer, and Sanofi Synthelabo; DWC has received consultancy fees, speaker fees, and reimbursement for attending conferences from Jannsen Cilag, GlaxoSmithKline, and Sanofi Synthelabo; PNC has received speaker fees from Jannsen Cilag, reimbursement for attending conferences from Jannsen Cilag, GlaxoSmithKline, Novartis, Pfizer, and Sanofi Synthelabo, and research funding from Sanofi Synthelabo; PJG, has received speaker fees and reimbursement for attending conferences from Eisai, GlaxoSmithKline, Jannsen Cilag, Pfizer, Cyberonics, and UCB; SJLH has received reimbursement for attending conferences from Jannsen Cilag, GlaxoSmithKline, Novartis, Pfizer, and Sanofi Synthelabo, and payment for research from Jannsen Cilag and GlaxoSmithKline; AH has received reimbursement for attending conferences from Jannsen Cilag and GlaxoSmithKline; MJ has received speaker fees and reimbursement for attending conferences from GlaxoSmithKline, Pfizer, and Jannsen Cilag; AJ has received funding from SanofiSynthelabo, GlaxoSmithKline, and Jannsen Cilag, and has acted as a research consultant to Johnson and Johnson Pharmaceuticals; MK has received speaker fees and reimbursement for attending conferences from Jannsen Cilag, GlaxoSmithKline, and Pfizer; GRL has received speaker fees and reimbursement for attending conferences from UCB and Jannsen Cilag; JPL has received speaker fees and reimbursement for attending conferences from Jannsen Cilag, GlaxoSmithKline, Pfizer, Eisai, UCB, and Sanofi Synthelabo, and research funding from GlaxoSmithKline; PN has received speaker fees and reimbursement for attending conferences from Jannsen Cilag, GlaxoSmithKline, Pfizer, and Sanofi Synthelabo; RR has received consultancy fees, speaker fees, and reimbursement for attending conferences from GlaxoSmithKline, Jannsen Cilag, Novartis, and Pfizer; DFS has received speaker fees and reimbursement for attending conferences from Jannsen Cilag, GlaxoSmithKline, Novartis, Pfizer, and Sanofi Synthelabo, and research funding from GlaxoSmithKline; PEMS has received speaker fees, consultancy fees, and reimbursement for attending conferences from UCB Pharma, Pfizer, Eisai, Novartis, and GlaxoSmithKline, and research funding from UCB Pharma and GlaxoSmithKline; the other authors declare no conflict of interest.
Contributor Information
Anthony G Marson, Division of Neurological Science, University of Liverpool, UK.
Asya M Al-Kharusi, Southport and Formby District General Hospital, Merseyside, UK.
Muna Alwaidh, St Helens and Knowsley Hospital NHS Trust, Whiston Hospital, Liverpool, UK.
Richard Appleton, Roald Dahl EEG Unit, Department of Neurology, Royal Liverpool Children's NHS Trust (Alder Hey), Liverpool, UK.
Gus A Baker, Division of Neurological Science, University of Liverpool, UK.
David W Chadwick, Division of Neurological Science, University of Liverpool, UK.
Celia Cramp, Royal Shrewsbury Hospital, Shrewsbury, UK.
Oliver C Cockerell, Royal London Hospital, London, UK.
Paul N Cooper, Royal Bolton Hospital, Bolton, UK.
Julie Doughty, School of Population and Health Sciences, University of Newcastle, UK.
Barbara Eaton, Division of Neurological Science, University of Liverpool, UK.
Carrol Gamble, Centre for Medical Statistics and Health Evaluation, University of Liverpool, UK.
Peter J Goulding, Leeds General Infirmary, Leeds, UK.
Stephen J L Howell, Royal Hallamshire Hospital, Sheffield, UK.
Adrian Hughes, Arrowe Park Hospital, Wirral, UK.
Margaret Jackson, Royal Victoria Infirmary, Newcastle-Upon-Tyne, UK.
Ann Jacoby, Division of Public Health, University of Liverpool, UK.
Mark Kellett, Royal Bolton Hospital, Bolton, UK.
Geoffrey R Lawson, Sunderland Royal Hospital, Sunderland, UK.
John Paul Leach, Southern General Hospital, Glasgow, UK.
Paola Nicolaides, Great Ormond Street Hospital, London, UK.
Richard Roberts, Ninewells Hospital, Dundee, UK.
Phil Shackley, School of Population and Health Sciences, University of Newcastle, UK.
Jing Shen, School of Population and Health Sciences, University of Newcastle, UK.
David F Smith, The Walton Centre for Neurology and Neurosurgery NHS Trust, Liverpool, UK.
Philip E M Smith, University Hospital of Wales, Cardiff, UK.
Catrin Tudur Smith, Centre for Medical Statistics and Health Evaluation, University of Liverpool, UK.
Alessandra Vanoli, School of Population and Health Sciences, University of Newcastle, UK.
Paula R Williamson, Centre for Medical Statistics and Health Evaluation, University of Liverpool, UK.
References
- 1.Hauser WA, Hesdorffer DC. Epilepsy: frequency, causes and consequences. New York: Demos Publications; 1990. [Google Scholar]
- 2.Annegers JF, Hauser WA, Elveback LR. Remission of seizures and relapse in patients with epilepsy. Epilepsia. 1979;20:729–37. doi: 10.1111/j.1528-1157.1979.tb04857.x. [DOI] [PubMed] [Google Scholar]
- 3.Cockerell OC, Johnson AL, Sander JW, Hart YM, Shorvon SD. Remission of epilepsy: results from the National General Practice Study of Epilepsy. Lancet. 1995;346:140–44. doi: 10.1016/s0140-6736(95)91208-8. [DOI] [PubMed] [Google Scholar]
- 4.London: National Institute for Clinical Excellence; 2004. The epilepsies: the diagnosis and management of the epilepsies in adults and children in primary and secondary care. (Clinical Guideline 20). [Google Scholar]
- 5.French JA, Kanner AM, Bautista J, et al. Efficacy and tolerability of the new antiepileptic drugs i: treatment of new onset epilepsy: report of the therapeutics and technology assessment subcommittee and quality standards subcommittee of the American Academy of Neurology and the American Epilepsy Society. Neurology. 2004;62:1252–60. doi: 10.1212/01.wnl.0000123693.82339.fc. [DOI] [PubMed] [Google Scholar]
- 6.Marson AG, Williamson PR, Clough H, Hutton JL, Chadwick DW. Carbamazepine versus valproate monotherapy for epilepsy: a meta-analysis. Epilepsia. 2002;43:505–13. doi: 10.1046/j.1528-1157.2002.20801.x. [DOI] [PubMed] [Google Scholar]
- 7.Marson AG, Kadir ZA, Chadwick DW. New antiepileptic drugs: a systematic review of their efficacy and tolerability. BMJ. 1996;313:1169–74. doi: 10.1136/bmj.313.7066.1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Marson AG, Hutton JL, Leach JP, et al. Levetiracetam, oxcarbazepine, remacemide and zonisamide for drug resistant localization-related epilepsy: a systematic review. Epilepsy Res. 2001;46:259–70. doi: 10.1016/s0920-1211(01)00287-x. [DOI] [PubMed] [Google Scholar]
- 9.Brodie MJ, Richens A, Yuen AWC. Double-blind comparison of lamotrigine and carbamazepine in newly diagnosed epilepsy. UK Lamotrigine/Carbamazepine Monotherapy Trial Group. Lancet. 1995;345:476–79. doi: 10.1016/s0140-6736(95)90581-2. [DOI] [PubMed] [Google Scholar]
- 10.Chadwick DW, Anhut H, Greiner MJ, et al. A double-blind trial of gabapentin monotherapy for newly diagnosed partial seizures. International Gabapentin Monotherapy Study Group 945–77. Neurology. 1998;51:1282–88. doi: 10.1212/wnl.51.5.1282. [DOI] [PubMed] [Google Scholar]
- 11.Dam M, Ekberg R, Loyning Y, Waltimo O, Jakobsen K. A double-blind study comparing oxcarbazepine and carbamazepine in patients with newly diagnosed, previously untreated epilepsy. Epilepsy Res. 1989;3:70–76. doi: 10.1016/0920-1211(89)90070-3. [DOI] [PubMed] [Google Scholar]
- 12.Reunanen M, Dam M, Yuen AW. A randomised open multicentre comparative trial of lamotrigine and carbamazepine as monotherapy in patients with newly diagnosed or recurrent epilepsy. Epilepsy Res. 1996;23:149–55. doi: 10.1016/0920-1211(95)00085-2. [DOI] [PubMed] [Google Scholar]
- 13.Kalviainen R, Aikia M, Saukkonen AM, Mervaala E, Riekkinen PJ., Sr Vigabatrin vs carbamazepine monotherapy in patients with newly diagnosed epilepsy. A randomized, controlled study. Arch Neurol. 1995;52:989–96. doi: 10.1001/archneur.1995.00540340081016. [DOI] [PubMed] [Google Scholar]
- 14.Chadwick D. Safety and efficacy of vigabatrin and carbamazepine in newly diagnosed epilepsy: a multicentre randomised double-blind study. Vigabatrin European Monotherapy Study Group. Lancet. 1999;354:13–19. doi: 10.1016/s0140-6736(98)10531-7. [DOI] [PubMed] [Google Scholar]
- 15.Brodie MJ, Overstall PW, Giorgi L. Multicentre, double-blind, randomised comparison between lamotrigine and carbamazepine in elderly patients with newly diagnosed epilepsy. The UK Lamotrigine Elderly Study Group. Epilepsy Res. 1999;37:81–87. doi: 10.1016/s0920-1211(99)00039-x. [DOI] [PubMed] [Google Scholar]
- 16.Brodie MJ, Bomhof MAM, Kalviainen R, et al. Double-blind comparison of tiagabine and carbamazepine monotherapy in newly diagnosed epilepsy. Epilepsia. 1997;38(suppl 3):66–67. [Google Scholar]
- 17.Brodie MJ, Wroe SJ, Dean ADP, Holdich TAH, Whitehead J, Stevens JW. Efficacy and safety of remacemide versus carbamazepine in newly diagnosed epilepsy: comparison by sequential analysis. Epilepsy Behav. 2002;3:140–46. doi: 10.1006/ebeh.2002.0337. [DOI] [PubMed] [Google Scholar]
- 18.Chadwick DW, Anhut H, Greiner MJ, et al. A double-blind trial of gabapentin monotherapy for newly diagnosed partial seizures. International Gabapentin Monotherapy Study Group 945–77. Neurology. 1998;51:1282–88. doi: 10.1212/wnl.51.5.1282. [DOI] [PubMed] [Google Scholar]
- 19.Privitera MD, Brodie MJ, Mattson RH, et al. Topiramate, carbamazepine and valproate monotherapy: double-blind comparison in newly diagnosed epilepsy. Acta Neurol Scand. 2003;107:165–75. doi: 10.1034/j.1600-0404.2003.00093.x. [DOI] [PubMed] [Google Scholar]
- 20.Bill PA, Vigonius U, Pohlmann H, et al. A double-blind controlled clinical trial of oxcarbazepine versus phenytoin in adults with previously untreated epilepsy. Epilepsy Res. 1997;27:195–204. doi: 10.1016/s0920-1211(97)00024-7. [DOI] [PubMed] [Google Scholar]
- 21.Christe W, Kramer G, Vigonius U, et al. A double-blind controlled clinical trial: oxcarbazepine versus sodium valproate in adults with newly diagnosed epilepsy. Epilepsy Res. 1997;26:451–60. doi: 10.1016/s0920-1211(96)01013-3. [DOI] [PubMed] [Google Scholar]
- 22.Guerreiro MM, Vigonius U, Pohlmann H, et al. A double-blind controlled clinical trial of oxcarbazepine versus phenytoin in children and adolescents with epilepsy. Epilepsy Res. 1997;27:205–13. doi: 10.1016/s0920-1211(97)00025-9. [DOI] [PubMed] [Google Scholar]
- 23.Steiner TJ, Dellaportas CI, Findley LJ, et al. Lamotrigine monotherapy in newly diagnosed untreated epilepsy: a double-blind comparison with phenytoin. Epilepsia. 1999;40:601–07. doi: 10.1111/j.1528-1157.1999.tb05562.x. [DOI] [PubMed] [Google Scholar]
- 24.Newer drugs for epilepsy in adults. London: National Institute for Clinical Excellence; 2004. (Technology Appraisal Guidance 76). [Google Scholar]
- 25.Newer drugs for epilepsy in children. London: National Institute for Clinical Excellence; 2004. (Technology Appraisal Guidance 79). [Google Scholar]
- 26.Gamble CL, Williamson PR, Marson AG. Lamotrigine versus carbamazepine monotherapy for epilepsy. Cochrane Database Syst Rev. 2006;1:CD001031. doi: 10.1002/14651858.CD001031.pub2. [DOI] [PubMed] [Google Scholar]
- 27.Medical Research Council . Guidelines for good clinical practice in clinical trials. London: MRC; 1998. [Google Scholar]
- 28.Commission on Classification and Terminology of the International League Against Epilepsy Proposal for revised classification of epilepsies and epileptic syndromes. Epilepsia. 1989;30:389–99. doi: 10.1111/j.1528-1157.1989.tb05316.x. [DOI] [PubMed] [Google Scholar]
- 29.Commission on Classification and Terminology of the International League Against Epilepsy Proposal for revised clinical and electroencephalographic classification of epileptic seizures. Epilepsia. 1981;22:489–501. doi: 10.1111/j.1528-1157.1981.tb06159.x. [DOI] [PubMed] [Google Scholar]
- 30.Williamson P, Hutton J, Marson A, Chadwick D. Individual patient data meta-analyses for time to event outcomes: an example from epilepsy. Control Clin Trials. 1997;18(3 suppl 1):S184. [Google Scholar]
- 31.Jones B, Jarvis P, Lewis JA, Ebutt AF. Trials to assess equivalence; the importance of rigorous methods. BMJ. 1996;313:36–39. doi: 10.1136/bmj.313.7048.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Donald MN. Implications of non-response for the interpretation of mail questionnaire data. Public Opin Q. 1960;24:99–114. [Google Scholar]
- 33.Goyden J. The Silent Minority: non-respondents to sample surveys. Cambridge: Polity Press; 1987. [Google Scholar]
- 34.Messenheimer JA, Guberman AH. Rash with lamotrigine: dosing guidelines. Epilepsia. 2000;41:488. doi: 10.1111/j.1528-1157.2000.tb00197.x. [DOI] [PubMed] [Google Scholar]
- 35.Wong IC, Mawer GE, Sander JW. Factors influencing the incidence of lamotrigine-related skin rash. Ann Pharmacother. 1999;33:1037–42. doi: 10.1345/aph.18422. [DOI] [PubMed] [Google Scholar]
- 36.Rowan AJ, Ramsay RE, Collins JF, et al. New onset geriatric epilepsy: a randomized study of gabapentin, lamotrigine, and carbamazepine. Neurology. 2005;64:1868–73. doi: 10.1212/01.WNL.0000167384.68207.3E. [DOI] [PubMed] [Google Scholar]
- 37.Brodie MJ, Chadwick DW, Anhut H, et al. Gabapentin versus lamotrigine monotherapy: a double-blind comparison in newly diagnosed epilepsy. Epilepsia. 2002;43:993–1000. doi: 10.1046/j.1528-1157.2002.45401.x. [DOI] [PubMed] [Google Scholar]