

Abstract
Amyloid beta protein (Aβ) levels are elevated in the brain of Alzheimer's disease patients. Anti-Aβ antibodies can reverse the histologic and cognitive impairments in mice which overexpress Aβ. Passive immunization appears safer than vaccination and treatment of patients will likely require human rather than xenogenic antibodies. Effective treatment will likely require antibody to cross the blood-brain barrier (BBB). Unfortunately, antibodies typically cross the BBB very poorly and accumulate less well in brain than even albumin, a substance nearly totally excluded from the brain. We compared the ability of two anti-Aβ human monoclonal IgM antibodies, L11.3 and HyL5, to cross the BBB of young CD-1 mice to that of young and aged SAMP8 mice. The SAMP8 mouse has a spontaneous mutation that induces an age-related, Aβ-dependent cognitive deficit. There was preferential uptake of intravenously administered L11.3 in comparison to HyL5, albumin, and a control human monoclonal IgM (RF), especially by hippocampus and olfactory bulb in aged SAMP8 mice. Injection of L11.3 into the brains of aged SAMP8 mice reversed both learning and memory impairments in aged SAMP8 mice, whereas IgG and IgM controls were ineffective. Pharmacokinetic analysis predicted that an intravenous dose 1000 times higher than the brain injection dose would reverse cognitive impairments. This predicted intravenous dose reversed the impairment in learning, but not memory, in the aged SAMP8 mice. It conclusion, an IgM antibody was produced that crosses the BBB to reverse cognitive impairment in a murine model of Alzheimer's disease.
Keywords: Alzheimer's disease, amyloid beta protein, therapeutics, monoclonal antibody, blood-brain barrier, IgM, passive immunization, cognition
The amyloid hypothesis (Hardy and Selkoe, 2002) states that amyloid beta protein (Aβ) plays a causal role in Alzheimer's disease (AD). Aβ is a major component of the amyloid plaques (Glenner and Wong, 1984) that are one of the hallmarks of AD. Numerous studies have shown that administration of Aβ directly into brain is neurotoxic and induces cognitive impairments (Watt, Pike et al., 1994; Morley, Farr et al., 2002b). Rodents that overexpress amyloid precursor peptide and so have elevated levels of Aβ, whether transgenic such as the APPsw or spontaneous mutations such as the SAMP8, have impairments in cognition, an altered cholinergic system, increased oxidative stress, and histologic changes, all reminiscent of AD (Flood, Morley et al., 1993; Hsiao, Chapman et al., 1996; Frautschy, Yang et al., 1998; Chen, Chen et al., 2000; Morley, 2002; Poon, Castegna et al., 2004).
The identification of Aβ as playing a major role in the pathogenesis of AD has led to using Aβ as a therapeutic target. Some therapies such as breaker peptides, antibodies, and antisense directly target Aβ (Bard, Cannon et al., 2000; Banks, Farr et al., 2001a; DeMattos, Bales et al., 2002; Permanne, Adessi et al., 2002; Boules, Williams et al., 2004) whereas others such as antioxidants and anti-inflammatory agents target the downstream effects of elevated Aβ levels (Butterfield, 2002; Farr, Poon et al., 2003; Townsend and Pratico, 2005). One of the currently most popular therapeutic approaches has been the use of antibodies which target Aβ (Schenk, Barbour et al., 1999; Bard, Cannon et al., 2000; Chen, Chen et al., 2000; Janus, Pearson et al., 2000; Morgan, Diamond et al., 2000; DeMattos, Bales et al., 2002; Morley, Farr et al., 2002a; Hock, Konietzko et al., 2003). Initial reports of toxicity with active immunization has shifted interest towards passive immunization (DeMattos, Bales et al., 2002; Pan, Solomon et al., 2002). To the extent that circulating Aβ contributes to brain and cerebrovascular levels of Aβ, antibodies would not have to enter the brain to exert important effects (DeMattos, Bales et al., 2002; Pan, Solomon et al., 2002). However, to bind to amyloid within the CNS, antibodies must negotiate the blood-brain barrier (BBB).
To date, few studies have examined the abilities of Aβ-directed antibodies to enter the brain from the blood (Kozlowski, Sterzl et al., 1992; Poduslo, Curran et al., 1994; Banks, Terrell et al., 2002; Banks, Pagliari et al., 2005). The studies which have examined the ability of Aβ-directed IgG antibodies to cross the BBB found that they did so modestly by using the extracellular pathways (Banks, 2004). Evidence suggests that a brain-to-blood efflux system may exist at the BBB for IgG molecules (Schlachetzki, Zhu et al., 2002); such an efflux system would hinder accumulation of IgG by the CNS.
Passive immunization of AD patients requires production of polyclonal or monoclonal anti-Aβ antibodies. In general, antibodies given as therapy, unless derived from humans, induce an immune response that rapidly eliminates the administered immunoglobulins. Therefore, there is a definite requirement to engineer anti-Aβ human monoclonal antibodies. These can be produced by a variety of molecular engineering methods or by immortalization of human B cells which produce these specific anti-Aβ antibodies. In the present paper, we analyzed the penetration into the brain and the cognitive effects induced in a murine model of AD by two monoclonal IgM anti-Aβ antibodies established recently in our laboratory.
We examined the ability of two targeted IgM molecules, L11.3 and HyL5, to cross the BBB. We compared these IgM antibodies to albumin which crosses the BBB by way of the extracellular pathways (the same pathway previously found to be used by Aβ-directed IgG antibodies), IgG, and a control human IgM antibody, RF, for their abilities to cross the BBB and to reverse the cognitive impairments in a the SAMP8 mouse model of AD.
Materials and Methods
Mice
CD-1 or SAMP8 mice were from our in-house colony housed at the Veterans Affairs Medical Center-St. Louis. All animals were used under protocols approved by our local animal use committee and were conducted in an AAALAC approved facility. The CD-1 is the standard outbred albino laboratory mouse originally derived from stock from Charles Rivers Laboratories (Wilmington, MA). The SAMP8 is used as a model for AD, as it has an age-related overexpression of Aβ that mediates an age-related development of cognitive defects (Flood and Morley, 1998; Morley, Farr et al., 2002b). The cognitive defects are reversed with antibody or antisense directed against Aβ (Banks, Farr et al., 2001a; Morley, Farr et al., 2002a). Our SAMP8 colony has been inbred for 19 years from stock obtained from Dr. Takeda of Kyoto University, Japan. Over this time, the behavioral phenotype has remained stable. Mice were housed on a 12 h light:12 h dark cycle with lights on at 0600. Food and water was available ad libitum. Sentinels from the colony have remained free from pathogens including mycoplasma, salmonella/shigella, ectoparasites, pnenomiavirus, Sendia virus, mouse hepatitis, REO 3 ectomelia, GBVII, and lymphcytic choriomeningitis.
Purification of Human MoAb
Anti-Aβ (Geylis, Kourilov et al., 2005) and RF (Steinitz, Izak et al., 1980) human MoAb's were isolated from cell culture supernatants derived from monoclonal antibody secreting EBV-immortalized lymphoblastoid cell lines. First, the immunoglobulins were precipitated with 50% saturated ammonium sulfate and this fraction was then dissolved in PBS and dialyzed against distilled water. The precipitate was finally solubilized in PBS and sterile-filtered.
Labeling of Antibodies and Albumin
The anti-Aβ monoclonal IgM antibodies L11.3 and HyL5, albumin, or the control IgM antibody RF (5 microg) were radioactively labeled with iodine (1 mCi of 125I or 131I; Perkin-Elmer, Shelton, CT) by the chloramine-T method and purified on a column of G-10 sephadex. Specific activities were approximately 90 Ci/mMol for all proteins.
Measurement of Blood-to-Brain Unidirectional Influx Rate
The blood-to-brain unidirectional influx rate (Ki, in units of microl/g-min) was determined by multiple-time regression analysis (Blasberg, Fenstermacher et al., 1983; Banks and Kastin, 1990). CD-1 (4 mo old) or SAMP8 (4 or 12 mo old) were anesthetized with urethane and the right jugular vein and the left carotid artery exposed. Labeled antibody in a volume of 0.2 ml of lactated Ringer's solution (LR) was injected into the jugular vein and blood from a cut in the carotid artery was obtained 10, 30, 60, 120, or 240 min later; blood was centrifuged at 5,000 g for 10 min and serum obtained. Immediately after taking a blood sample, mice were decapitated, the hippocampus and olfactory bulb dissected from the brain, and the weight and level of radioactivity determined for the hippocampus, olfactory bulb, remainder of the brain, and the serum. The brain region/serum ratio was calculated for the olfactory bulb and for the hippocampus in units of microl/g. The whole brain/serum ratio was calculated by adding the weight and level of radioactivity of the hippocampus and olfactory bulb to those for the remainder of the brain. The brain/serum ratios were plotted against exposure time (in units of min) calculated as previously described (Blasberg, Fenstermacher et al., 1983; Patlak, Blasberg et al., 1983) and the slope of the linear portion of this relation taken as the Ki.
Calculation of Antibody/Albumin and Antibody/Antibody Ratios
Mice were anesthetized and prepared as above and given an injection into the jugular vein containing either a radioactive antibody and albumin or two radioactively labeled antibodies. The test antibody was labeled with 125I and the comparative substance (i.e., albumin or a second antibody) was labeled with 131I. Arterial blood and brain were harvested as above 120 min after the intravenous injection. Brain/serum ratios were calculated as above and the results were expressed as the ratio of antibody to albumin or as the ratio of L11.3 to HyL5.
Calculation of Percent of Injected Dose Taken up per g of Hippocampus
First, the percent of the injected dose of L11.3 present per ml of serum (%Inj/ml) in the 12 mo old SAMP8 mice was calculated. Then, a value of 10 microl was subtracted from the hippocampal/serum ratio for these mice to correct for vascular space. Finally, this corrected hippocampal/serum ratio was multiplied by %Inj/ml and divided by 1000 to yield %Inj/g of hippocampus.
Capillary Depletion
In other mice (n = 4), the relative distribution of L11.3 between the cerebral cortex and capillaries was assessed by the method of Triguero et al (Triguero, Buciak et al., 1990) as modified for mice by Gutierrez (Gutierrez, Banks et al., 1993). Anesthetized mice received an injection into the jugular vein of 0.2 ml of LR-BSA containing 1×106 cpm of radioactively labeled L11.3. Two h later, the abdomen was opened and blood was collected from the abdominal aorta. The thorax was opened, the thoracic descending aorta clamped, the left and right jugular veins severed, and the brain flushed of its intravascular contents by injecting 20 ml lactated Ringer’s solution over 1 min into the left ventricle of the heart. The mouse was decapitated and the brain harvested. The cerebral cortex was isolated and weighed and placed in ice cold physiologic buffer (10 mM HEPES, 141 mM NaCl, 4 mM KCl, 2.8 mM CaCl2, 1 mM MgSO4, 1 mM NaH2PO4, and 10 mM D-glucose adjusted to pH 7.4). The cortex was then homogenized using a glass tissue grinder (10 strokes) in 0.8 ml physiologic buffer. Dextran solution, 1.6 ml of a 26% solution in physiologic buffer, was added to the homogenate, mixed vigorously, and homogenized (3 strokes). The homogenate was centrifuged at 5400 × g for 15 min at 4°C in a swing bucket rotor. The pellet which contains the brain vasculature and the supernatant which contains the brain parenchyma were carefully separated and the radioactivity of each component determined using a gamma counter. The parenchyma /serum and capillary/serum ratios (ul/g) were calculated by the equation:
where cpm Fr is the cpm in either the parenchyma or supernatant fraction, w is the weight of the cortex, and cpm/ µl serum is the level cpm in a µl of serum.
Stability of L11.3 in Blood and Brain as Assessed by Acid Precipitation
Other mice (n = 4) were anesthetized and radioactive L11.3 (3×106 cpm/mouse) injected into the left jugular vein in a volume of 0.2 ml of lactated Ringer’s solution. Serum from the carotid artery and the whole brain were obtained two h later as described above. Serum (50 µl) was mixed thoroughly with 0.25 ml of LR-BSA and 0.25 ml of 30% TCA, centrifuged at 5000 g at 4 °C for 10 min, and the resulting pellet and supernatant counted. Each whole brain was mechanically homogenized in 2 ml of 0.25 M phosphate buffer. The homogenate was centrifuged at 5,000 g for 10 min and the supernatant collected. Brain supernatant (0.25 ml) was vigorously mixed with 0.25 ml of 30% TCA, centrifuged at 5,000 g × 10 min, and the resulting supernatant and pellet collected. To determine degradation of antibody that occurred ex vivo (processing controls), 100 ul of radioactive antibody in lactated Ringer’s and BSA solution was placed on the surface of a nonradioactive mouse brain or in a tube used to obtain carotid blood and the samples processed as above. The results were expressed as the percent of total radioactivity (supernatant cpm + pellet cpm) that was found in the pellet. Results were corrected for degradation during processing by dividing the percent precipitated for the biological samples by the percent precipitated in the processing controls and multiplying by 100.
Measurement of Acquisition and Retention
A dose response curve for L11.3 was constructed following its intracerebroventricular (icv) injection in 12 mo old SAMP8 mice. Forty-eight hours prior to testing, the mice were anesthetized with 2,2,2-tribromoethanol, placed in a stereotaxic instrument, and the scalp was deflected. A unilateral hole was drilled 0.5 mm anterior to and 1.0 mm to the right of the bregma. Twenty four hours prior to training, the mice were again placed under light anesthesia, and received an inject into the lateral ventricle of 0.5 µl of either rabbit anti-mouse IgG (Sigma, St. Louis, MO), L11.3, or RF.
We calculated an approximate intravenous dose of L11.3 based on the %Inj/g and the icv dose needed to affect acquisition and retention. This dose of L11.3 or rabbit antimouse IgG was given by tail vein to 12 mo old SAMP8 mice in a volume of 0.1 ml to lightly anesthetized mice 24 h prior to training.
Twenty four hours after the iv or icv injection of antibody, 12 mo old mice were trained in T-maze footshock avoidance. Mice were trained and tested between 0700 and 1500 hours. The T-maze consisted of a black plastic alley with a start box at one end and two goal boxes at the other. The start box was separated from the alley by a plastic guillotine door, which prevented movement down the alley until training began. An electrifiable stainless steel rod floor ran throughout the maze to deliver scrambled footshock. Mice were not permitted to explore the maze prior to training. A block of training trials began when a mouse was placed into the start box. The guillotine door was raised and a buzzer sounded simultaneously; 5 sec later footshock was applied. The goal box entered on the first trial was designated "incorrect" and the footshock was continued until the mouse entered the other goal box, which in all subsequent trials was designated as "correct" for the particular mouse. At the end of each trial, the mouse was returned to its home cage until the next trial.
Mice were trained until they made one avoidance. Training used an intertrial interval of 60 sec, the buzzer was of the door-bell type, sounded at 55 dB and shock was set at 0.35 mA (Coulbourn Instruments scrambled grid floor shocker model E13-08). Retention was tested one week later by continuing training until mice reach a criterion (5 avoidances in 6 consecutive trials). The results were reported as the number of trails to first avoidance for acquisition and number of trials to criterion for the retention test.
Statistics
Means are presented with their standard error and the number of mice in the statistical cell (n). Means were compared by analysis of variance followed by Newman-Keuls range test. Antibody/albumin ratios were compared to the theoretical value of 1.0 with the software in Prism 4.0 (GraphPad, Inc., San Diego, CA). Regression lines were calculated by the least squares method and compared statistically with the software package in Prism 4.0.
Results
Blood-to-Brain Uptake Rates
Figure 1 shows representative curves for CD-1 mice given L11.3 antibody. Table 1 summarizes the results for brain uptake of L11.3 and HL5 in CD-1 and SAMP8 mice. It shows Ki values for those groups that had a statistically significant relation between brain/serum ratios and exposure time. By this criterion, L11.3 had many more occurrences of statistically significant uptakes than did HyL5. Among the statistically significant relations for L11.3, whole brain in CD-1 mice had the slowest uptake and olfactory bulb in 12 mo old SAMP8 mice had the fastest rate.
Figure 1.
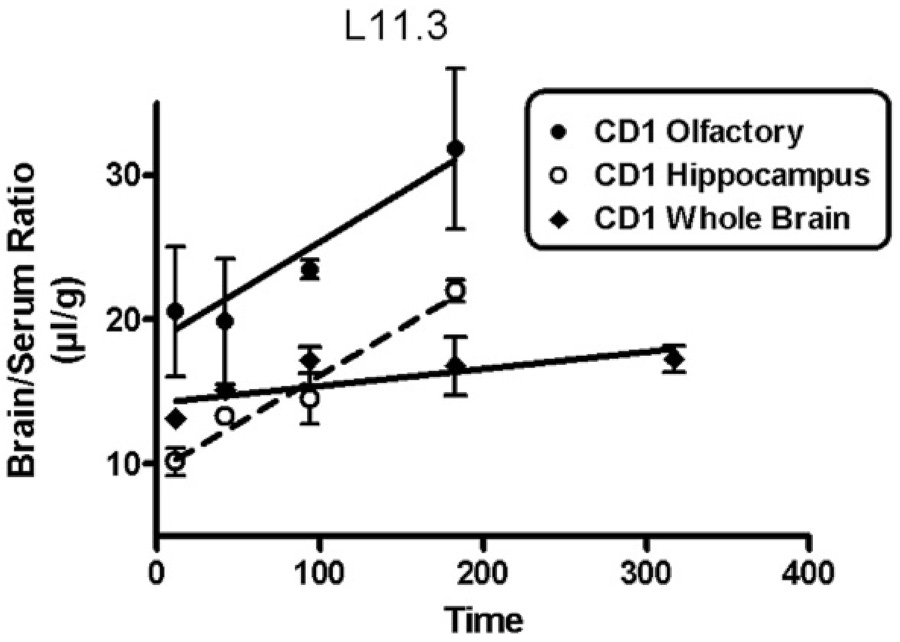
Representative blood-to-brain uptake curves for L11.3 in CD-1 mice. All three regions showed a statistically significant uptake of L11.3. See Table 1 for Ki and statistical parameters.
Table 1.
Antibody Transport Rates into Various Brain Regions in CD-1 and SAMP8 mice. Ki (± SE of the mean) and p values for significant relations between brain/serum ratios and exposure time. n = number of mice on which analysis is based. OB= olfactory bulb; Hippo= Hippocampus; WBr = Whole Brain. NS = nonsignificant
| Antibody L11.3 | ||||
|---|---|---|---|---|
| Strain | Region | Ki (µl/g-min) | p value | n |
| CD-1 | OB | 0.0686 ± 0.0314 | 0.048 | 15 |
| Hippo | 0.0660 ± 0.0082 | <0.0001 | 14 | |
| W Br | 0.0118 ± 0.0052 | 0.036 | 20 | |
| SAM 4mo | OB | NS | ||
| Hippo | 0.0558 ± 0.014 | 0.0019 | 13 | |
| W Br | 0.0231 ± 0.0099 | 0.0367 | 15 | |
| SAM 12 mo | OB | 0.106 ± 0.0478 | 0.047 | 14 |
| Hippo | 0.0714 ± 0.0290 | 0.0316 | 13 | |
| W Br | 0.0253 ± 0.0098 | 0.0205 | 17 | |
| Antibody HYL5 | ||||
| Strain | Region | Ki (µl/g-min) | p value | n |
| CD-1 | OB | 0.074 ± 0.028 | .026 | 12 |
| Hippo | NS | |||
| W Br | NS | |||
| SAM 4mo | OB | NS | ||
| Hippo | 0.0295 ± 0.011 | 0.0204 | 13 | |
| W Br | NS | |||
| SAM 12 mo | OB | NS | ||
| Hippo | NS | |||
| W Br | NS | |||
Clearance from Blood
Clearance from blood was biphasic for both the L11.3 and HL5 antibodies and in all 3 groups of mice (CD-1, young SAMP8, aged SAMP8). Blood levels were higher for HyL5 than for L11.3 and CD-1 mice tended to have lower blood levels than 12 mo old SAMP8 which tended to have blood levels a little lower than 4 mo old. Representative curves are shown in Figure 2.
Figure 2.
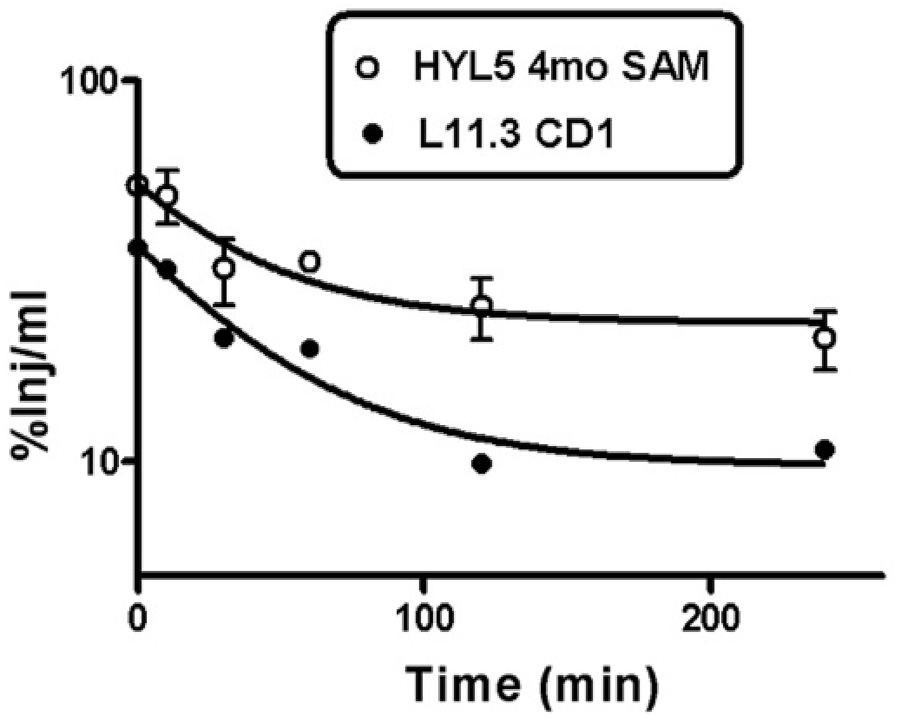
Clearance of antibody from blood. The highest values for %Inj/ml were found in 4 mo old SAMP8 mice given HyL5 and the lowest values in CD-1 mice given L11.3. All other clearance curves fell between these two curves.
Comparison of Antibodies and Albumin
The uptakes by brain of L11.3 and HyL5 were compared by co-injecting them together or individually with albumin or the control IgM antibody RF. Blood and brain were obtained 120 min after injection and the brain/serum ratios expressed as a ratio to the reference substance. Values greater than 1.0 demonstrated selective uptake relative to the reference substance. Figure 3 shows that L11.3/albumin (upper left panel) ratios tended to be higher than HyL5/albumin ratios (upper right panel) and that ratios tended to be higher in aged SAMP8 mice. Two way ANOVAs showed no differences among brain regions or interactions but did show differences among mouse groups for the L11.3/albumin ratios [F(2,81) = 4.95, p<0.01] and for the HyL5 ratios [F(2,81) = 5.24, p<0.01]. When compared to a theoretical value of 1.0, L11.3/albumin ratios for total brain in all three groups of mice and for hippocampus in 4 and 12 mo old SAMP8 mice were statistically significant (p<0.05). None of the HyL5/albumin ratios were statistically greater than 1.0. In other 12 mo old SAMP8 mice, the L11.3/HyL5 ratio (Figure 3, lower right panel) was greater than 1.0 for hippocampus and for olfactory bulb (p<0.05). The RF/albumin ratios (Figure 3, lower left panel) showed statistical differences for brain region, mouse group, and interactions all at p<0.05, but the trends tended to be opposite those seen with L11.3, with 12 mo old SAMP8 having lower RF/albumin ratios. Only the 4 mo old SAMP8 total brain value was greater than 1.0. In 12 mo old SAMP8 mice, L11.3 had a preferential penetration compared to HyL5 into olfactory bulb and hippocampus at p<0.05 (Figure 3, lower right panel).
Figure 3.
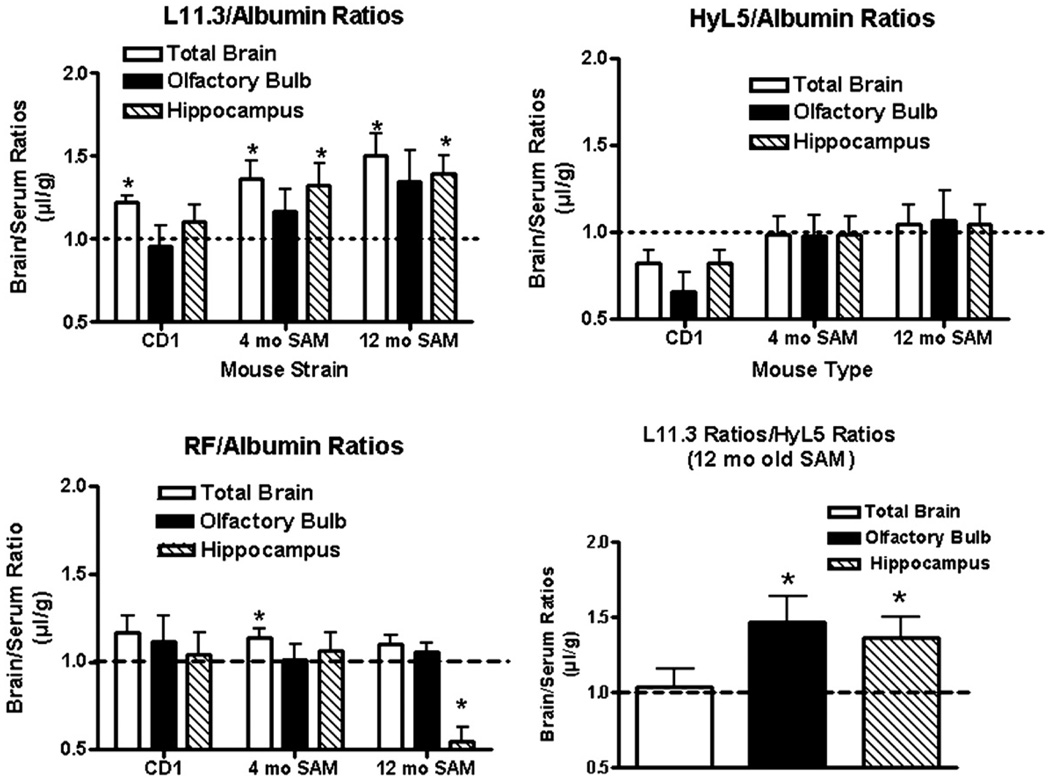
Comparison of antibodies and albumin. The brain/serum ratios for L11.3 (upper left panel), HyL5 (upper right panel), and control IgM antibody RF (lower left panel) were divided by the brain/serum ratios for simultaneously injected albumin. The lower right panel shows the brain/serum ratios for L11.3 divided by the brain/serum ratios for simultaneously injected HyL5. Data were collected 2h after the simultaneous iv injection of an antibody and albumin so that a ratio of greater than 1.0 indicates an uptake in excess of that for albumin. *indicates value different from the theoretical value of 1.0 at p<0.05 level.
Capillary Depletion
We performed capillary depletion to determine whether the L11.3 was sequestered by capillaries as opposed to crossing the full width of the capillary wall to enter brain interstitial fluid. We found that the parenchyma/serum ratio was 2.49 ± 0.64 µl/g, while the capillary/serum ratio was 0.03 ± 0.01 µl/g. Therefore, 98.8% of L11.3 taken up by brain had entered its parenchymal space.
Stability of L11.3 in Blood and Brain as Assessed by Acid Precipitation
No degradation of L11.3 was found in blood 2 h after its injection. For brain, 89.6 ± 2.5% of the radioactivity recovered precipitated with acid.
Acquisition and Retention
The upper panel of figure 4 shows a dose response curve for the effect of icv administered of L11.3 on acquisition (learning) and retention (memory). The ANOVA for acquisition showed a significant effect [F(4,35) = 21.3, p<<0.01] and the Newman-Keuls posttest showed that the 24 ng and 240 ng doses significantly improved learning, both at the p<<0.01 level when compared to either the IgG or RF antibody controls. The ANOVA for retention showed a significant effect [F(4,32) = 9.39, p<0.01] and the Newman-Keuls post-test showed that the 24 ng (p<0.05) and 240 ng (p<0.01) doses significantly improved memory when compared to either the of the control antibodies.
Figure 4.
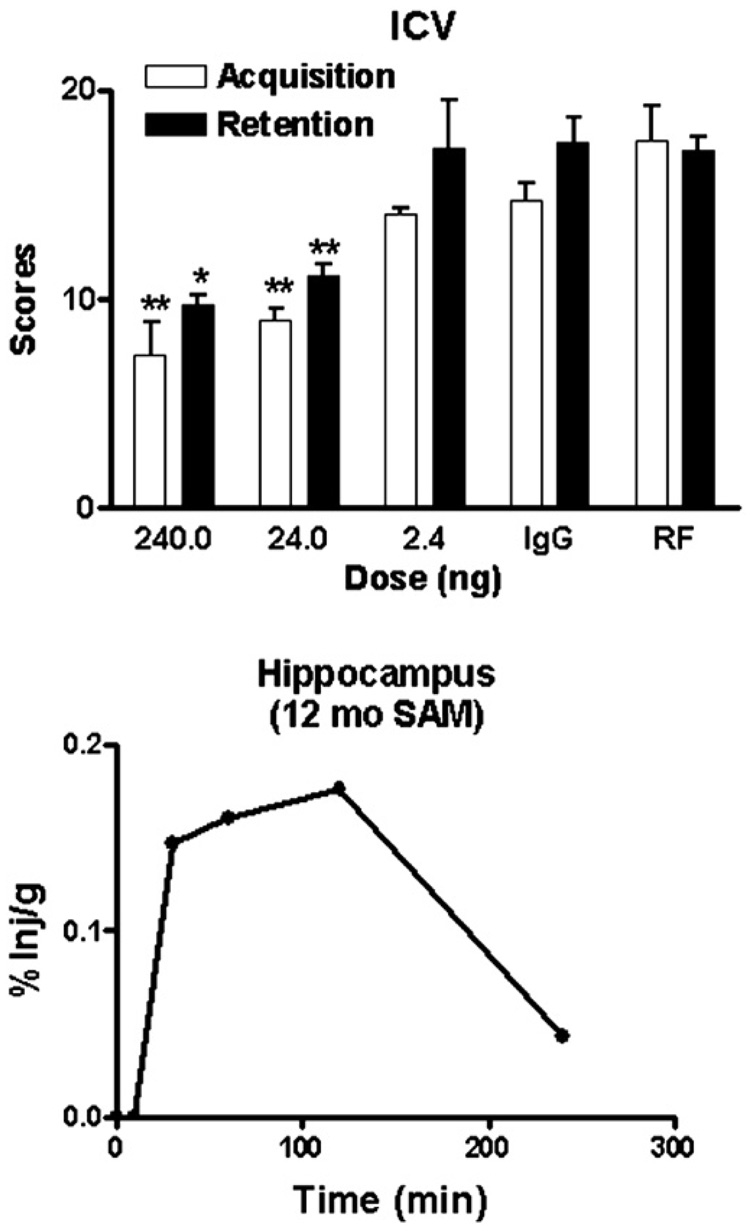
Upper panel shows a dose response curve for the effect of L11.3 given by intracerebroventricular administration on acquisition and retention in 12 mo old SAMP8 mice. Both acquisition and retention were improved (* = p<0.05;** = p<0.01) by the 24 and 240 ng dose of Ly11.3 when compared to administration to either the IgG or RF antibody controls. These doses were also significantly different from the 2.4 ng dose (p<0.05).
The lower panel of figure 4 shows the percent of an iv injection of L11.3 calculated to be taken up by the hippocampus in a 12 mo old SAMP8 mouse. Based on previous examples with similar levels of hippocampal uptake (Uchida, Arimura et al., 1996; Banks, Farr et al., 2001a; Banks, Farr et al., 2001b), we predicted that an iv dose of about 100–1000 times that of the icv dose of L11.3 would be needed to have an effect on learning and memory. As figure 5 shows, an iv dose of 25 microg/mouse improved acquisition [F(3,24) = 17.5, p<<0.01] but not retention in 12 mo old SAMP8 mice. For acquisition, treatment with L11.3 returned acquisition to those levels seen in young 4 mo old SAMP8 mice.
Figure 5.
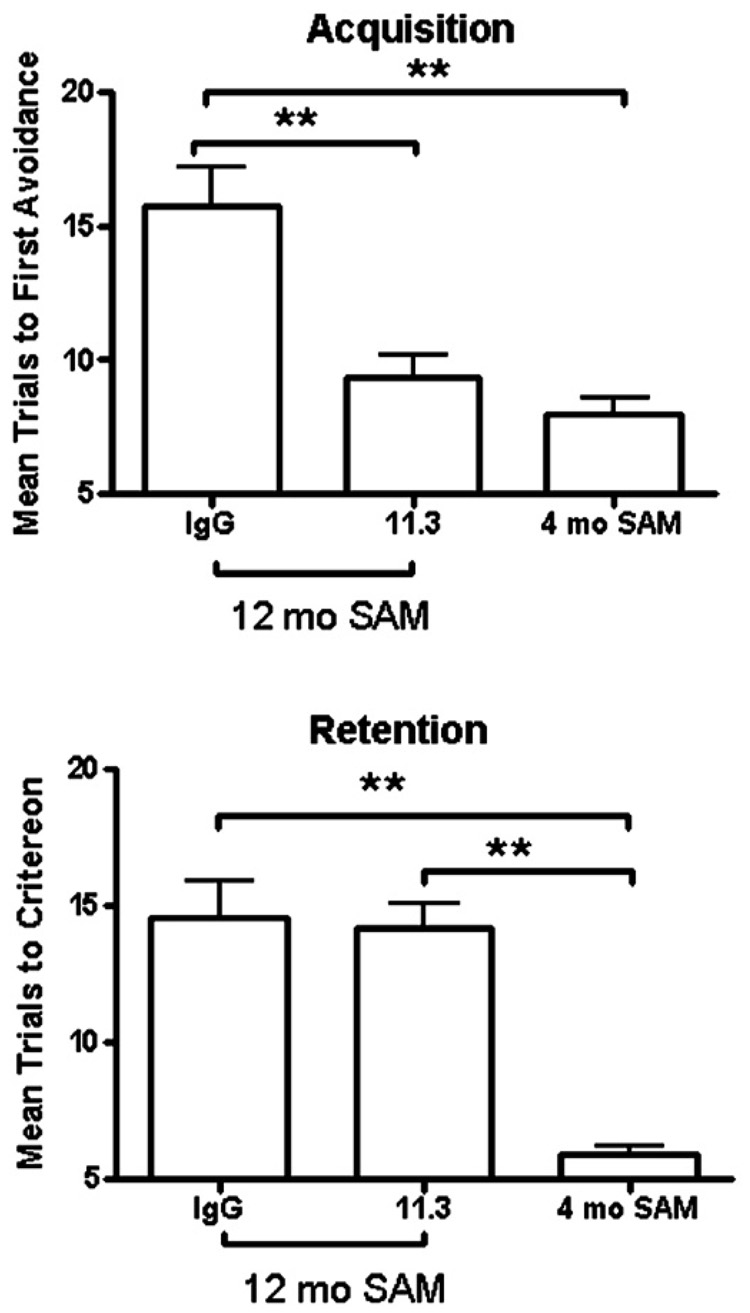
Effect of iv L11.3 (25 microg/mouse) on acquisition and retention in 12 mo old SAMP8 mice. Acquisition, but not retention, was improved by L11.3. 4 mo old SAMP8 mice are shown for comparison.
Discussion
Immunotherapy shows promise as a treatment for Alzheimer's disease. However, it is probable that circulating antibody must enter the brain in order to be effective. This would require the antibody to negotiate the BBB. Little work has been done on the abilities of antibodies to cross the BBB, and most of that has used IgG antibodies. In general, penetration of the BBB by IgG is poor and accumulation is low (Kozlowski, Sterzl et al., 1992; Poduslo, Curran et al., 1994; Banks, Terrell et al., 2002; Banks, Pagliari et al., 2005). This is in part because the IgG antibodies studied to date have no saturable blood-to-brain transport system and so must rely on the extracellular pathways to enter the CNS (Banks, 2004). It may also be in part because of a brain-to-blood transport of IgG antibodies (Schlachetzki, Zhu et al., 2002).
It should not be assumed that all antibodies will behave as do the IgG antibodies studied to date. For example, evidence suggests that Fab can cross the BBB (Chekhonin, Kabanov et al., 1991; Broadwell, Bakder et al., 1994). Here, we studied two human IgM antibodies specifically raised against AβP. The monoclonal antibodies were engendered by two lymphoblastoid cell lines derived from Ebstein-Barr virus (EBV)-immortalized peripheral blood lymphocytes. The fact that both are IgM antibodies is probably due to the fact that in blood, IgM positive cells constitute the vast majority of the B lymphocytes. Our major findings show that one of the antibodies crossed the BBB to a greater degree than the other AβP-directed antibody, albumin, or a control human IgM antibody and that it could reverse learning impairments in an animal model of Alzheimer's disease after administration either directly into brain or into blood.
We first measured the ability of the two AβP-directed IgM antibodies, L11.3 and HyL5, to cross the BBB of the whole brain as well as at the olfactory bulb and hippocampus. The olfactory bulb and hippocampus are the two brain regions found to be capable of neurogenesis in the adult (Elder, De Gasperi et al., 2006) and so an interesting therapeutic target. The olfactory bulb, like the hippocampus, is also an area of early changes in Alzheimer's disease(Christen-Zaech, Kraftsik et al., 2003; Attems and Jellinger, 2006) and a seat of learning and memory in the rodent (Hallam, Horgan et al., 2004). We used multiple-time regression analysis to measure the CNS uptake of the radioactively labeled antibodies after their intravenous injection. This method is very sensitive and can often detect the uptake of even albumin when studied over a 4 h time-course as we did here. The unidirectional influx rate, or Ki, into whole brain as measured for albumin by this method is typically 0.005–0.01 µl/g-min (Blasberg, Fenstermacher et al., 1983; Patlak, Blasberg et al., 1983). The L11.3 antibody exceeded these values for whole brain in young CD-1 mice and even more so for olfactory bulb and hippocampus, whereas HyL5 uptake was only measurable in the olfactory bulb of CD-1 mice (Table 1).
Uptake of L11.3, but not of HyL5, was greater in the aged SAMP8 mice than in the CD-1 or 4 mo old SAMP8 mice. The SAMP8 mouse has a spontaneous mutation and has an age-related overexpression of APP, elevated levels of AβP, and cognitive defects (Morley, 2002; Morley, Farr et al., 2002b). At 4 mo of age, the SAMP8 has brain levels of APP, AβP, and learning abilities similar to the control CD-1 mice. The 12 mo old SAMP8 mouse has APP and AβP levels that are about twice that of the 4 mo old SAMP8 and has developed severe learning and memory problems (Kumar, Farr et al., 2000; Morley, Kumar et al., 2000). These learning and memory problems can be reversed by treating mice with either antibody or antisense directed against AβP (Kumar, Farr et al., 2000; Morley, Farr et al., 2002a). Thus, the SAMP8 represents a model of age-related, AβP-dependent cognitive decline. We postulate that the increase in uptake of L11.3 by aged SAMP8 is a result of those mice having more AβP which can bind and retain the antibody. This is also consistent with the higher uptakes by hippocampus, an area important in learning and memory, and the olfactory bulb, an area of increased permeability in the SAMP8 mouse (Ueno, Dobrogowska et al., 1996). Similar retention has been noted for intravenously administered ligands in brain areas that have a high concentration of the ligand's receptor (Weber, Greene et al., 1991; Weber, Greene et al., 1992). The results cannot be explained by disruption of the BBB as such disruption would equally effect albumin and all the antibodies. Furthermore, this strain of SAMP8 mice do not develop regional defects of the BBB with aging (Banks, Farr et al., 2000).
Further evidence for the selectivity of L11.3 uptake was found by co-injecting radioactively labeled albumin and dividing the brain/serum ratio for L11.3 by the brain/serum ratio for albumin. Values greater than 1.0 indicate an uptake that is selective relative to that of uptake of albumin. This analysis again showed that L11.3 was taken up better than HyL5 or RF, especially by total brain and hippocampus. The L11.3/HyL5 ratio was also studied in 12 mo old SAMP8 mice and showed a selective uptake by olfactory bulb and hippocampus.
Overall, the results of the kinetics studies show that intravenously administered L11.3 had a greater uptake than albumin or other IgM antibodies, especially by hippocampus and in 12 mo old SAMP8 mice. We showed that L11.3 was effective when given directly into brain in reversing the cognitive impairments in 12 mo old SAMP8 mice. The kinetic findings raised the possibility that L11.3 might also be effective in reversing cognitive impairments after intravenous administration. We calculated that uptake by hippocampus of L11.3 exceeded 0.1%/g for over 3 h after a single iv dose. In comparison, this exceeded the uptake of an AβP-directed IgG previously tested (Banks, Terrell et al., 2002). Based on these calculations, we administered 25 microg/mouse of L11.3 by tail vein injection and tested acquisition (learning) 24 h later and retention (memory) 1 week after that. We found an effect on acquisition about equal to the 24 or 240 ng icv dose of L11.3, but there was no effect on retention. It may be that the short residence time in brain of L11.3 was too short to affect memory which was tested a week after antibody administration.
In conclusion, we tested two human IgM antibodies directed against AβP for their abilities to cross the BBB. One of them, L11.3, had a greater uptake than expected based on BBB leakage, especially in hippocampus and 12 mo old SAMP8 mice. L11.3 was effective in improving cognition in the aged SAMP8 mouse, a model of Alzheimer's disease, after either direct administration into brain or the circulation. The results suggest that L11.3 or other human anti-Aβ antibodies may be effective treatments for Alzheimer's disease. Passive immunization of AD patients with human MoAb's would circumvent the harmful effects induced by xenogenic antibodies.
Acknowledgments
Supported by VA Merit Reviews (WAB, SAF), R01NS050547 (WAB), R01NS051334 (WAB), and the Abisch-Frenkel Foundation (VG)
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- Attems J, Jellinger KA. Olfactory tau pathology in Alzheimer's disease and mild cognitive impairment. Clinical Neuropathology. 2006;25:265–271. [PubMed] [Google Scholar]
- Banks WA. Are the extracellular pathways a conduit for the delivery of therapeutics to the brain? Current Pharmaceutical Design. 2004;10:1365–1370. doi: 10.2174/1381612043384862. [DOI] [PubMed] [Google Scholar]
- Banks WA, Farr SA, Butt W, Kumar VB, Franko MW, Morley JE. Delivery across the blood-brain barrier of antisense directed againt amyloid β: reversal of learning and memory deficits in mice overexpressing amyloid precursor protein. J. Pharmacol. Exp. Ther. 2001a;297:1113–1121. [PubMed] [Google Scholar]
- Banks WA, Farr SA, La Scola ME, Morley JE. Intravenous human interleukin-1α impairs memory processing in mice: Dependence on blood-brain barrier transport into posterior division of the septum. J. Pharmacol. Exp. Ther. 2001b;299:536–541. [PubMed] [Google Scholar]
- Banks WA, Farr SA, Morley JE. Permeability of the blood-brain barrier to albumin and insulin in the young and aged SAMP8 mouse. Journal of Gerontology: Biological Science. 2000;55A:B601–B606. doi: 10.1093/gerona/55.12.b601. [DOI] [PubMed] [Google Scholar]
- Banks WA, Kastin AJ. In: Exchange of peptides between the circulation and the nervous system: role of the blood-brain barrier. Porter JC, Jezova D, editors. New York: Plenum Press; 1990. pp. 59–69. [DOI] [PubMed] [Google Scholar]
- Banks WA, Pagliari P, Nakaoke R, Morley JE. Effects of a behaviorally active antibody on the brain uptake and clearance of amyloid beta proteins. Peptides. 2005;26:287–294. doi: 10.1016/j.peptides.2004.09.016. [DOI] [PubMed] [Google Scholar]
- Banks WA, Terrell B, Farr SA, Robinson SM, Nonaka N, Morley JE. Transport of amyloid β protein antibody across the blood-brain barrier in an animal model of Alzheimer's disease. Peptides. 2002;23:2223–2226. doi: 10.1016/s0196-9781(02)00261-9. [DOI] [PubMed] [Google Scholar]
- Bard F, Cannon C, Barbour R, Burke RL, Games D, Grajeda H, Guido T, Hu K, Huang J, Johnson-Wood K, Khan K, Kholodenko D, Lee M, Lieberburg I, Motter R, Nguyen M, Soriano F, Vasquez N, Weiss K, Welch B, Seubert P, Schenk D, Yednock T. Peripherally administered antibodies against amyloid β-peptide enter the central nervous system and reduce pathology in a mouse model of Alzheimer's disease. Nature Medicine. 2000;6:916–919. doi: 10.1038/78682. [DOI] [PubMed] [Google Scholar]
- Blasberg RG, Fenstermacher JD, Patlak CS. Transport of α-aminoisobutyric acid across brain capillary and cellular membranes. J. Cereb. Blood Flow Metab. 1983;3:8–32. doi: 10.1038/jcbfm.1983.2. [DOI] [PubMed] [Google Scholar]
- Boules M, Williams K, Gollatz E, Fauq A, Richelson E. Down-regulation of amyloid precursor protein by peptide nucleic acid in vivo. Journal of Molecular Neuroscience. 2004;24:123–128. doi: 10.1385/JMN:24:1:123. [DOI] [PubMed] [Google Scholar]
- Broadwell RD, Bakder BJ, Banks WA, Friden PM, Moran M, Oliver C, Villegas JC, Flanagan TR, Emerich DF, Winn SR. Methods in Neuroscience. Vol. 21. 1994. Ferrotransferrin and antibody against the transferrin receptor as potential vehicles for drug delivery across the mammalian blood-brain barrier into the central nervous system; pp. 93–117. Ref Type: Serial (Book,Monograph) [Google Scholar]
- Butterfield DA. Amyloid beta-peptide (1–42)-induced oxidative stress and neurotoxicity: implications for neurodegeneration in Alzheimer's disease brain. A review. Free Radic Res. 2002;36:1307–1313. doi: 10.1080/1071576021000049890. [DOI] [PubMed] [Google Scholar]
- Chekhonin VP, Kabanov AV, Zhirkov YA, Morozov GV. Fatty acid acylated Fab-fragments of antibodies to neurospecific proteins as carriers for neuroleptic targeted delivery in brain. FEBS Lett. 1991;287:149–152. doi: 10.1016/0014-5793(91)80037-4. [DOI] [PubMed] [Google Scholar]
- Chen G, Chen KS, Knox J, Inglis J, Bernard A, Martin SJ, Justice A, McConlogue L, Games D, Freedman SB, Morris RGM. A learning deficit related to age and β-amyloid plaques in a mouse model of Alzheimer's disease. Nature. 2000;408:975–979. doi: 10.1038/35050103. [DOI] [PubMed] [Google Scholar]
- Christen-Zaech S, Kraftsik R, Pillevuit O, Kiraly M, Martins R, Khalili K, Miklossy J. Early olfactory involvement in Alzheimer's disease. Canadian Journal of Neurological Sciences. 2003;30:20–25. doi: 10.1017/s0317167100002389. [DOI] [PubMed] [Google Scholar]
- DeMattos RB, Bales KR, Cummins DJ, Paul SM, Holtzman DM. Brain to plasma amyloid-β efflux: a measure of brain amyloid burden in a mouse model of Alzheimer's disease. Science. 2002;295:2264. doi: 10.1126/science.1067568. [DOI] [PubMed] [Google Scholar]
- Elder GA, De Gasperi R, Gama Sosa MA. Research update: neurogenesis in adult brain and neuropsychiatric disorders. Mount Sinai Journal of Medicine. 2006;73:931–940. [PubMed] [Google Scholar]
- Farr SA, Poon HF, Dogrukol-Ak D, Drake J, Banks WA, Eyerman E, Butterfield DA, Morley JE. The antioxidants alpha-lipoic acid and N-acetylcysteine reverse memory impairment and brain oxidative stress in aged SAMP8 mice. Journal of Neurochemisrty. 2003;84:1173–1183. doi: 10.1046/j.1471-4159.2003.01580.x. [DOI] [PubMed] [Google Scholar]
- Flood JF, Morley JE. Learning and memory in the SAMP8 mouse. Neurosci. Biobehav. Rev. 1998;22:1–20. doi: 10.1016/s0149-7634(96)00063-2. [DOI] [PubMed] [Google Scholar]
- Flood JF, Morley JE, La Reginna M. Age-related changes in the pharmacological improvement of retention in senescence accelerated mouse (SAM) Neurobiol. Aging. 1993;14:159–166. doi: 10.1016/0197-4580(93)90092-p. [DOI] [PubMed] [Google Scholar]
- Frautschy SA, Yang F, Irrizarry M, Hyman B, Saido TC, Hsiao K, Cole GM. Microglial response to amyloid plaques in APPsw transgenic mice. Am J Pathology. 1998;152:307–317. [PMC free article] [PubMed] [Google Scholar]
- Geylis V, Kourilov B, Meiner Z, Nennesmo I, Bogdanovic N, Steinitz M. Human monoclonal antibody against amyloid-beta engendered by EBV-immortalized lymphocytes from healthy adults. Neurobiol Aging. 2005;26:597–606. doi: 10.1016/j.neurobiolaging.2004.06.008. [DOI] [PubMed] [Google Scholar]
- Glenner GG, Wong CW. Alzheimer's disease: initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem. Biophys. Res. Commun. 1984;120:885–890. doi: 10.1016/s0006-291x(84)80190-4. [DOI] [PubMed] [Google Scholar]
- Gutierrez EG, Banks WA, Kastin AJ. Murine tumor necrosis factor alpha is transported from blood to brain in the mouse. J. Neuroimmunol. 1993;47:169–176. doi: 10.1016/0165-5728(93)90027-v. [DOI] [PubMed] [Google Scholar]
- Hallam KT, Horgan JE, McGrath C, Norman TR. An investigation of the effect of tacrine and physostigmine on spatial working memory deficits in the olfactory bulbectomized rat. Behavioral Brain Research. 2004;153:481–486. doi: 10.1016/j.bbr.2004.01.005. [DOI] [PubMed] [Google Scholar]
- Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science. 2002;297:353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- Hock C, Konietzko U, Streffer JR, Tracy J, Signorell A, Muller-Tillmanns B, Lemke U, Henke K, Moritz E, Garcia E, Wollmer MA, Umbricht D, de Quervain DJ, Hofmann M, Maddalena A, Papassotiropoulos A, Nitsch RM. Antibodies against beta-amyloid slow cognitive decline in Alzheimer's disease. Neuron. 2003;38:547–554. doi: 10.1016/s0896-6273(03)00294-0. [DOI] [PubMed] [Google Scholar]
- Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, Yang F, Cole G. Correlative memory deficits, Aβ elevation, and amyloid plaques in transgenic mice. Science. 1996;274:99–102. doi: 10.1126/science.274.5284.99. [DOI] [PubMed] [Google Scholar]
- Janus C, Pearson J, McLaurin J, Mathews PM, Jiang Y, Schmidt SD, Chishti MA, Horne P, Heslin D, French J, Mount HTJ, Nixon RA, Mercken M, Bergeron C, Fraser PE, George-Hyslop P, Westaway D. Aγ peptide immunization reduces behavioral impairment and plaques in a model of Alzheimer's disease. Nature. 2000;408:979–982. doi: 10.1038/35050110. [DOI] [PubMed] [Google Scholar]
- Kozlowski GP, Sterzl I, Nilaver G. In: Localization patterns for immunoglobulins and albumins in the brain suggest diverse mechanisms for their transport across the blood-brain barrier (BBB) Ermisch A, Landgraf R, Rühle HJ, editors. Amsterdam: Elsevier; 1992. pp. 149–154. [DOI] [PubMed] [Google Scholar]
- Kumar VB, Farr SA, Flood JF, Kamlesh V, Franko M, Banks WA, Morley JE. Site-directed antisense oligonucleotide decreases the expression of amyloid precursor protein and reverses deficits in learning and memory in aged SAMP8 mice. Peptides. 2000;21:1769–1775. doi: 10.1016/s0196-9781(00)00339-9. [DOI] [PubMed] [Google Scholar]
- Morgan D, Diamond DM, Gottschall PE, Ugen KE, Dickey C, Hardy J, Duff K, Jantzen P, DiCarlo G, Wilcock D, Connor K, Hatcher J, Hope C, Gordon M, Arendash GW. Aβ peptide vaccination prevents memory loss in an animal model of Alzheimer's disease. Nature. 2000;408:982–985. doi: 10.1038/35050116. [DOI] [PubMed] [Google Scholar]
- Morley JE. The SAMP8 mouse: a model of Alzheimer's disease? Biogerontology. 2002;31:57–60. doi: 10.1023/a:1015207429786. [DOI] [PubMed] [Google Scholar]
- Morley JE, Farr SA, Flood JF. Antibody to amyloid beta protein alleviates impaired acquisition, retention, and memory processing in SAMP8 mice. Neurobiology of Learning and Memory. 2002a;78:125–138. doi: 10.1006/nlme.2001.4047. [DOI] [PubMed] [Google Scholar]
- Morley JE, Farr SA, Kumar VB, Banks WA. Alzheimer's disease through the eye of a mouse: Acceptance lecture for the 2001 Gayle A. Olson and Richard D. Olson prize. Peptides. 2002b;23:589–599. doi: 10.1016/s0196-9781(01)00630-1. [DOI] [PubMed] [Google Scholar]
- Morley JE, Kumar VB, Bernardo AF, Farr SA, Uezu K, Tumosa N, Flood JF. β-Amyloid precursor polypeptide in SAMP8 mice affects learning and memory. Peptides. 2000;21:1761–1767. doi: 10.1016/s0196-9781(00)00342-9. [DOI] [PubMed] [Google Scholar]
- Pan W, Solomon B, Maness LM, Kastin AJ. Antibodies to β-amyloid decrease the blood-to-brain transfer of β-amyloid peptide. Exp Biol Med. 2002;227:609–615. doi: 10.1177/153537020222700808. [DOI] [PubMed] [Google Scholar]
- Patlak CS, Blasberg RG, Fenstermacher JD. Graphical evaluation of blood-to-brain transfer constants from multiple-time uptake data. J. Cereb. Blood Flow Metab. 1983;3:1–7. doi: 10.1038/jcbfm.1983.1. [DOI] [PubMed] [Google Scholar]
- Permanne B, Adessi C, Saborio GP, Fraga S, Frossard MJ, Van Dorpe J, Dewachter I, Banks WA, Van Leuven F, Soto C. Reduction of amyloid load and cerebral damage in a transgenic mouse model of Alzheimer's disease by treatment with a beta-sheet breaker peptide. FASEB J. 2002;16:860–862. doi: 10.1096/fj.01-0841fje. [DOI] [PubMed] [Google Scholar]
- Poduslo JF, Curran GL, Berg CT. Macromolecular permeability across the blood-nerve and blood-brain barrier. Proc. Natl. Acad. Sci. USA. 1994;91:5705–5709. doi: 10.1073/pnas.91.12.5705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poon HF, Castegna A, Farr SA, Thongboonkerd V, Lynn BC, Banks WA, Morley JE, Klein JB, Butterfield DA. Quantitative proteomics analysis of specific protein expression and oxidative modification in aged senescence-accelerated-prone 8 mice brain. Neuroscience. 2004;126:915–926. doi: 10.1016/j.neuroscience.2004.04.046. [DOI] [PubMed] [Google Scholar]
- Schenk D, Barbour R, Dunn W, Gordon G, Grajeda H, Guido T, Hu K, Huang J, Johnson-Wood K, Khan K, Kholodenko D, Lee M, Liao A, Lieberburg I, Motter R, Mutter L, Soriano F, Shopp G, Vasquez N, Vandevert C, Walker S, Wogulis M, Yednock T, Games D, Seubert P. Immunization with amyloid-β attenuates Alzheimer-disease-like pathology in the PDAPP mouse. Nature. 1999;400:173–177. doi: 10.1038/22124. [DOI] [PubMed] [Google Scholar]
- Schlachetzki F, Zhu C, Pardridge WM. Expression of the neonatal Fc receptor (FcRn) at the blood-brain barrier. J. Neurochem. 2002;81:203–206. doi: 10.1046/j.1471-4159.2002.00840.x. [DOI] [PubMed] [Google Scholar]
- Steinitz M, Izak G, Cohen S, Ehrenfeld M, Flechner I. Continuous production of monoclonal rheumatoid factor by in vitro EBV-transformed lymphocytes. Nature. 1980;287:443–445. doi: 10.1038/287443a0. [DOI] [PubMed] [Google Scholar]
- Townsend KP, Pratico D. Novel therapeutic opportunities for Alzheimer's disease: focus on nonsteroidal anti-inflammatory drugs. FASEB J. 2005;19:1592–1601. doi: 10.1096/fj.04-3620rev. [DOI] [PubMed] [Google Scholar]
- Triguero D, Buciak J, Pardridge WM. Capillary depletion method for quantification of blood-brain barrier transport of circulating peptides and plasma proteins. J. Neurochem. 1990;54:1882–1888. doi: 10.1111/j.1471-4159.1990.tb04886.x. [DOI] [PubMed] [Google Scholar]
- Uchida D, Arimura A, Somogyvari-Vigh A, Shioda S, Banks WA. Prevention of ischemia-induced death of hippocampal neurons by pituitary adenylate cyclase activating polypeptide. Brain Res. 1996;736:280–286. doi: 10.1016/0006-8993(96)00716-0. [DOI] [PubMed] [Google Scholar]
- Ueno M, Dobrogowska DH, Vorbrodt AW. Immunocytochemical evaluation of the blood-brain barrier to endogenous albumin in the olfactory bulb and pons of senescence-accelerated mice (SAM) Histochem. Cell Biol. 1996;105:203–212. doi: 10.1007/BF01462293. [DOI] [PubMed] [Google Scholar]
- Watt JA, Pike CJ, Walencewicz-Wasserman AJ, Cotman CW. Ultrastructural analysis of β-amyloid-induced apoptosis in cultured hippocampal neurons. Brain Res. 1994;661:147–156. doi: 10.1016/0006-8993(94)91191-6. [DOI] [PubMed] [Google Scholar]
- Weber SJ, Greene DL, Hruby VJ, Yamamura HI, Porreca F, Davis TP. Whole body and brain distribution of [³H]cyclic[D-Pen²,D-Pen5] enkephalin after intraperitoneal, intravenous, oral and subcutaneous administration. J. Pharmacol. Exp. Ther. 1992;263:1308–1316. [PubMed] [Google Scholar]
- Weber SJ, Greene DL, Sharma SD, Yamamura HI, Kramer TH, Burks TF, Hrub VJ, Hersh LB, Davis TP. Distribution and analgesia of [³H][D-Pen²,D- Pen5]enkephalin and two halogenated analogs after intravenous administration. J. Pharmacol. Exp. Ther. 1991;259:1109–1117. [PubMed] [Google Scholar]