

Abstract
Phosphatidylinositol‐specific phospholipase C (PI‐PLC) is activated in cell nuclei during the cell cycle progression. We have previously demonstrated two peaks of an increase in the nuclear PI‐PLC activities in nocodazole‐synchronized HL‐60 cells. In this study, the activity of nuclear PI‐PLC was investigated in serum‐stimulated HL‐60 cells. In serum‐starved HL‐60 cells, two peaks of the activity of nuclear PI‐PLC were detected at 30 minutes and 11 h after the re‐addition of serum with no parallel increase in PLC activity in cytosol, postnuclear membranes or total cell lysates. An increase in the serine phosphorylation of b splicing variant of PI‐PLCβ1 was detected with no change in the amount of PI‐PLCβ1b in nuclei isolated at 30 min and 11h after the addition of serum. PI‐PLC inhibitor ET‐18‐OCH3 and MEK inhibitor PD 98059 completely abolished serum‐mediated increase at both time‐points. The addition of inhibitors either immediately or 6 h after the addition of serum had inhibitory effects on the number of cells entering S phase. These results demonstrate that two waves of nuclear PI‐PLCβ1b activity occur in serum‐stimulated cells during G1 phase of the cell cycle and that the later increase in the PLC activity is equally important for the progression into the S phase.
Keywords: Phospholipase Cβ1b, Nuclei, Serum, Cell cycle, G1 phase, HL‐60 cells
1. Introduction
Over the last decades, several studies have demonstrated the existence of a distinct nuclear inositol lipid signaling system that is regulated independently from the phosphoinositide cycle at the plasma membrane (1, 2). The major phosphoinositide‐specific phospholipase C (PI‐PLC) isoform that was shown to be activated in nuclei of various cells early (5–30 min) after an addition of a mitogen is PI‐PLCβ1 that is known to exist in two alternatively spliced forms; PI‐PLCβ1a is equally distributed in cytosol and nuclei and PI‐PLCβ1b predominates in nuclei (3–8). The mechanism of the activation of the nuclear PI‐PLCβ1b differs from the classical mechanism of G‐protein‐mediated activation of the PI‐PLCβ1 at the cell membrane and involves the mitogen‐activated protein kinase (MAPK)‐mediated phosphorylation of the enzyme at the serine residue (6–8). A possible role for the nuclear PI‐PLCβ1 activation in the cell proliferation has been documented in Swiss3T3 cells as the mitogenic response of the cells to insulin‐like growth factor (IGF) was attenuated by the ablation of the isoform through antisense RNA (4). Furthermore, the possible role of the nuclear PI‐PLCβ1 in cell cycle progression was indicated in murine erythroleukemia (MEL) cells transfected with PI‐PLCβ1 splice variants and a mutant lacking the nuclear localization signal (9).
The two wave hypothesis has been proposed to explain the role of temporally distinct activation of Ras, phosphatidylinositol 3‐kinase (PI3K) or protein kinase C (PKC) that was observed at the cell membrane or total lysates of various cell types during the growth factor‐induced progression through G1 phase (10–13). The hypothesis suggests that a commitment to cell‐cycle progression requires more than commonly studied growth factor‐stimulated signaling that occurs as G0 cells are re‐entering G1; to enter the S phase cells must be either exposed continuously to growth factor for 8–10 h, or the growth factor must be present at the G0/G1 transition, and then again in mid‐G1 (10). In human myeloblastic HL‐60 leukemia cells, the activation of the nuclear phospholipase C has been observed at several time points of the cell cycle. IGF‐mediated early increase in the level of nuclear DAG in serum starved HL‐60 cells that was sensitive to the presence of a PI‐PLC inhibitor probably corresponds to the activation of the nuclear enzyme at G0 exit (14). In aphidicolin‐synchronized HL‐60 cells, the increase in the level of the nuclear diacylglycerol (DAG) occurs 8 h after the release from the block and correlates with G2/M phase of the cell cycle (15). Our recent study demonstrated two peaks of an increase in the nuclear activity of PI‐PLCβ1b in nocodazole‐synchronized cells at 1 and 8,5 h after release from the block that correlated with G2/M and late G1 phase of the cell cycle (16). To further discriminate between genuine cell‐cycle‐related events and possible cycle‐independent effects that might be the consequence of a nocodazole‐synchronization procedures, we perform measurements in serum‐starved HL‐60 cells stimulated to progress through the G1 phase of the cycle by a re‐addition of serum.
In this study, two waves of nuclear PI‐PLCβ1b activity in serum‐stimulated cells were detected during the progression through the G1 phase of the cell cycle and the later increase in the activity of the enzyme was found to be equally important for the progression into the S phase.
2. Materials and methods
2.1 Materials
Reagents were obtained from the following sources: EGTA, EDTA, HEPES, Tris, leupeptin, phenylmethylsulfonyl fluoride (PMSF), RNase A, propidium iodide, Triton X‐100, Na+deoxycholate, sodium metavanadate, 2‐mercaptoethanol, protein G‐Sepharose, aprotinin, SDS, phosphatidylinositol 4,5‐bisphosphate (PtdIns(4,5)P2), Nonidet P‐40 (NP‐40), bromodeoxyridine (BrdU) and monoclonal anti‐β‐tubulin antibody (T0198) from Sigma, St. Louis, MO, USA; anti‐P‐ser antibody (clone 16B4) from Alexis Biochemicals, Laufelfingen, Switzerland; fetal bovine serum (FBS) from Gibco, Invitrogen, UK, 1‐O‐octadecyl‐2‐Omethyl‐sn‐glycero‐3‐phosphocholine (ET‐18‐OCH3), LY 294002 from Calbiochem, Nottingham, U.K.; PD 98059 from Cell Signaling Technology, Beverly, MA, USA; [³H] PtdIns(4,5)P2 from Perkin Elmer Life Sciences, Boston, MA, USA, BrdU FITC set (556028) from BD Biosciences, San Diego, CA, USA; enhanced chemiluminescence kit from Amersham Pharmacia Biotech., Uppsala, Sweden, Lake Placid, NY, USA. All other chemicals were of analytical grade.
2.2 Cell culture, cell cycle synchronization, and flow cytometric analysis
HL‐60 cells (ECCACC no. 88112501) were obtained from the European Collection of Animal Cell Cultures, PHLS, Porton, Salisbury, UK. The cells were maintained in exponential growth in RPMI 1640 medium with 10% heat‐inactivated FBS, 100 U/ml penicillin and 100 µg/ml streptomycin in a 5% CO2 humidified atmosphere at 37 °C. For synchronization, exponentially growing cells were washed and maintained in medium containing no serum for 24 h and then stimulated with FBS (10%, v/v). In some cases, the phospholipase inhibitor ET‐18‐OCH3, PI3K‐inhibitor LY 294002, and MAP kinase kinase (MEK) inhibitor PD 98059 were added to the cultures at the times and concentrations indicated in the figure legends.
Cells were sampled at the indicated time points after the addition of FBS, washed twice with PBS and stained directly with propidium iodide solution (20 µg/ml of propidium iodide and 20 µg/ml RNase A in PBS) for 30 min. Monoparametric DNA analyses were performed on at least 15,000 cells for each sample with the FACSCalibur system (Becton Dickinson), and the data were analyzed using CellQuest and ModFit software (Becton Dickinson). For the purpose of analysis, acquired events were gated to eliminate cell aggregates and debris.
2.3 Preparation of nuclei, postnuclear membranes, cytosolic fractions and total cell lysates, and determination of PLC activity
At the indicated time points, cells were sampled, washed two times with cold PBS and different cellular fractions were isolated as previously described (15–17). Briefly, whole nuclei were isolated by homogenization in STM buffer (50 mM Tris‐HCl, pH 7.4, 250 mM sucrose, 5 mM MgSO4) containing no detergents, homogenates were layered over a cushion (2.1 M sucrose, 50 mM Tris‐HCl, pH 7.4, 5 mM MgSO4), spun at 70 000 x g for 60 min and the resulting pellet represents the whole nuclei containing nuclear envelopes, as previously described (7, 17). In some preparations of nuclei, NP‐40 (0.02%, w/v) was added to STM buffer as previously described (15). The postnuclear membranes, cytosolic fractions, and total cell lysates were prepared as previously described (16, 17).
Equal amounts of proteins from nuclei, cytosol, postnuclear membranes and cell lysates (100 µg) were subjected to PLC assay using [³H] (PtdIns(4,5)P2) as previously described (16).
2.4 Immunoprecipitation and Western blot analysis for the presence and serine phosphorylation of PLCβ1b
PI‐PLCβ1b was immunoprecipitated from nuclear fractions containing 100 µg of protein by incubating in the presence of 5 µg of antibody specific for PLCβ1b residues 1164–1173 (5, 18) and protein G‐sepharose at 4°C for 1 hour. Immunoprecipitates were washed once in incubation buffer (50 mM Tris, pH 7.6, 150 mM NaCl, 1% Triton X‐100 (w/v), 0.5% Na+deoxycholate (w/v), 0.1% SDS (w/v), 2 mM PMSF, 1 µg/ml aprotinin, and 1 µg/ml leupeptin) and three times with 5 mM HEPES/ 2 mM EDTA (pH 7.5). The immunoprecipitates were subjected to Western blot analysis using anti‐P‐serine antibodies or anti‐PLCβ1b antibodies, as previously described (16, 19).
2.5. BrdU incorporation assay
HL‐60 cells were cultured in the presence of FBS for 17 h and then pulsed with 20 µM BrdU for 1 hour at 37 °C. After incubation, cells were washed with PBS, fixed in 70% ethanol at room temperature for 20 min and then washed with washing buffer (PBS containing 0,5% BSA). To denature DNA, the pellet was resuspended in 0.3 ml of 2 M HCl and incubated at room temperature for 20 minutes. After washing and neutralization of any remaining acid by 0.1 M sodium tetraborate (pH 8.5), cells were resuspended in PBS containing 0.5% BSA and incubated with fluorescein isothiocyanate (FITC)‐conjugated anti‐BrdU antibody or isotypic control for 30 minutes at room temperature. After the incubation, the cells were washed and the pellet resuspended in 0.5 ml propidium iodide (10 µg/ml in PBS). Fluorescence was analyzed on at least 15,000 cells for each sample with the FACSCalibur system (Becton Dickinson), and the data were analyzed using CellQuest software (Becton Dickinson). For the purpose of analysis, acquired events were gated to eliminate cell aggregates and debris.
2.6 Statistical evaluation
The data are shown as means ± S. E. M. For statistical analyses, the Student's t‐test for unpaired samples at the level of significance of 0.05 was used.
3. Results
When exponentially growing HL‐60 cells were harvested, washed and resuspended in medium containing no FBS, the percentage of cells in G0/G1 increased with no significant increase in the number of subG1‐events even after 48 h of incubation in medium alone (Fig. 1A). The re‐addition of FBS (10% v/v) caused a progressive increase in the number of cells in S phase. Nuclear fractions were isolated from cells at different times after the addition of serum and the activity of phospholipase C was measured using tritium‐labeled PtdIns(4,5)P2 as a substrate. The PLC‐activity in nuclei isolated from cells after the 24 h‐starvation did not differ from the activity measured in nuclei of cells growing exponentially in culture (3.03 ± 0.09 vs. 3.15 ± 0.13 nmol Ins(1,4,5)P3 released/mg protein/h incubation). It is important to note that nuclei were isolated through sucrose cushion using buffer containing no detergents and therefore pellets represent nuclei retaining their nuclear envelopes as measured by the presence of β‐tubulin (7). As some amount of β‐tubulin could be detected even after the addition of Nonidet P‐40 in concentration previously used in aphidicolin‐synchronized HL‐60 cells (15), all subsequent measurements of nuclear PLC activity were performed on the whole nuclei isolated in the absence of detergents (Fig. 1 B). As shown in Fig. 1C, the maximal activity of nuclear PLC was measured 30 min after the addition of serum, the activity was still significantly increased at 60 min and returned to the baseline levels at 2 h. No further increase in the activity of nuclear PLC was detected up to 10.5 h after the addition of serum. The second peak of an increase was measured at 11 h after the addition of serum and at 12 h the activity did not differ from the activity in control (Fig. 1D). Further experiments were performed in order to confirm that two peaks of PLC‐activation in serum‐stimulated cells are specific for nuclear compartments. No increase in the level of PLC activity was detected in cytosol, postnuclear membranes or total cell lysates at either 30 min or 11 h after the addition of serum (Fig. 2).
Fig. 1. The activity of PLC in nuclei isolated from HL‐60 cells after addition of FBS.
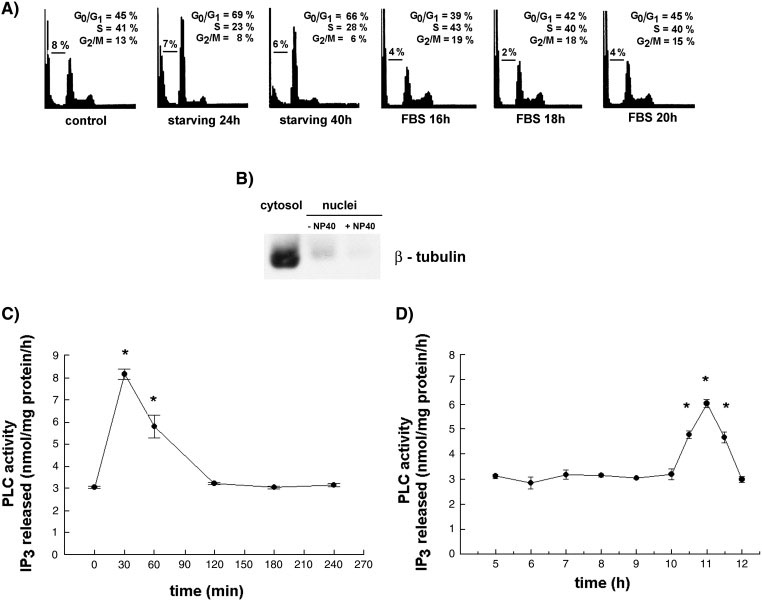
A) Representative histograms of control, serum‐starved and FBS‐stimulated HL‐60 cells that were stained with propidium iodide and assessed for cell cycle distribution by flow cytometric analysis. B) Western blot analysis for the presence of β‐tubulin in cytosol, nuclei isolated in the absence of NP‐40, and nuclei isolated in the presence of 0.02% NP‐40. Equal amounts of cytosolic and nuclear proteins (30µg) were loaded. (C) PLC activity in nuclei isolated from HL‐60 cells early after addition of FBS. (D) PLC activity in nuclei isolated from HL‐60 cells 5‐12 h after addition of FBS. Results are means ± S.E.M. for three different experiments, each performed in duplicate. *P < 0.05 (Student’s t‐test) with respect to the control.
Fig. 2. The activity of PLC in HL‐60 cells subcellular fractions at different times after addition of FBS.

Cytosolic fractions, nuclei, postnuclear membranes (PNM) and total cell lysates (TCL) were isolated as described in Materials and methods from HL‐60 cells immediately after (0 h) or 30’ and 11 h after addition of FBS. PLC activity was assayed using PtdIns(4,5)P2 as substrate. Results are means ± S.E.M. for three different experiments, each performed in duplicate. *P < 0.05 (Student’s t‐test) with respect to the control.
Previous studies have shown that the activation of nuclear PLC early after mitogen stimulation (14) or during G2/M in aphidicolin (15) and nocodazole‐synchronized (16) HL‐60 cells is sensitive to the presence of PI‐PLC inhibitor ET‐18‐OCH3. As shown at Fig. 3, the addition of 10 µM ET‐18‐OCH3 early after addition of serum (A) or 6 h after the stimulation (B) completely inhibited the increase in the activity of PLC. The presence of PD 98059, a MEK‐inhibitor that inhibited MAPK‐mediated phosphorylation of PI‐PLCβ1 (6–8) abolished serum‐mediated increase at both time‐points, too. In contrast, the addition of PI3K inhibitor LY 294002 30 min before the addition of serum and at 6 h after the stimulation had no effects on early and late increase in the nuclear PLC‐activity, respectively (Fig. 3A, Fig. 3B). To further test for possible serine phosphorylation of the activated enzyme, nuclear fractions were isolated at time 0 h, 30 min and 11 h, immunoprecipitated using PI‐PLCβ1b specific antibody, and immunoprecipitates were subjected to Western blot analysis using an anti‐P‐serine antibody. As shown in Fig. 4, no change in the overall amount of PI‐PLCβ1b was detected in nuclei isolated at any time points. However, an increase in the level of serine‐phosphorylated band of the same molecular size as PI‐PLCβ1b was observed in nuclei isolated at 30 min and 11 h after the addition of serum in comparison to control.
Fig. 3. The effect of inhibitors on the increase in nuclear PLC‐activity at 30 min (A) and 11 h (B) after addition of FBS.
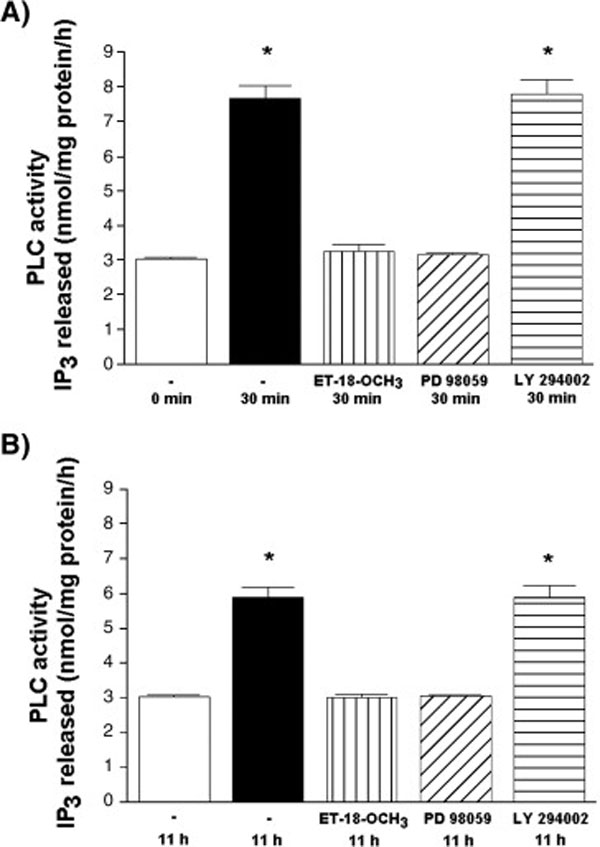
Exponentially growing HL‐60 cells were washed and released into serum‐free medium. After 24 h, cells were stimulated with 10% FBS (v/v.). PI‐PLC inhibitor ET‐18‐OCH3 (10 µM), MEK inhibitor PD 98059 (20 µM) or PI3K‐inhibitor LY 294002 (25 µM) were added either immediately (A) or 6 h after addition of FBS (B). Nuclei were isolated from cells harvested at 30 min and 11 h after addition of FBS, and PLC‐activity was assayed using PtdIns(4,5)P2 as substrate. Results are means ± S.E.M. for three different experiments, each performed in duplicate. *P < 0.05 (Student’s t‐test) with respect to the control.
Fig. 4. The serine‐phosphorylation of PLCβ1b immunoprecipitated from the nuclear fraction of HL‐60 cells at different times after addition of FBS.

Nuclear fractions were isolated from HL‐60 cells at 0 h, 0,5 h and 11 h after addition of FBS and incubated in the presence of antibodies to PLCβ1b. The immunoprecipitated enzyme was subjected to SDS‐PAGE, transferred to nitrocellulose, and then probed with anti‐P‐serine antibodies or anti‐PLCβ1b antibodies.
To test for the possible role of both early and late nuclear PLC activation in serum‐stimulated G1 progression, the effects of the inhibitors added either immediately (Fig. 5) or 6 h after the addition of serum (Fig. 6) on the cell cycle distribution were further investigated. As shown in Fig. 5A, Fig. 5B and Fig. 6A, the presence of ET‐18‐OCH3 (10 µM), LY 294002 (25 µM) and PD 98059 (20 µM) decreased the percentage of cells in S phase at 16 h after serum treatment. The presence of ET‐18‐OCH3 increased the proportion of sub‐G1 events and the percentage of cells in G2/M phase. A decrease in the proportion of cells in S phase of the cell cycle that was measured by propidium iodide staining and ModFit analysis correlates well with a decrease in the level of BrdU incorporation determined in cells treated with inhibitors immediately (Fig. 5C) or 6 h after the addition of serum (Fig. 6B).
Fig. 5. The cell cycle progression of HL‐60 cells in the presence of inhibitors added immediately after addition of FBS.
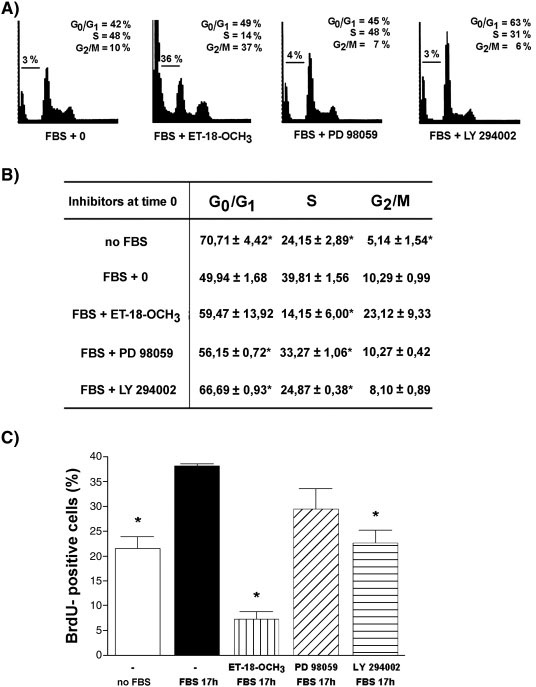
HL‐60 cells were incubated in serum‐free medium for 24 h and then stimulated with 10% FBS (v/v). PI‐PLC inhibitor ET‐18‐OCH3 (10 µM), MEK inhibitor PD 98059 (20 µM) or PI3K‐inhibitor LY 294002 (25 µM) were added immediately after addition of FBS. Cells were either harvested at 16 h after addition of FBS, stained with propidium iodide and assessed for cell cycle distribution by flow cytometric analysis. (A and B), or incubated in the presence of BrdU (10 µM) for 1 h and then analyzed for the level of BrdU incorporation as described in Materials and methods section (C). Results are means ± S.E.M. for three different experiments. *P < 0.05 (Student’s t‐test) with respect to the control.
Fig. 6. The cell cycle progression of HL‐60 cells in the presence of inhibitors added 6 hours after addition of FBS.
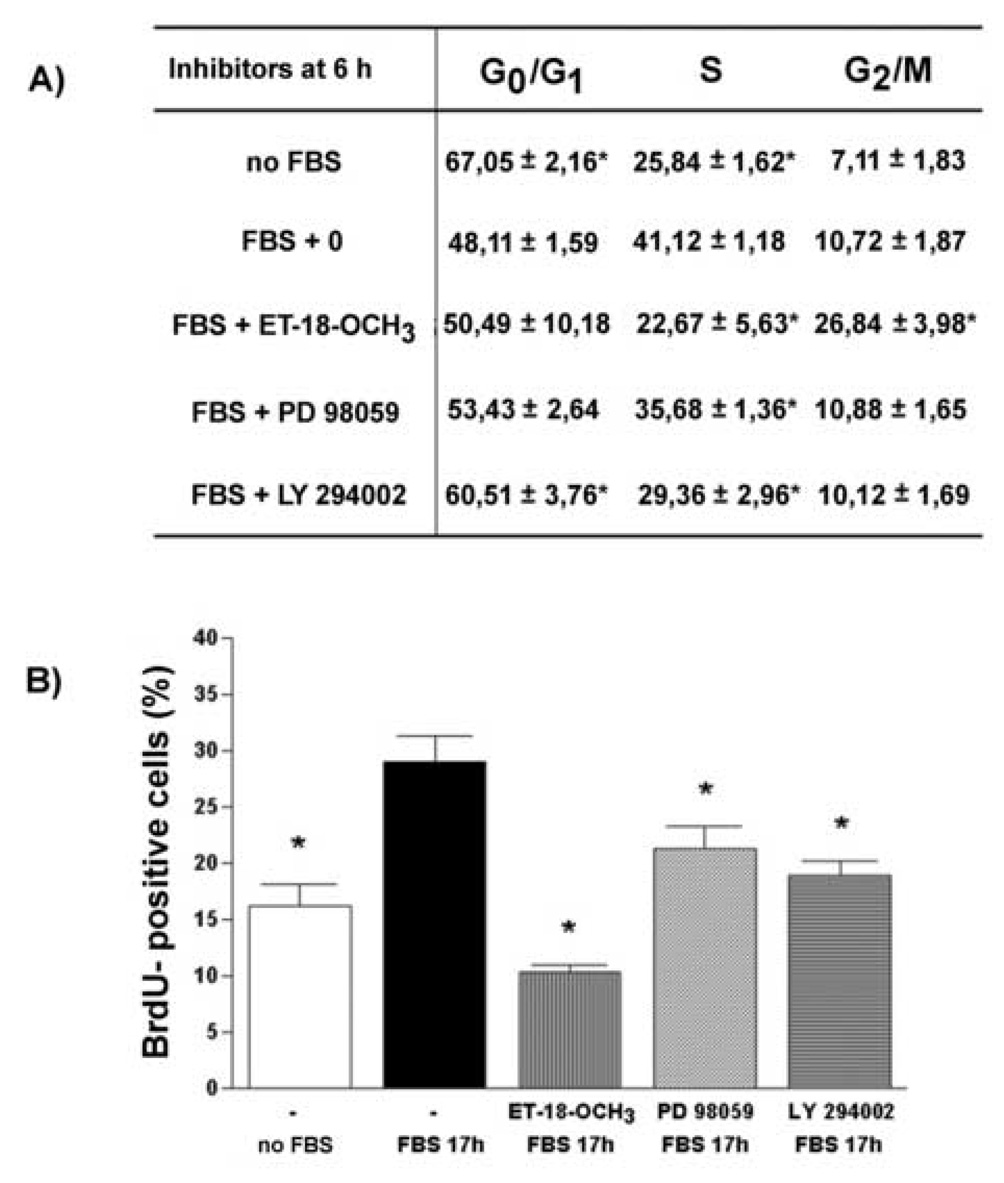
HL‐60 cells were incubated in serum‐free medium and then stimulated with 10% FBS (v/v). PI‐PLC inhibitor ET‐18‐OCH3 (10 µM), MEK inhibitor PD 98059 (20 µM) or PI3K‐inhibitor LY 294002 (25 µM) were added 6 h after addition of FBS. Cells were either harvested at 16 h after addition of FBS, stained with propidium iodide and assessed for cell cycle distribution by flow cytometric analysis. (A), or incubated in the presence of BrdU (10 µM) for 1 h and then analyzed for the level of BrdU incorporation as described in Materials and methods section (B). Results are means ± S.E.M. for three different experiments. *P < 0.05 (Student’s t‐test) with respect to the control.
4. Discussion
Early studies investigating the changes in the phospholipids levels that are specific for a particular phase of the cell cycle pointed to the beginning of S phase as the increase in the level of nuclear DAG was detected somewhere before the onset of DNA synthesis in the regenerating rat liver (20) or a decrease in nuclear PtdIns levels was measured in HeLa cells at S phase (21). A recent analysis of PI‐PLC isoforms in the nuclei of regenerating rat liver showed two peaks of the enzyme activity that preceded the increase in [methyl‐³H]thymidine incorporation into DNA; the activity of PI‐PLCβ1b, an isoform that accounts for 60% of the total PLC‐activity in the membrane‐depleted nuclei was found to increase at 6 and 20 h after partial hepatectomy (22). In HL‐60 cells, a pulse of nuclear DAG generation was detected in HL‐60 cells 8 h after aphidicolin block which corresponds to G2 phase of the cell cycle (15). When HL‐60 cells were blocked in G2/M phase by nocodazole and released into a fresh medium, two peaks of the PI‐PLC activity were measured at 1 and 8.5 h after release that were due to MEK‐inhibitor‐sensitive serine phosphorylation of the enzyme (16). In present study, the activation of PI‐PLC was investigated in serum‐stimulated HL‐60 cells. Two peaks of the nuclear PI‐PLC activity occurs at 0.5 and 11.5 h after serum‐stimulation; both peaks are associated with serine‐phosphorylation of PI‐PLCβ1b and inhibited by PI‐PLC and MEK inhibitors.
An early (30 min) increase in the activity of PI‐PLC in HL‐60 cells stimulated with serum was expected as the previous study demonstrated an increase in the level of nuclear DAG in HL‐60 cells at 30 min after addition of IGF, although the isoform that was responsible for the increase in the level of DAG was not investigated (14). However, the increase in the level of DAG was sensitive to the presence of ET‐18‐OCH3 similar to the effects of the inhibitor on the increase in nuclear DAG levels in aphidicolin‐synchronized cells (15), the increase in the activity of PI‐PLC described in nocodazole (16) or serum‐treated cells in present study. Our previous study confirmed the presence of both splice variants of PLCβ1 in HL‐60 cells; PLCβ1a was predominantly cytosolic, PLCβ1b was more abundant in nuclei, and the later variant was responsible for two peaks of PI‐PLC activities in nocodazole‐synchronized cells (16). The mechanism of PLCβ1b activation involved a PD 98059‐sensitive phosphorylation of nuclear enzyme on serine (16) similar to the one described in IGF‐treated Swiss 3T3 (6), insulin‐stimulated NIH3T3 (7), IL‐2 treated NK cells (8), and rat liver nuclei isolated 6 h after partial hepatectomy (22). The results of the present study show that two peaks of PD 98059‐sensitive PI‐PLC activity in serum‐stimulated HL‐60 cells correspond to the increase in serine phosphorylation of nuclear PI‐PLCβ1b splice variant.
Signaling events detected early after mitogen‐mediated stimulation of serum‐starved cells correspond to G0/G1 transition phase of the cell cycle. The second peak of PI‐PLC activity measured at 11 h in serum‐stimulated cells corresponds to the peak of the PLC activity detected at 8.5 h in continuously cycling nocodazole‐released cells (16). Previous studies performed on clones of MEL cells transfected with PI‐PLCβ splice variants and a mutant lacking the nuclear localization signal (M2b clones) confirmed the role of the nuclear PI‐PLCβ1 in the G1 phase progression (9). MEL clones overexpressing PI‐PLCβ1a and PI‐PLCβ1b grew in the absence of serum, had an increase in the level of cyclinD/cdk4, phosphorylation of retinoblastoma protein (Rb) on Ser‐795, activation of E2F (9) and an increase in the expression of CD24 surface marker (23), and none of these effects were observed in M2b clones. Although PLC M2b clones show 3‐fold higher PLC activity in cytoplasm, the nuclear PLC activity in M2b did not differ from the nuclear activity in wild type cells and the presence of the endogenous nuclear enzyme is sufficient to allow normal growth of clones in the presence of serum (9) or normal expression of CD24 (23).
Neither the use of siRNA or transfected cells can distinguish the possible roles of two temporally distinct peaks of the activity in the progression of cells through the G1 phase. ET‐18‐OCH3, a nonspecific inhibitor of PI‐PLC that has a toxic effect on HL‐60 cells incubated in medium lacking serum (24) and increases a percentage of sub‐G1 events (25), and PD 98059, a specific inhibitor of MEK (26) that non‐specifically inhibits all downstream effects mediated by MAPK pathway, were used to dissect the possible physiological role of the second peak of the nuclear PI‐PLC activity in G1 progression. The addition of inhibitors immediately after the serum had inhibitory effects on the number of cells entering S phase. When the addition of the inhibitors was delayed so that only the second peak of the activity could be inhibited, the proportion of cells in different phases of the cell cycle was similar to the one observed in cells treated continuously with the inhibitors suggesting that the second peak of the nuclear PI‐PLC activity is equally important for the progression of the cells through G1 phase of the cell cycle.
It is less clear how the various products of PLC activation might interact with the cell cycle machinery to cause G1 progression. Putative targets of the nuclear DAG involve several PKC isoforms that were shown to translocate to nuclei (14, 27–29), and phosphorylate lamins (30, 31). The phosphorylation of the nuclear Ins(1,4,5)P3 is known to generate nuclear inositol phosphates that have been described to regulate mRNA export from the nucleus (32), transcription (33), telomere length (34) and RNA editing (35). Recently, the connection between Rb protein, a key regulator of G1/S phase transition, and nuclear phospholipid signaling has been described as pRb was shown to regulate the levels of both the nuclear PtdIns(4,5)P2 through its interaction with PtdInsP(4)P 5‐kinase (36) and the level of the nuclear DAG by activation of the nuclear DAG kinase (37).
Altogether, the results of the present study show that serum‐mediated re‐entry of starving HL‐60 cells into the cell cycle is associated with two peaks of the nuclear PI‐PLC activity; an early peak that occurs 30 min after the addition of serum and that was previously described in IGF‐stimulated cells, and the late increase in the nuclear PI‐PLC activity that occurs at 11 h and corresponds to late G1 phase PLC activation observed in nocodazole‐synchronized cells. As described in nocodazole‐synchronized cells, the mechanism of the PI‐PLC activation involves serine‐mediated phosphorylation of PI‐PLCβ1b splice variant. Both peaks of the activities of nuclear PI‐PLC are shown to be important for serum‐stimulated progression through G1 into S phase of the cell cycle further confirming hypothesis that two temporally distinct waves of mitogenic signaling occurs in cells stimulated to proliferate. Our results show that temporally distinct activation of several signaling pathways occurs not only at the cell membrane, but involves the activation of autonomous nuclear phospholipase, too.
Acknowledgements
We thank Alan P. Fields for valuable help and advice. This work was supported by the Ministry for Science of the Republic of Croatia (to D. V. and H. B.), and by the Fogarty International Research Collaboration Award (to H. B.)
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Irvine RF. Nuclear lipid signalling. Nat. Rev. Mol. Cell Biol. 2003;4:349–360. doi: 10.1038/nrm1100. [DOI] [PubMed] [Google Scholar]
- [2].Cocco L, Faenza I, Fiume R, Billi AM, Gilmour RS, Manzoli FA. Phosphoinositide‐specific phospholipase C (PI‐PLC) beta1 and nuclear lipid‐dependent signalling. Biochim. Biophys. Acta. 2006;1761:509–521. doi: 10.1016/j.bbalip.2006.03.001. [DOI] [PubMed] [Google Scholar]
- [3].Martelli AM, Gilmour RS, Bertagnolo V, Neri LM, Manzoli L, Cocco L. Nuclear localization and signalling activity of phosphoinositidase C beta in Swiss 3T3 cells. Nature. 1992;358:242–245. doi: 10.1038/358242a0. [DOI] [PubMed] [Google Scholar]
- [4].Manzoli L, Billi AM, Rubbini S, Bavelloni A, Faenza I, Gilmour RS, Rhee SG, Cocco L. Essential role for nuclear phospholipase C beta1 in insulin‐like growth factor I‐induced mitogenesis. Cancer Res. 1997;57:2137–2139. [PubMed] [Google Scholar]
- [5].Bahk YY, Lee YH, Lee TG, Seo J, Ryu SH, Suh PG. Two forms of phospholipase C‐beta1 generated by alternative splicing. J. Biol. Chem. 1994;269:8240–8245. [PubMed] [Google Scholar]
- [6].Xu A, Suh PG, Marmy‐Conus N, Pearson RB, Seok OY, Cocco L, Gilmour RS. Phosphorylation of nuclear phospholipase C beta1 by extracellular signal‐regulated kinase mediates the mitogenic action of insulin‐like growth factor I. Mol. Cell. Biol. 2001;21:2981–2990. doi: 10.1128/MCB.21.9.2981-2990.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Martelli AM, Billi AM, Manzoli L, Faenza I, Aluigi M, Falconi M, De Pol A, Gilmour RS, Cocco L. Insulin selectively stimulates nuclear phosphoinositide‐specific phospholipase C (PI‐PLC) beta1 activity through a mitogen‐activated protein (MAP) kinase‐dependent serine phosphorylation. FEBS Lett. 2000;486:230–236. doi: 10.1016/s0014-5793(00)02313-9. [DOI] [PubMed] [Google Scholar]
- [8].Vitale M, Matteucci A, Manzoli L, Rodella L, Mariani AR, Zauli G, Falconi M, Billi AM, Martelli AM, Gilmour RS, Cocco L. Interleukin 2 activates nuclear phospholipase C beta by mitogen‐activated protein kinase‐dependent phosphorylation in human natural killer cells. FASEB J. 2001;15:1789–1791. doi: 10.1096/fj.01-0008fje. [DOI] [PubMed] [Google Scholar]
- [9].Faenza I, Matteucci A, Manzoli L, Billi AM, Aluigi M, Peruzzi D, Vitale M, Castorina S, Suh PG, Cocco L. A role for nuclear phospholipase C beta 1 in cell cycle control. J. Biol. Chem. 2000;275:30520–30524. doi: 10.1074/jbc.M004630200. [DOI] [PubMed] [Google Scholar]
- [10].Jones SM, Kazlauskas A. Growth factor‐dependent signaling and cell cycle progression. FEBS Lett. 2001;490:110–116. doi: 10.1016/s0014-5793(01)02113-5. [DOI] [PubMed] [Google Scholar]
- [11].Kazlauskas A. The priming/completion paradigm to explain growth factor‐dependent cell cycle progression. Growth Factors. 2005;23:203–210. doi: 10.1080/08977190500096020. [DOI] [PubMed] [Google Scholar]
- [12].Jones SM, Klinghoffer R, Prestwich GD, Toker A, Kazlauskas A. PDGF induces an early and a late wave of PI 3‐kinase activity, and only the late wave is required for progression through G1. Curr. Biol. 1999;9:512–521. doi: 10.1016/s0960-9822(99)80235-8. [DOI] [PubMed] [Google Scholar]
- [13].Jones SM, Kazlauskas A. Growth‐factor‐dependent mitogenesis requires two distinct phases of signalling. Nat. Cell. Biol. 2001;3:165–172. doi: 10.1038/35055073. [DOI] [PubMed] [Google Scholar]
- [14].Neri LM, Bortul R, Borgatti P, Tabellini G, Baldini G, Capitani S, Martelli AM. Proliferating or differentiating stimuli act on different lipid‐dependent signaling pathways in nuclei of human leukemia cells. Mol. Biol. Cell. 2002;13:947–964. doi: 10.1091/mbc.01-02-0086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Sun B, Murray NR, Fields AP. A role for nuclear phosphatidylinositol‐specific phospholipase C in the G2/M phase transition. J. Biol. Chem. 1997;272:26313–26317. doi: 10.1074/jbc.272.42.26313. [DOI] [PubMed] [Google Scholar]
- [16].Lukinovic‐Skudar V, Donlagic L, Banfic H, Visnjic D. Nuclear phospholipase C‐beta1b activation during G2/M and late G1 phase in nocodazole‐synchronized HL‐60 cells. Biochim. Biophys. Acta. 2005;1733:148–156. doi: 10.1016/j.bbalip.2004.12.009. [DOI] [PubMed] [Google Scholar]
- [17].Visnjic D, Crljen V, Curic J, Batinic D, Volinia S, Banfic H. The activation of nuclear phosphoinositide 3‐kinase C2beta in all‐trans‐retinoic acid‐differentiated HL‐60 cells. FEBS Lett. 2002;529:268–274. doi: 10.1016/s0014-5793(02)03357-4. [DOI] [PubMed] [Google Scholar]
- [18].Lee SB, Rhee SG. Molecular cloning, splice variants, expression, and purification of phospholipase C‐delta 4. J. Biol. Chem. 1996;271:25–31. doi: 10.1074/jbc.271.1.25. [DOI] [PubMed] [Google Scholar]
- [19].Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- [20].Banfic H, Zizak M, Divecha N, Irvine RF. Nuclear diacylglycerol is increased during cell proliferation in vivo. Biochem. J. 1993;290:633–636. doi: 10.1042/bj2900633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].York JD, Majerus P. Nuclear phosphatidylinositols decrease during S phase of the cell cycle in HeLa cells. J. Biol. Chem. 1994;269:7847–7850. [PubMed] [Google Scholar]
- [22].Crljen V, Visnjic D, Banfic H. Presence of different phospholipase C isoforms in the nucleus and their activation during compensatory liver growth. FEBS Lett. 2004;571:35–42. doi: 10.1016/j.febslet.2004.06.051. [DOI] [PubMed] [Google Scholar]
- [23].Fiume R, Faenza I, Matteucci A, Astolfi A, Vitale M, Martelli AM, Cocco L. Nuclear phospholipase C beta1 (PLCbeta1) affects CD24 expression in murine erythroleukemia cells. J. Biol. Chem. 2005;280:24221–24226. doi: 10.1074/jbc.M411833200. [DOI] [PubMed] [Google Scholar]
- [24].Heesbeen EC, Rijksen G, van Heugten HG, Verdonck LF. Influence of serum levels on leukemic cell destruction by the ether lipid ET‐18‐OCH3. Leuk. Res. 1995;19:417–425. doi: 10.1016/0145-2126(95)00008-c. [DOI] [PubMed] [Google Scholar]
- [25].Gajate C, Molinedo F. The antitumor ether lipid ET‐18‐OCH3 induces apoptosis through translocation and capping of Fas/CD95 into membrane rafts in human leukemic cells. Blood. 2001;98:3860–3863. doi: 10.1182/blood.v98.13.3860. [DOI] [PubMed] [Google Scholar]
- [26].Gille H, Downward J. Multiple Ras effector pathways contribute to G1 cell cycle progression. J. Biol. Chem. 1999;274:22033–22040. doi: 10.1074/jbc.274.31.22033. [DOI] [PubMed] [Google Scholar]
- [27].Divecha N, Banfic H, Irvine RF. The polyphosphoinositide cycle exists in the nuclei of Swiss 3T3 cells under the control of a receptor (for IGF‐I) in the plasma membrane, and stimulation of the cycle increases nuclear diacylglycerol and apparently induces translocation of protein kinase C to the nucleus. EMBO J. 1991;10:3207–3214. doi: 10.1002/j.1460-2075.1991.tb04883.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Xu A, Wang Y, Xu LY, Gilmour RS. Protein kinase C alpha‐mediated negative feedback regulation is responsible for the termination of insulin‐like growth factor I‐induced activation of nuclear phospholipase C beta1 in Swiss 3T3 cells. J. Biol. Chem. 1999;276:14980–14986. doi: 10.1074/jbc.M009144200. [DOI] [PubMed] [Google Scholar]
- [29].Martelli AM, Evangelisti C, Nyakern M, Manzoli FA. Nuclear protein kinase C. Biochim. Biophys. Acta. 2006;1761:542–551. doi: 10.1016/j.bbalip.2006.02.009. [DOI] [PubMed] [Google Scholar]
- [30].Goss VL, Hocevar BA, Thompson LJ, Stratton CA, Burns DJ, Fields AP. Identification of nuclear beta II protein kinase C as a mitotic lamin kinase. J. Biol. Chem. 1994;269:19074–19080. [PubMed] [Google Scholar]
- [31].Thompson LJ, Fields AP. Beta II protein kinase C is required for the G2/M phase transition of cell cycle. J. Biol. Chem. 1996;271:15045–15053. doi: 10.1074/jbc.271.25.15045. [DOI] [PubMed] [Google Scholar]
- [32].York JD, Odom AR, Murphy R, Ives EB, Wente SR. A phospholipase C‐dependent inositol polyphosphate kinase pathway required for efficient messenger RNA export. Science. 1999;285:96–100. doi: 10.1126/science.285.5424.96. [DOI] [PubMed] [Google Scholar]
- [33].Odom AR, Stahlberg A, Wente SR, York JD. A role for nuclear inositol 1,4,5‐triphosphate kinase in transcriptional control. Science. 2000;287:2026–2029. doi: 10.1126/science.287.5460.2026. [DOI] [PubMed] [Google Scholar]
- [34].York SJ, Armbruster BN, Greenwell P, Petes TD, York JD. Inositol diphosphate signaling regulates telomere length. J. Biol. Chem. 2005;280:4264–4269. doi: 10.1074/jbc.M412070200. [DOI] [PubMed] [Google Scholar]
- [35].Macbeth MR, Schubert HL, Vandemark AP, Lingam AT, Hill CP, Bass BL. Inositol hexakisphosphate is bound in the ADAR2 core and required for RNA editing. Science. 2005;309:1534–1539. doi: 10.1126/science.1113150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Divecha N, Roefs M, Los A, Halstead J, Bannister RA, D’Santos C. Type I PIPkinases interact with and are regulated by the retinoblastoma susceptibility gene product‐pRb. Curr. Biol. 2002;12:582–587. doi: 10.1016/s0960-9822(02)00769-8. [DOI] [PubMed] [Google Scholar]
- [37].Los A, Vinke FP, deVidt J, Topham MK, van Blitterswijk WJ, Divecha N. The retinoblastoma family proteins bind to and activate diacylglycerol kinase ζ. J. Biol. Chem. 2006;281:858–866. doi: 10.1074/jbc.M502693200. [DOI] [PubMed] [Google Scholar]