

Summary
Class A Scavenger Receptors (SR-A) participate in multiple macrophage functions including adhesion to modified extracellular matrix proteins present in various inflammatory disorders such as atherosclerosis and diabetes. By mediating macrophage adhesion to modified proteins and increasing macrophage retention, SR-A may contribute to the inflammatory process. Eicosanoids produced following PLA2–catalyzed release of arachidonic acid (AA) are important regulators of macrophage function and inflammatory responses. The potential roles of AA release and metabolism in SR-A-mediated macrophage adhesion were determined using macrophages adherent to modified protein. SR-A-dependent macrophage adhesion was abolished by selectively inhibiting calcium-independent PLA2 (iPLA2) activity and absent in macrophages isolated from iPLA2β-/- mice. Our results further demonstrate that 12/15lipoxygenase (12/15-LOX)-derived (but not cyclooxygenase (COX)- or CytP450 epoxygenase-derived) AA metabolites are specifically required for SR-A-dependent adhesion. Because of their role in regulating actin polymerization and cell adhesion, Rac and Cdc42 activation were also examined and shown to be increased via an iPLA2- and LOX-dependent pathway. Together, our results identify a novel role for iPLA2-catalyzed AA release and its metabolism by 12/15-LOX in coupling SR-A-mediated macrophage adhesion to Rac and Cdc42 activation.
Introduction
Cell adhesion to the substratum involves cell surface receptors that bind components of the extracellular matrix (ECM) and initiate intracellular signaling cascades that regulate actin polymerization and focal adhesion formation [reviewed in (1-4)]. Class A macrophage scavenger receptors (SR-A) are homotrimeric membrane glycoproteins that mediate multiple functions including calcium-independent macrophage adhesion to modified ECM components. Adhesion substrates for SR-A include glycated and cigarette smoke-modified collagen type IV, denatured collagen type I, and β-amyloid fibrils (5-9). In addition, proteoglycans that are upregulated during inflammation, such as biglycan and decorin, are ligands for SR-A (10). Macrophages isolated from transgenic mice overexpressing SR-A display increased spreading in culture and enhanced macrophage accumulation in carageenan-induced granulomas in vivo (11). Furthermore, macrophage activation increases SR-A expression and SR-A-dependent macrophage adhesion (12,13). In contrast, SR-A deficient macrophages fail to acquire spread morphology when plated on modified protein (14). Taken together, these observations suggest an important role for SR-A-mediated macrophage adhesion in various inflammatory processes characterized by macrophage activation and modification of the extracellular matrix.
Macrophages respond to various physiological and pathological stimuli via the activation of intracellular signaling cascades including phospholipase A2 (PLA2)-catalyzed hydrolysis of arachidonic acid (AA) from membrane phospholipids (15). Based on their location and Ca2+ requirements for enzymatic activity, PLA2s can be classified into three groups: secretory (sPLA2s) that are secreted from cells and require millimolar Ca2+; cytosolic (cPLA2s) that require micromolar Ca2+; and Ca2+-independent (iPLA2s) that reside in the cytosol of resting cells but do not require Ca2+ for enzymatic activity (16). Several members of the iPLA2 family are now recognized, and they are designated Group VI PLA2 enzymes (17). The first recognized and best characterized is the Group VIA PLA2 (18-20), which is also designated iPLA2β (21,22).
Most cellular AA is esterified to the glycerol backbone of phospholipids. The free AA that is accessible to AA metabolizing oxygenases is generally thought to be released by the action of either cPLA2 or iPLA2. Whether AA release is involved in regulating SR-A function is not yet known, but this possibility is suggested by the finding that acetylated-LDL (AcLDL) promotes TNFα production in macrophages via a pathway that depends on Ca2+ and PLA2 activation, although the specific receptor mediating this response has not been identified (23). In addition, a role for specific PLA2 isoforms in regulating the calcium-independent SR-A adhesion has not been investigated.
PLA2-derived AA is subsequently metabolized to produce a variety of biologically active eicosanoids. Enzymes that metabolize AA include cyclooxygenases (COX) that catalyze the production of prostaglandins (PGs) and thromboxanes (Txs), lipoxygenases (LOX) that catalyze the production of hydroxyeicosatetraenoic acid (HETEs), lipoxins, and leukotrienes (LTs), and cytochrome P450-dependent epoxygenases (CytP450) that synthesize epoxyeicosatrienoic acids (EETs) (24-26). Although AA-derived metabolites are involved in intercellular signaling, recent evidence indicates that AA metabolites also participate in regulating intracellular signaling processes including those involved in actin polymerization (15,27-29).
Activation of Rho-GTPases, including Rac and Cdc42, plays a key role in coordinating cell adhesion (30-32). Binding of cell surface receptors to the ECM promotes Rac and Cdc42 activation, which, in turn, activates additional signaling molecules that ultimately result in the projection of actin-containing fibers and changes in the cytoskeleton that are required for acquiring a spread morphology and firm cell adhesion. In contrast, Rho activation promotes fiber retraction and detachment that are characteristic of migrating cells (33,34). We have previously shown that SR-A mediates macrophage spreading on modified protein (14), but the potential involvement of Rac and Cdc42 in coupling surface SR-A binding to modified protein with increased macrophage adhesion has not been investigated.
The aim of the current study was to determine the potential role for specific PLA2 isoforms and AA metabolites in regulating Rac and Cdc42 activation and SR-A-mediated macrophage adhesion to modified protein. For this, we used pharmacologic inhibitors of specific AA signaling pathways and macrophages isolated from mice lacking specific AA metabolic enzymes. Our results indicate that iPLA2-catalyzed release of AA and its subsequent metabolism by 12/15-LOX couples SR-A to Rac and Cdc42 activation and macrophage adhesion.
Experimental Procedures
Chemicals
Dulbecco's modified Eagle's medium (DMEM) with L-glutamine, DMEM with 25 mM HEPES without phenol red, penicillin, streptomycin, heat-inactivated fetal bovine serum (FBS), and human plasma fibronectin were purchased from GibcoBRL (Grand Island NY). Ultra low attachment polystyrene 6-well culture plates were purchased from Costar (Corning, NY). SKF525A was purchased from Biomol Research Laboratories, Inc (Plymouth, PA). AACOCF3 (arachidonyltrifluoromethyl ketone), BEL (6E-[bromoethylene]-tetrahydro-3-(1-naphthalenyl)- 2H-pyran-2-one), cPLA2α inhibitor, MK-886 (3-[p-Chlorobenzyl)-5-(isopropyl)-3-t-butylthioindol-2-yl]-2,2-dimethilpropanoic acid, Na), and indomethacin (1-(p-chlorobenzoyl)-5-methoxy-2-methyl-1H-indole-3-acetic acid) were purchased from Calbiochem (La Jolla, CA). NDGA (nordihydroguaiaretic acid) and prostaglandin E2 enzyme-linked immunoassay kits were purchased from Cayman Chemical (Ann Arbor, MI). Malondialdehyde bis (dimethyl acetal), was purchased from Sigma Aldrich (St Louis, MO).
Malondialdehyde modification of bovine serum albumin
MDA-BSA was prepared using malondialdehyde bis (dimethyl acetal) as previously described (35). Protein modification was confirmed by immunoblotting with anti-MDA-specific antibody (Academy Bio-Medical Company, Inc; Houston, TX).
Cell isolation and culture
Mouse peritoneal macrophages (MPM) were harvested from wild-type mice (Jackson Laboratory, Bar Harbor, ME), SR-A-/- (University of Kentucky), iPLA2β-/- mice [prepared and characterized elsewhere (36-39)], 5-LOX-/- mice or 12/15-LOX-/- mice (Jackson Laboratory, Bar Harbor, ME), via peritoneal lavage with ice-cold sterile saline and cultured in DMEM containing FBS (10% v/v), penicillin, and streptomycin as previously described (40). Animal care and use for all procedures was done according to protocols reviewed and approved by the Institutional Animal Care and Use Committee at the University of Kentucky.
Cell adhesion assays
To assess cell spreading, MPM were plated (30,000 cells/well) into four-chambered LAB-TEK slides (Nalge Nunc International; Naperville, IL) precoated with either MDA-BSA or fibronectin and treated with inhibitors as described in the figure legends. Trypan blue exclusion was used to confirm that the treatments did not affect cell viability. Following treatments, macrophages were gently washed with warm PBS, fixed with 4% paraformaldehyde, and permeabilized with 0.1% Triton X-100. The cells were then blocked with 1% BSA for 30 min prior to staining with Alexa–Fluor568-conjugated phalloidin and DAPI (Molecular Probes; Eugene, OR). Cells were mounted in Prolong® Antifade reagent (Molecular Probes; Eugene OR) and random images of at least twenty-five cells from at least three independent experiments were digitally captured using a Leica TCS SP confocal microscope. Individual cells were outlined and total cell area quantified using Metamorph® software.
Rac/Cdc42 Activation Assay
To examine Rac and Cdc42 activation during SR-A-dependent macrophage adhesion, cells were treated as described in figure legends and then lysed in RIPA buffer containing phosphatase and protease inhibitors. Protein concentration was determined using the BioRad DC assay (Hercules, CA). Equal amounts of cell lysate protein were incubated with the p21-binding domain of PAK1 fused to GST (GST-PAK) beads for 1 hour at 4°C. The p21-binding domain of PAK binds the GTP-bound (active) form of Rac and Cdc42, but not the GDP-bound state (41). GST-PAK beads were isolated and the amount of GTP-bound (active) Rac and Cdc42 quantified by immunoblotting isolated proteins with Rac1 and Cdc42 specific antibodies (Transduction Labs; Lexington, KY). Proteins were visualized by enhanced chemiluminesence using anti-mouse HRP-coupled secondary antibody. Images were digitally captured and quantified using a Kodak Image Station 4000MM. The amount of GTP-bound Rac was normalized to the total amount of Rac or Cdc42 detected in each cell lysate.
Statistical analysis
Experiments were repeated at least three times and significance among treatment groups determined by one-way ANOVA with appropriate post-hoc tests using GraphPad Prism. Values with p<0.05 were considered to be statistically significant.
Results
SR-A-mediated macrophage adhesion requires PLA2-mediated arachidonic acid (AA) release
Macrophages respond to various stimuli via activation of intracellular signaling cascades including the phospholipase A2 (PLA2)-catalyzed hydrolysis of AA from membrane phospholipids. Therefore, we examined the potential role for AA in regulating SR-A-dependent macrophage adhesion to malondialdehyde-modified BSA (MDA-BSA). Malondialdehyde-modified proteins are SR-A ligands and have been previously used to study SR-A function (35,42).
To determine whether PLA2-mediated AA release plays a role in SR-A-dependent adhesion, peritoneal macrophages isolated from SR-A+/+ or SR-A-/- mice were pretreated with AACOCF3 (30 μM), an inhibitor of cPLA2 and iPLA2, prior to plating on MDA-BSA-coated slides. After 2 hrs, macrophage morphology was assessed by staining polymerized actin (F actin) with fluorescently-conjugated phalloidin. Consistent with our previous report (14), control (SR-A+/+) macrophages that were allowed to adhere to MDA-BSA for 2 hrs exhibited enhanced spreading compared to SR-A-/- macrophages (Figure 1). Importantly, SR-A+/+ and SR-A-/- macrophages spread to the same extent when allowed to adhere to an integrin ligand (14). Like SR-A-/- macrophages, macrophages treated with AACOCF3 remained rounded when allowed to adhere to MDA-BSA, which indicates that PLA2 activation is required for SR-A-mediated macrophage adhesion. The inhibitory effect of AACOCF3 on macrophage spreading was significantly reversed adding exogenous AA (10 μM), which confirms that AA is required for SR-A-dependent macrophage adhesion. Maximal reversal of the effects of PLA2 inhibition was achieved by adding exogenous AA at a concentration of 10 μM, and no further reversal occurred at higher AA concentrations (data not shown).
Figure 1. SR-A-mediated macrophage adhesion requires PLA2-mediated arachidonic acid (AA) release.

MPM isolated from SR-A+/+ (control) or SR-A-/- mice were pretreated as indicated with AACOCF3 (30 μM) for 25 min in suspension. Arachidonic acid (AA; 10 μM) was added to pretreated cells immediately prior to plating into MDA-BSA coated slides. Cells were allowed to adhere for 2 hr at 37°C, fixed, and stained with Alexa–Fluor568-conjugated phalloidin. Nuclei were stained with DAPI. Confocal images were digitally captured and cell surface area quantified. The scale bar represents 50 μm. The graph depicts the mean ± SEM of at least three separate experiments. * denotes significantly different (p<0.05) from SR-A+/+ control; # denotes significantly different (p<0.05) from SR-A-/- cells.
SR-A-mediated macrophage adhesion requires calcium-independent PLA2 (iPLA2)-mediated arachidonic acid release
To examine the potential involvement of different PLA2 enzymes in SR-A-mediated adhesion, macrophages were pretreated with bromoenol lactone (BEL), a suicide substrate inhibitor of iPLA2 (43), or with a specific pyrrolidine inhibitor of cPLA2α (44), and then plated on MDA-BSA (Figure 2). Like macrophages treated with AACOCF3, macrophages treated with BEL (3.0 μM; 30 min) prior to plating on MDA-BSA remained rounded compared to untreated control macrophages. Furthermore, adding exogenous AA (10 μM) restored SR-A-mediated macrophage spreading after iPLA2 inhibition with BEL. The specific requirement for iPLA2β activity was confirmed using macrophages isolated from iPLA2β-/- mice which exhibited significantly less spreading on MDA-BSA than did untreated iPLA2β+/+ control macrophages. The spreading of iPLA2β-/- macrophages plated on MDA-BSA was increased by adding exogenous AA (data not shown). In contrast to the effect of inhibiting iPLA2, treating macrophages with the pyrrolidine cPLA2 inhibitor at a concentration (5 μM) that abolishes calcium-mediated prostaglandin formation [data not shown and (44)] had no effect on SR-A mediated cell spreading. This result indicates that cPLA2 activity is not required for SR-A-mediated cell adhesion. Taken together, these results demonstrate that iPLA2β-catalyzed AA release is specifically required for SR-A-dependent macrophage adhesion to modified protein.
Figure 2. Calcium-independent PLA2β (iPLA2β)-catalyzed AA release is required for SR-A-mediated macrophage adhesion.

MPM isolated from wild-type or iPLA2β-/- mice were pretreated as indicated with the specific iPLA2 suicide substrate inhibitor BEL (3 μM) or the cPLA2α inhibitor (5 μM) for 25 min in suspension. AA (10 μM) was added to pretreated cells immediately prior to plating into MDA-BSA coated slides. Cells were allowed to adhere for 2 hr at 37°C, fixed, and stained with Alexa–Fluor568-conjugated phalloidin. Nuclei were stained with DAPI. Confocal images were digitally captured and cell surface area quantified. The scale bar represents 50 μm. The graph depicts the mean ± SEM of at least three separate experiments. * denotes significantly different (p<0.05) from control; # denotes significantly different (p<0.05) from iPLA2β-/- macrophages.
SR-A-mediated macrophage adhesion requires 12/15-LOX-derived AA metabolites
To examine the potential role of AA metabolism in SR-A-mediated adhesion, SR-A+/+ macrophages were pretreated with NDGA, an inhibitor of both 12/15-LOX and 5-LOX enzymes, and the ability of macrophages to spread on MDA-BSA was assessed. Pretreatment with NDGA abolished macrophage spreading on MDA-BSA (Figure 3). The addition of exogenous AA (10 μM) did not restore the spreading of NDGA-treated macrophages plated on MDA-BSA, which indicates that LOX-derived AA metabolites rather than AA itself are required for SR-A-dependent cell adhesion/spreading.
Figure 3. SR-A-mediated macrophage adhesion requires 12/15-LOX-derived products of AA.
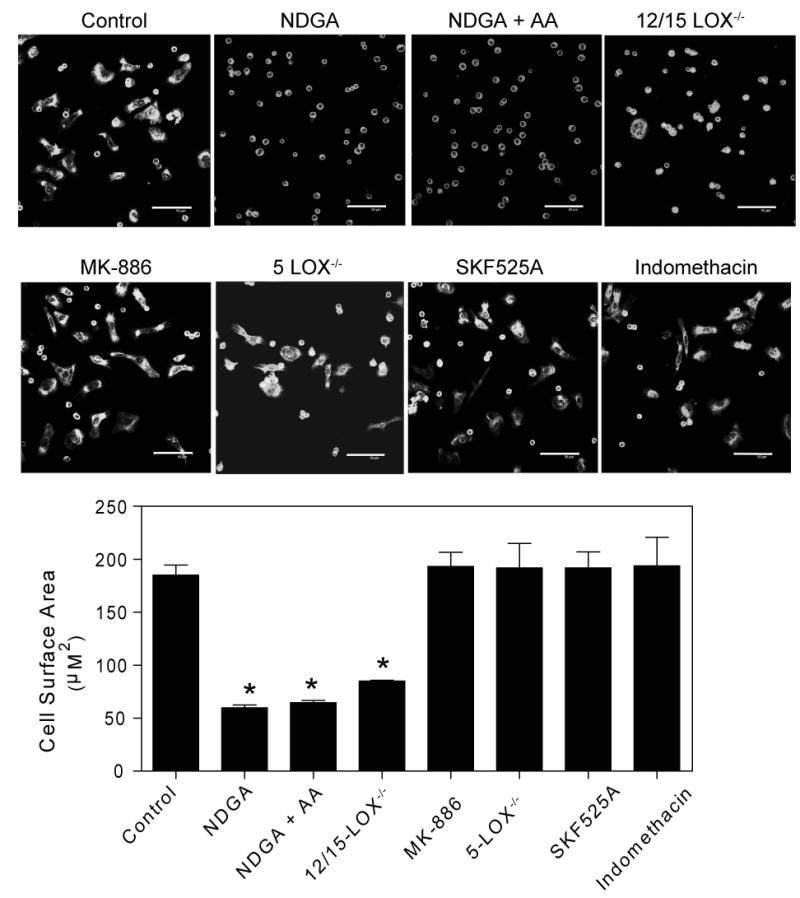
MPM isolated from wild type, 12/15-LOX-/-, or 5-LOX-/- mice were pretreated as indicated with the non-selective LOX inhibitor NDGA (50 μM), the specific 5-LOX inhibitor MK-866 (5 μM), the CytP450 inhibitor SKF525A (10 μM), or the COX inhibitor indomethacin (10 μM) for 25 min in suspension. AA (10 μM) was added to pretreated cells immediately prior to plating into MDA-BSA coated slides. Cells were allowed to adhere for 2 hr at 37°C, fixed, and stained with Alexa–Fluor568-conjugated phalloidin. Nuclei were stained with DAPI. Confocal images were digitally captured and cell surface area quantified. The scale bar represents 50 μm. The graph depicts the mean ± SEM of at least three separate experiments. * denotes significantly different (p<0.05) from control.
The inhibition of SR-A-mediated macrophage spreading by NDGA suggests a role for LOX-derived products in that process. To determine the specific LOX enzyme(s) required for SR-A-mediated adhesion we used macrophages isolated from 12/15-LOX-/- mice or from 5-LOX-/- mice, or macrophages pretreated with the specific 5-LOX inhibitor MK-866 (Figure 3). Macrophages isolated from 12/15-LOX-/- mice showed diminished ability to spread on MDA-BSA, which indicates that 12/15-LOX activity is required. In contrast, macrophages that were isolated from 5-LOX-/- mice or treated with the selective 5-LOX inhibitor (MK-886) at a concentration (5 μM) previously shown to inhibit macrophage production of LTB4 and 5-HETE (45) spread normally on MDA-BSA. This finding indicates that SR-A-dependent macrophage adhesion does not require 5-LOX-derived AA metabolites.
In addition to the LOX pathway, cytochrome p450-dependent monooxygenases (CytP450) and COX isoenzymes catalyze the metabolism of AA to biologically active compounds (46). To examine the potential role for a CytP450 pathway, cells were treated with an inhibitor of CytP450 (SKF525A) at a concentration (10 μM) that inhibits epoxyeicosatrienoic acid (EET) production (47), and the ability of cells to spread was assessed. Treating macrophages with SKF525A (Figure 3) did not affect SR-A-dependent macrophage spreading. To examine the potential involvement of the COX pathway, macrophages were treated with a nonselective inhibitor of COX-1 and COX-2 (indomethacin) at a concentration (10 μM) that inhibits prostaglandin production in macrophages (48,49). Treating macrophages with indomethacin also failed to affect SR-A-dependent spreading (Figure 3). Taken together, the data in Figure 3 indicate that 12/15-LOX-derived products of AA are specifically required for SR-A-dependent macrophage adhesion but that AA metabolites produced by CytP450 or COX are not.
LOX metabolism of iPLA2-derived AA couples SR-A to Rac and Cdc42 activation
The Rho-family GTPases Rac and Cdc42 are important regulators of actin polymerization and formation of the cytoskeletal structures (e.g., lamellipodia and filopodia) involved in cell adhesion. To investigate whether Rac and Cdc42 are activated by SR-A-dependent macrophage adhesion to modified protein, macrophages were plated for various intervals on MDA-BSA, and cell lysates were then prepared. The active forms of Rac and Cdc42 were isolated from cell lysates using a fusion protein that binds only the active, GTP-bound form of these GTPases. As illustrated in Figure 4A, Rac and Cdc42 were both rapidly activated in macrophages that adhere to MDA-BSA. Activation increased significantly after 10 minutes of adhesion to MDA-BSA and tended to remain above basal after 120 minutes. Importantly, neither Rac nor Cdc42 activation was detected in SR-A-/- macrophages plated on MDA-BSA (Figure 4B), which confirms that Rac and Cdc42 activation are SR-A-dependent.
Figure 4. Rac and Cdc42 are rapidly activated in SR-A-dependent manner during macrophage adhesion to modified protein.

MPM isolated from wild type or SR-A-/- mice were either kept in suspension or plated into MDA-BSA coated dishes for the indicated times at 37°C. Cell lysates were then prepared and Rac/Cdc42 activation was assessed. Total GTPase was detected in whole cell lysates using the same antibody and the amounts of GTP-bound Rac/Cdc42 were normalized to the total GTPase for each sample. A representative blot and the mean ± SEM of at least 3 separate experiments are shown. * denotes significantly different (p<0.05) from non-adhered (suspension) SR-A+/+ cells.
To determine whether iPLA2-catalyzed AA release and subsequent metabolism by LOX are required for SR-A-dependent Rac activation, cells were pretreated with inhibitors of iPLA2 or of LOX before assessing Rac activation. Pretreating macrophages with inhibitors of iPLA2 (AACOCF3 or BEL) or LOX (NDGA) prevented activation of both Rac and Cdc42 during SR-A-dependent macrophage adhesion/spreading (Figure 5). Taken together, these results indicate that the Rac and Cdc42 activation during SR-A-dependent macrophage adhesion requires metabolism of iPLA2-derived AA by LOX.
Figure 5. SR-A-dependent Rac and Cdc42 activation requires LOX-mediated metabolism of iPLA2-derived AA.

MPM isolated from wild type or SR-A-/- mice were pretreated as indicated with AACOCF3 (30 μM), BEL (3 μM), or NDGA (50 μM) for 30 min in suspension. Following pretreatments, cells were either maintained in suspension or plated into MDA-BSA coated dishes for 35 min at 37°C. Cell lysates were then prepared and Rac/Cdc42 activation was assessed. Total GTPase was detected in whole cell lysates using the same antibody, and the amounts of GTP-bound Rac/Cdc42 were normalized to the total GTPase for each sample. A representative blot and the mean ± SEM of at least 3 separate experiments are shown. * denotes significantly different (p<0.05) from cells in suspension.
Discussion
Specific roles for intracellular signaling cascades in regulating SR-A function continue to be elucidated (14,23,40,50-54). In the current study, we examined the potential role for PLA2-catalyzed AA release and subsequent AA metabolism in SR-A-mediated cell adhesion using pharmacological inhibitors and macrophages isolated from genetically modified mice lacking iPLA2β or specific AA oxygenases.
Both calcium-dependent (cPLA2) and calcium-independent (iPLA2) PLA2 enzymes are involved in intracellular signaling in macrophages (55-57). We found that AACOCF3, an inhibitor of both cPLA2 and iPLA2, abolished SR-A-mediated macrophage spreading on modified protein. Using a specific pyrrolidine cPLA2α inhibitor and the specific iPLA2 inhibitor (BEL) and macrophages isolated from iPLA2β-/- mice, we further show that iPLA2β activity is specifically required for SR-A-mediated macrophage adhesion/spreading. PLA2-catalyzed hydrolysis of membrane phospholipids produces a free fatty acid (e.g., AA) and a 2-lysophospholipid, each of which can have biologic activity. The ability of exogenous AA to restore SR-A-mediated spreading to iPLA2β-/- macrophages or to macrophages treated with BEL confirms that AA is an important mediator of SR-A-mediated macrophage adhesion.
Intracellular AA can be rapidly metabolized to bioactive eicosanoids by various oxygenases, including those of the LOX, COX, and CytP450 families. Inhibition of LOX with NDGA abolished macrophage spreading on MDA-BSA, but neither the COX inhibitor indomethacin nor the CytP450 inhibitor SKF525A affected macrophage spreading. These findings suggest that AA metabolites produced by LOX are specifically required for SR-A-mediated adhesion/spreading. A specific requirement for 12/15-LOX activity was demonstrated using macrophages isolated from 12/15-LOX-/- mice, in which SR-A-mediated spreading was abolished. In contrast, macrophages isolated from 5-LOX-/- mice spread normally as did macrophages isolated from wild-type mice and treated with the 5-LOX inhibitor MK-866. These data indicate that 5-LOX is not involved in SR-A-mediated macrophage spreading. Exogenous AA failed to restore spreading to wild-type macrophages in which 12/15-LOX was inhibited by NDGA or to macrophages from 12/15-LOX-/- mice, which confirms that a metabolite of AA produced by 12/15-LOX rather than AA itself is required for the spreading response.
Activation of Rac and Cdc42, members of the Rho-like GTPase family, coordinates changes in cell morphology during cell adhesion by promoting assembly and organization of the actin cytoskeleton (30-32). Like other G proteins, activation of Rac and Cdc42 requires GDP/GTP exchange, which is mediated by guanine nucleotide exchange factors (GEFs). Several GEFs have been identified, and mechanisms that regulate these GEFs continue to be elucidated [reviewed in (58,59)]. In addition to GEFs, RhoGTPases are regulated by interactions with guanine-nucleotide dissociation inhibitors [(GDIs); reviewed in (60,61)]. The ability of AA metabolites to regulate Rac and Cdc42 has been suggested previously (62-64). For example, it has been reported that AA promotes Rac and Cdc42 activation in a COX-2 dependent manner (63). It has also been suggested that various biologically active lipids, including AA, can disrupt the interaction of Rac with Rac-GDI (62). In addition to regulating Rac/Cdc42 activation, other studies have demonstrated that Rac activation promotes cPLA2-catalyzed AA release (65,66). The findings that Rac activation may either precede or follow AA release suggests a complex relationship between RhoGTPase activation and AA release. To our knowledge, ours is the first study to demonstrate that 12/15-LOX metabolism of AA released by iPLA2 mediates Rac or Cdc42 activation. Thus, our findings indentify a novel pathway for regulating Rac and Cdc42 activation.
Diverse chronic inflammatory diseases including diabetes, atherosclerosis, and Alzheimer's disease are characterized by modifications of extracellular matrix components. Such modifications result in formation of SR-A ligands, and SR-A may contribute to macrophage accumulation at specific inflammatory sites. For example, SR-A recognizes glycated proteins formed as a result of the hyperglycemia accompanying diabetes (7). The finding that SR-A contributes to enhanced glomerular macrophage accumulation in diabetic animals suggests a role for SR-A in diabetes-induced complications, such as nephropathy (67). Similarly, extracellular matrix proteoglycans that are upregulated in atherosclerotic plaques are adhesion substrates for SR-A, which suggests that SR-A may contribute to macrophage adhesion and retention in atherosclerotic lesions (10). An additional role for SR-A in Alzheimer's disease is suggested by the finding that SR-A mediates adhesion of microglia to β-amyloid fibril-coated surfaces (6). Accumulation of SR-A ligands at sites where macrophage accumulate suggests that SR-A-mediated adhesion may participate in chronic inflammatory diseases.
Macrophage expression of both SR-A and 12/15-LOX can be altered during chronic inflammation, such as that associated with atherosclerosis and diabetes (68-72). Changes in SR-A and 12/15-LOX expression can have important effects on the inflammatory process. For example, the involvement of 12/15-LOX in lipid peroxidation, cytokine expression, and monocyte recruitment indicates that 12/15-LOX-derived metabolites are proinflammatory (73-76). This notion is supported by the observation that 12/15-LOX-/- mice have reduced atherosclerosis and brain oxidative stress (73,77,78) and reduced sensitivity to diabetes in models associated with islet inflammation (79). Similarly, SR-A-/- mice are protected from atherosclerosis and diabetic nephropathy (80,81). Thus, inhibiting SR-A or components of its downstream adhesion signaling pathway, including iPLA2β and 12/15-LOX, might be therapeutically beneficial in chronic inflammatory diseases.
Overall, our results identify a novel role for iPLA2β and 12/15-LOX in coupling SR-A to Rac and Cdc42 activation and macrophage adhesion. An implication of our results is that SR-A-mediated adhesion might promote inflammation by increasing macrophage retention at sites of extracellular matrix modification and by activating signaling pathways in which AA and its metabolites participate.
Acknowledgments
The authors would like to acknowledge Cecelia Gass and Leah Allen for their technical assistance and members of the Cardiovascular Research Center and the Kentucky Pediatrics Research Institute at the University of Kentucky for their helpful comments and suggestions. This work was supported by grants from the NIH (HL075241) and an Established Investigator Award from the American Heart Association. D.N. was a recipient of a predoctoral fellowship from the American Heart Association (Ohio Valley Affiliate). J.T. is supported by United States Public Health Service Grants R37-DK34388, P41-RR00954, P60-DK20579, and P30-DK56341.
References
- 1.Schwartz MA, Ginsberg MH. Nat Cell Biol. 2002;4:E65–68. doi: 10.1038/ncb0402-e65. [DOI] [PubMed] [Google Scholar]
- 2.Berton G, Lowell CA. Cellular Signalling. 1999;11:621–635. doi: 10.1016/s0898-6568(99)00003-0. [DOI] [PubMed] [Google Scholar]
- 3.DeMali KA, Burridge K. J Cell Sci. 2003;116:2389–2397. doi: 10.1242/jcs.00605. [DOI] [PubMed] [Google Scholar]
- 4.Zamir E, Geiger B. J Cell Sci. 2001;114:3583–3590. doi: 10.1242/jcs.114.20.3583. [DOI] [PubMed] [Google Scholar]
- 5.Gowen BB, Borg TK, Ghaffar A, Mayer EP. Matrix Biology. 2000;19:61–71. doi: 10.1016/s0945-053x(99)00052-9. [DOI] [PubMed] [Google Scholar]
- 6.El Khoury J, Hickman SE, Thomas CA, Cao L, Silverstein SC, Loike JD. Nature. 1996;382:716–719. doi: 10.1038/382716a0. [DOI] [PubMed] [Google Scholar]
- 7.el Khoury J, Thomas CA, Loike JD, Hickman SE, Cao L, Silverstein SC. J Biol Chem. 1994;269:10197–10200. [PubMed] [Google Scholar]
- 8.Kirkham PA, Spooner G, Ffoulkes-Jones C, Calvez R. Free Radic Biol Med. 2003;35:697–710. doi: 10.1016/s0891-5849(03)00390-3. [DOI] [PubMed] [Google Scholar]
- 9.Santiago-Garcia J, Mas-Oliva J, Innerarity TL, Pitas RE. J Biol Chem. 2001;276:30655–30661. doi: 10.1074/jbc.M102879200. [DOI] [PubMed] [Google Scholar]
- 10.Santiago-Garcia J, Kodama T, Pitas RE. J Biol Chem. 2003;278:6942–6946. doi: 10.1074/jbc.M208358200. [DOI] [PubMed] [Google Scholar]
- 11.Daugherty A, Kosswig N, Cornicelli JA, Whitman SC, Wolle S, Rateri DL. Journal of lipid research. 2001;42:1049–1055. [PubMed] [Google Scholar]
- 12.Haworth R, Platt N, Keshav S, Hughes D, Darley E, Suzuki H, Kurihara Y, Kodama T, Gordon S. Journal of Experimental Medicine. 1997;186:1431–1439. doi: 10.1084/jem.186.9.1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.van Velzen AG, Suzuki H, Kodama T, van Berkel TJ. Exp Cell Res. 1999;250:264–271. doi: 10.1006/excr.1999.4530. [DOI] [PubMed] [Google Scholar]
- 14.Nikolic DM, Cholewa J, Gass C, Gong MC, Post SR. Am J Physiol Cell Physiol. 2007;292:C1450–1458. doi: 10.1152/ajpcell.00401.2006. [DOI] [PubMed] [Google Scholar]
- 15.Akiba S, Ohno S, Chiba M, Kume K, Hayama M, Sato T. Biochem Pharmacol. 2002;63:1969–1977. doi: 10.1016/s0006-2952(02)00988-7. [DOI] [PubMed] [Google Scholar]
- 16.Schaloske RH, Dennis EA. Biochimica et Biophysica Acta (BBA) - Molecular and Cell Biology of Lipids. 2006;1761:1246–1259. doi: 10.1016/j.bbalip.2006.07.011. [DOI] [PubMed] [Google Scholar]
- 17.Six DA, Dennis EA. Biochimica et Biophysica Acta (BBA) - Molecular and Cell Biology of Lipids. 2000;1488:1–19. doi: 10.1016/s1388-1981(00)00105-0. [DOI] [PubMed] [Google Scholar]
- 18.Tang J, Kriz RW, Wolfman N, Shaffer M, Seehra J, Jones SS. J Biol Chem. 1997;272:8567–8575. doi: 10.1074/jbc.272.13.8567. [DOI] [PubMed] [Google Scholar]
- 19.Balboa MA, Balsinde J, Jones SS, Dennis EA. J Biol Chem. 1997;272:8576–8580. doi: 10.1074/jbc.272.13.8576. [DOI] [PubMed] [Google Scholar]
- 20.Ma Z, Ramanadham S, Kempe K, Chi XS, Ladenson J, Turk J. J Biol Chem. 1997;272:11118–11127. [PubMed] [Google Scholar]
- 21.Jenkins CM, Mancuso DJ, Yan W, Sims HF, Gibson B, Gross RW. J Biol Chem. 2004;279:48968–48975. doi: 10.1074/jbc.M407841200. [DOI] [PubMed] [Google Scholar]
- 22.Mancuso DJ, Jenkins CM, Gross RW. J Biol Chem. 2000;275:9937–9945. doi: 10.1074/jbc.275.14.9937. [DOI] [PubMed] [Google Scholar]
- 23.Pollaud-Cherion C, Vandaele J, Quartulli F, Seguelas MH, Decerprit J, Pipy B. European journal of biochemistry / FEBS. 1998;253:345–353. doi: 10.1046/j.1432-1327.1998.2530345.x. [DOI] [PubMed] [Google Scholar]
- 24.Natarajan R, Nadler JL. Arterioscler Thromb Vasc Biol. 2004;24:1542–1548. doi: 10.1161/01.ATV.0000133606.69732.4c. [DOI] [PubMed] [Google Scholar]
- 25.Fleming I. Prostaglandins & Other Lipid Mediators. 2007;82:60–67. doi: 10.1016/j.prostaglandins.2006.05.003. [DOI] [PubMed] [Google Scholar]
- 26.Phillis JW, Horrocks LA, Farooqui AA. Brain Research Reviews. 2006;52:201–243. doi: 10.1016/j.brainresrev.2006.02.002. [DOI] [PubMed] [Google Scholar]
- 27.Miller YI, Chang MK, Funk CD, Feramisco JR, Witztum JL. J Biol Chem. 2001;276:19431–19439. doi: 10.1074/jbc.M011276200. [DOI] [PubMed] [Google Scholar]
- 28.Miller YI, Worrall DS, Funk CD, Feramisco JR, Witztum JL. Mol Biol Cell. 2003;14:4196–4206. doi: 10.1091/mbc.E03-02-0063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Akiba S, Mizunaga S, Kume K, Hayama M, Sato T. J Biol Chem. 1999;274:19906–19912. doi: 10.1074/jbc.274.28.19906. [DOI] [PubMed] [Google Scholar]
- 30.Nobes CD, Hall A. Cell. 1995;81:53–62. doi: 10.1016/0092-8674(95)90370-4. [DOI] [PubMed] [Google Scholar]
- 31.D'Souza-Schorey C, Boettner B, Van Aelst L. Mol Cell Biol. 1998;18:3936–3946. doi: 10.1128/mcb.18.7.3936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Price LS, Leng J, Schwartz MA, Bokoch GM. Mol Biol Cell. 1998;9:1863–1871. doi: 10.1091/mbc.9.7.1863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Worthylake RA, Lemoine S, Watson JM, Burridge K. J Cell Biol. 2001;154:147–160. doi: 10.1083/jcb.200103048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Worthylake RA, Burridge K. J Biol Chem. 2003;278:13578–13584. doi: 10.1074/jbc.M211584200. [DOI] [PubMed] [Google Scholar]
- 35.Haberland ME, Fogelman AM, Edwards PA. Proc Natl Acad Sci U S A. 1982;79:1712–1716. doi: 10.1073/pnas.79.6.1712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bao S, Miller DJ, Ma Z, Wohltmann M, Eng G, Ramanadham S, Moley K, Turk J. J Biol Chem. 2004;279:38194–38200. doi: 10.1074/jbc.M406489200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bao S, Song H, Wohltmann M, Ramanadham S, Jin W, Bohrer A, Turk J. J Biol Chem. 2006;281:20958–20973. doi: 10.1074/jbc.M600075200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jacobson DA, Weber CR, Bao S, Turk J, Philipson LH. J Biol Chem. 2007;282:7442–7449. doi: 10.1074/jbc.M607858200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Moran JM, Buller RML, McHowat J, Turk J, Wohltmann M, Gross RW, Corbett JA. J Biol Chem. 2005;280:28162–28168. doi: 10.1074/jbc.M500013200. [DOI] [PubMed] [Google Scholar]
- 40.Whitman SC, Daugherty A, Post SR. Journal of lipid research. 2000;41:807–813. [PubMed] [Google Scholar]
- 41.Bagrodia S, Taylor SJ, Jordon KA, Van Aelst L, Cerione RA. J Biol Chem. 1998;273:23633–23636. doi: 10.1074/jbc.273.37.23633. [DOI] [PubMed] [Google Scholar]
- 42.Haberland ME, Fogelman AM. Proceedings of the National Academy of Sciences of the United States of America. 1985;82:2693–2697. doi: 10.1073/pnas.82.9.2693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hazen SL, Zupan LA, Weiss RH, Getman DP, Gross RW. J Biol Chem. 1991;266:7227–7232. [PubMed] [Google Scholar]
- 44.Seno K, Okuno T, Nishi K, Murakami Y, Watanabe F, Matsuura T, Wada M, Fujii Y, Yamada M, Ogawa T, Okada T, Hashizume H, Kii M, Hara S, Hagishita S, Nakamoto S, Yamada K, Chikazawa Y, Ueno M, Teshirogi I, Ono T, Ohtani M. J Med Chem. 2000;43:1041–1044. doi: 10.1021/jm9905155. [DOI] [PubMed] [Google Scholar]
- 45.Peters-Golden M, Feyssa A. Am J Physiol Lung Cell Mol Physiol. 1993;264:L438–447. doi: 10.1152/ajplung.1993.264.5.L438. [DOI] [PubMed] [Google Scholar]
- 46.Needleman P, Truk J, Jakschik BA, Morrison AR, Lefkowith JB. Annual Review of Biochemistry. 1986;55:69–102. doi: 10.1146/annurev.bi.55.070186.000441. [DOI] [PubMed] [Google Scholar]
- 47.Rosolowsky M, Campbell WB. Am J Physiol Heart Circ Physiol. 1993;264:H327–335. doi: 10.1152/ajpheart.1993.264.2.H327. [DOI] [PubMed] [Google Scholar]
- 48.Zamora R, Bult H, Herman AG. European Journal of Pharmacology. 1998;349:307–315. doi: 10.1016/s0014-2999(98)00211-8. [DOI] [PubMed] [Google Scholar]
- 49.Yamada M, Niki H, Yamashita M, Mue S, Ohuchi K. J Pharmacol Exp Ther. 1997;281:1005–1012. [PubMed] [Google Scholar]
- 50.Post SR, Gass C, Rice S, Nikolic D, Crump H, Post GR. Journal of lipid research. 2002;43:1829–1836. doi: 10.1194/jlr.m200231-jlr200. [DOI] [PubMed] [Google Scholar]
- 51.Miki S, Tsukada S, Nakamura Y, Aimoto S, Hojo H, Sato B, Yamamoto M, Miki Y. FEBS Lett. 1996;399:241–244. doi: 10.1016/s0014-5793(96)01332-4. [DOI] [PubMed] [Google Scholar]
- 52.Coller SP, Paulnock DM. J Leukoc Biol. 2001;70:142–148. [PubMed] [Google Scholar]
- 53.Fong LG, Le D. J Biol Chem. 1999;274:36808–36816. doi: 10.1074/jbc.274.51.36808. [DOI] [PubMed] [Google Scholar]
- 54.Hsu HY, Hajjar DP, Khan KM, Falcone DJ. J Biol Chem. 1998;273:1240–1246. doi: 10.1074/jbc.273.2.1240. [DOI] [PubMed] [Google Scholar]
- 55.Teslenko V, Rogers M, Lefkowith JB. Biochimica et Biophysica Acta (BBA) - Lipids and Lipid Metabolism. 1997;1344:189–199. doi: 10.1016/s0005-2760(96)00137-3. [DOI] [PubMed] [Google Scholar]
- 56.Lefkowith JB, Lennartz MR, Rogers M, Morrison AR, Brown EJ. J Immunol. 1992;149:1729–1735. [PubMed] [Google Scholar]
- 57.Dennis EA. J Biol Chem. 1994;269:13057–13060. [PubMed] [Google Scholar]
- 58.Takai Y, Sasaki T, Matozaki T. Physiol Rev. 2001;81:153–208. doi: 10.1152/physrev.2001.81.1.153. [DOI] [PubMed] [Google Scholar]
- 59.Rossman KL, Der CJ, Sondek J. Nature Reviews Molecular Cell Biology. 2005;6:167–180. doi: 10.1038/nrm1587. [DOI] [PubMed] [Google Scholar]
- 60.DerMardirossian C, Bokoch GM. Trends in Cell Biology. 2005;15:356–363. doi: 10.1016/j.tcb.2005.05.001. [DOI] [PubMed] [Google Scholar]
- 61.Dransart E, Olofsson B, Cherfils J. Traffic. 2005;6:957–966. doi: 10.1111/j.1600-0854.2005.00335.x. [DOI] [PubMed] [Google Scholar]
- 62.Chuang TH, Bohl BP, Bokoch GM. J Biol Chem. 1993;268:26206–26211. [PubMed] [Google Scholar]
- 63.Dormond O, Foletti A, Paroz C, Ruegg C. Nat Med. 2001;7:1041–1047. doi: 10.1038/nm0901-1041. [DOI] [PubMed] [Google Scholar]
- 64.Shin EA, Kim KH, Han SI, Ha KS, Kim JH, Kang KI, Kim HD, Kang HS. FEBS Letters. 1999;452:355–359. doi: 10.1016/s0014-5793(99)00657-2. [DOI] [PubMed] [Google Scholar]
- 65.Woo CH, Kim BC, Kim KW, Yoo MH, Eom YW, Choi EJ, Sun Na D, Kim JH. Biochemical and Biophysical Research Communications. 2000;268:231–236. doi: 10.1006/bbrc.2000.2102. [DOI] [PubMed] [Google Scholar]
- 66.Peppelenbosch MP, Qiu RG, de Vries-Smits AMM, Tertoolen LGJ, de Laat SW, McCormick F, Hall A, Symons MH, Bos JL. Cell. 1995;81:849–856. doi: 10.1016/0092-8674(95)90005-5. [DOI] [PubMed] [Google Scholar]
- 67.Horiuchi S, Unno Y, Usui H, Shikata K, Takaki K, Koito W, Sakamoto YI, Nagai R, Makino K, Sasao A, Wada JUN, Makino H. Ann NY Acad Sci. 2005;1043:671–675. doi: 10.1196/annals.1333.076. [DOI] [PubMed] [Google Scholar]
- 68.Fukuhara-Takaki K, Sakai M, Sakamoto Yi, Takeya M, Horiuchi S. J Biol Chem. 2004:M408715200. doi: 10.1074/jbc.M408715200. [DOI] [PubMed] [Google Scholar]
- 69.Hiltunen TP, Luoma JS, Nikkari T, Yla-Herttuala S. Circulation. 1998;97:1079–1086. doi: 10.1161/01.cir.97.11.1079. [DOI] [PubMed] [Google Scholar]
- 70.Li Sl, Reddy MA, Cai Q, Meng L, Yuan H, Lanting L, Natarajan R. Diabetes. 2006;55:2611–2619. doi: 10.2337/db06-0164. [DOI] [PubMed] [Google Scholar]
- 71.Hatley ME, Srinivasan S, Reilly KB, Bolick DT, Hedrick CC. J Biol Chem. 2003;278:25369–25375. doi: 10.1074/jbc.M301175200. [DOI] [PubMed] [Google Scholar]
- 72.Hiltunen T, Luoma J, Nikkari T, Yla-Herttuala S. Circulation. 1995;92:3297–3303. doi: 10.1161/01.cir.92.11.3297. [DOI] [PubMed] [Google Scholar]
- 73.Cyrus T, Pratico D, Zhao L, Witztum JL, Rader DJ, Rokach J, FitzGerald GA, Funk CD. Circulation. 2001;103:2277–2282. doi: 10.1161/01.cir.103.18.2277. [DOI] [PubMed] [Google Scholar]
- 74.Sun D, Funk CD. J Biol Chem. 1996;271:24055–24062. [PubMed] [Google Scholar]
- 75.Bolick DT, Orr AW, Whetzel A, Srinivasan S, Hatley ME, Schwartz MA, Hedrick CC. Arterioscler Thromb Vasc Biol. 2005;25:2301–2307. doi: 10.1161/01.ATV.0000186181.19909.a6. [DOI] [PubMed] [Google Scholar]
- 76.Huber J, Furnkranz A, Bochkov VN, Patricia MK, Lee H, Hedrick CC, Berliner JA, Binder BR, Leitinger N. J Lipid Res. 2006;47:1054–1062. doi: 10.1194/jlr.M500555-JLR200. [DOI] [PubMed] [Google Scholar]
- 77.Cyrus T, Witztum JL, Rader DJ, Tangirala R, Fazio S, Linton MF, Funk CD. J Clin Invest. 1999;103:1597–1604. doi: 10.1172/JCI5897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Chinnici CM, Yao Y, Ding T, Funk CD, Pratico D. Am J Pathol. 2005;167:1371–1377. doi: 10.1016/S0002-9440(10)61224-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Bleich D, Chen S, Zipser B, Sun D, Funk CD, Nadler JL. J Clin Invest. 1999;103:1431–1436. doi: 10.1172/JCI5241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Suzuki H, Kurihara Y, Takeya M, Kamada N, Kataoka M, Jishage K, Ueda O, Sakaguchi H, Higashi T, Suzuki T, Takashima Y, Kawabe Y, Cynshi O, Wada Y, Honda M, Kurihara H, Aburatani H, Doi T, Matsumoto A, Azuma S, Noda T, Toyoda Y, Itakura H, Yazaki Y, Horiuchi S, Takahashi K, Kruijt JK, van Berkel TJC, Steinbrecher UP, Ishibashi S, Maeda N, Gordon S, Kodama T. Nature. 1997;386:292–296. doi: 10.1038/386292a0. [DOI] [PubMed] [Google Scholar]
- 81.Usui HK, Shikata K, Sasaki M, Okada S, Matsuda M, Shikata Y, Ogawa D, Kido Y, Nagase R, Yozai K, Ohga S, Tone A, Wada J, Takeya M, Horiuchi S, Kodama T, Makino H. Diabetes. 2007;56:363–372. doi: 10.2337/db06-0359. [DOI] [PubMed] [Google Scholar]