

Abstract
De novo proteins from designed combinatorial libraries were bound to heme terminated gold electrodes. The novel heme proteins were shown to possess peroxidase activity, and this activity was compared to that of horseradish peroxidase and bovine serum albumin when immobilized in a similar fashion. The various designed proteins from the libraries displayed distinctly different levels of peroxidase activity, thereby demonstrating that the sequence and structure of a designed protein can exert a substantial effect on the peroxidase activity of immobilized heme.
Keywords: Protein Design, Binary Patterning, Cyclic Voltammetry, Redox Protein, Heme Protein, Peroxidase activity, Horseradish peroxidase, Chronocoulometry
1. Introduction
The design of novel proteins that fold into native-like structures capable of molecular recognition and catalytic activity is emerging as an important goal of protein chemistry. Efforts are being made to design proteins with similar functions and activities to those of natural enzymes [1–11]. Such work is motivated both by a desire to understand the fundamental features necessary for catalytic activity, and by expectations of future applications in biotechnology. One natural enzyme that has received considerable attention is horseradish peroxidase (HRP) [12–14]. In this paper, the peroxidase activity of de novo designed proteins immobilized on a heme modified electrode are compared to HRP immobilized in a similar fashion.
The de novo proteins described in the current study were chosen from a library of binary patterned sequences designed to fold into four-helix bundles [15–17]. Previously, we demonstrated that several proteins from the first generation (74-residue) library, and all the proteins from a second generation (102-residue) library bind heme and show heme-dependent activities such as reversible binding of carbon monoxide (CO) and catalysis of peroxidase chemistry [18–22].
In the current study, we exploited the affinity of the de novo proteins for heme to immobilize them on self assembled monolayers on a gold electrode. The method of attaching the proteins to the heme modified electrode is shown in Fig. 1. A similar method of heme protein reconstitution on self-assembled monolayers was described previously for HRP [23]. Analogous studies have been carried out to assemble apo- glucose oxidase onto FAD terminated monolayers immobilised on a surface. The work described in the current report extends the application of the technique by demonstrating the generality of the approach [24, 25].
Fig. 1.

Schematic representation of reconstitution of protein on heme modified electrodes. Initially the gold electrode was coated with mixed thiol monolayer consisting of a 1:5 ratio of longer alkyl-thiols bearing an activated functional group and short unsubstituted alkyl-thiols. Incubation was in DMSO for 90 min at room temperature. The coated gold electrode was then reacted with 20 mM 1,12 diaminododecane which, reacts only with the thiols bearing activated functional group. The resulting electrode has amine terminated groups which are covalently coupled to the carboxyl groups on Heme (Fe(III) protoporphyrin IX), using EDC (1-ethyl-3-(3-dimethylaminopropyl)carbodiimide) coupling agent. Finally, the heme terminated electrode is reconstituted with the protein to form heme protein on surface.
The peroxidase activity of an immobilized enzyme can be measured electrochemically as explained below [26]. The normal catalytic cycle for peroxidase activity is described in scheme 1.
Scheme 1.

P and Q are the oxidized and reduced forms of a mediator, which is a redox cosubstrate that shuttles electrons from the electrode to the redox center in the protein.
As shown in the scheme 1, a heme-protein immobilized on an electrode can be oxidized by hydrogen peroxide to form an oxyferryl π-cation radical heme intermediate. Subsequently, the enzyme is converted back to its native resting state, ferric porphyrin, by a one-electron/two-proton reduction by a mediator. The oxidized counterpart of the mediator can be reduced by electrons from the electrode. The charge obtained from the catalytic reduction of mediator is correlated to the concentration of the oxyferryl species in the solution. Hence, this charge can be used to quantify the peroxidase activity of the immobilized protein-heme system using electrochemical methods such as chronocoulometry or chronoamperometry. Here we describe the use of chronocoulometry to quantify the peroxidase activity of the immobilized protein-heme system. This approach enables the direct measurement of the charge versus time response to an applied potential step [27].
2. Results
2.1 Immobilization and characterization of the de novo proteins bound to heme terminated gold electrodes
We exploited the affinity of the de novo proteins for heme to immobilize them on self assembled monolayers of heme on a gold electrode. The method of attaching the proteins to the heme modified electrode is shown in Fig. 1. Previously, a similar method of heme protein reconstitution on self-assembled monolayers was described for HRP [23]. Cyclic voltammetry was used to electrochemically characterize the electrode surface as shown in Fig. 2. The surface coverage of heme on a gold surface was estimated by integrating the area under the cathodic or anodic peak in the cyclic voltammogram of the heme modified electrode (before attachment of protein) [28]. Surface coverage was estimated to be ~10 pmol cm−2.
Fig. 2.
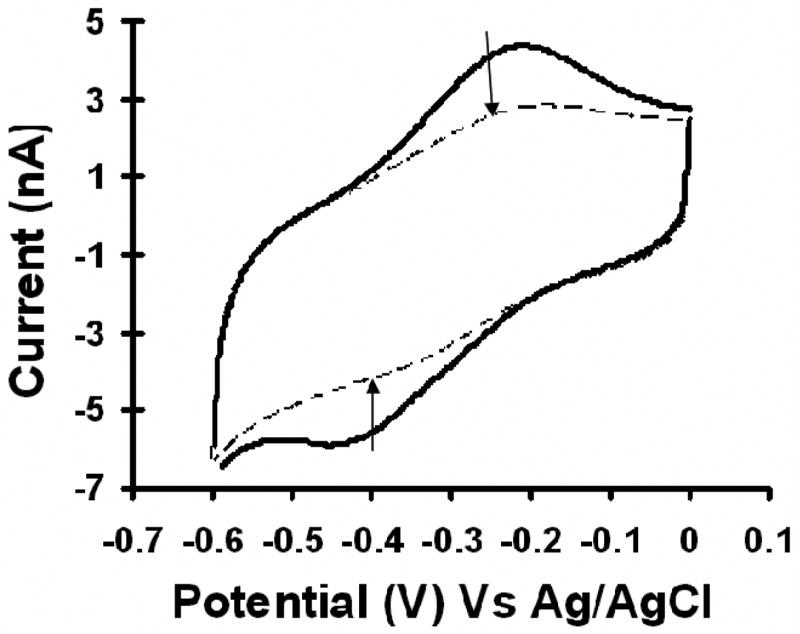
Cyclic voltammogram (CV) of the heme electrode reconstituted with protein. Note that the peaks of the heme modified electrode (solid dark line) decrease on treatment with protein S-824 (inner dashed line).
The heme modified electrode was then treated with a solution of protein – either HRP or one of the de novo designed proteins. (We assume that binding of HRP or the de novo protein to the heme coated electrode leads to the complete reconstitution of a holo-protein capable of enzymatic activity.) Treatment of the heme electrode with de novo protein S-824 caused the heme peak in the cyclic voltammogram to diminish significantly. Similar results were obtained with the other de novo proteins from the binary patterned collection, and for bovine serum albumin (BSA) and HRP.
There is a positive shift of ~30 mV in midpoint potential upon binding of protein S-824 to the immobilized heme. In solution, the redox potentials of the de-novo heme proteins are approximately −175 mV, and the redox potential of heme is −220mV (both vs SHE) [21]. Thus in solution, a shift in redox potential of approximately + 45 mV is observed upon binding protein to heme. However, on the surface of an electrode, the corresponding shift is only + 30 mV. This difference in behavior could be due to incomplete reconstitution of the heme-protein and/or electrostatic interactions due to protein binding on the surface as compared to reconsitution in solution.
The diminished peaks and shift in midpoint potential are indicative of interaction of heme with the ligating residues in the proteins [23]. Binding of protein also causes a decrease in peak-to-peak separation, which might indicate faster kinetics.
2.2 Determination of peroxidase activity of proteins bound to a heme modified surface
Chronocoulometry can be used to detect the peroxidase activity of a heme-protein. From the catalytic cycle for peroxidase activity (Scheme 1), it can be seen that in step 3, the enzyme oxidized by peroxide is converted back to ferric porphyrin by a two electron reduction by the mediator. The oxidized counterpart of the mediator gets reduced by electrons from the electrode. Therefore, the charge obtained on catalytic reduction of mediator can be used to quantify the peroxidase activity of the immobilized enzyme. The total charge is measured by chronocoulometry. Fig. 3 shows the chronocoulograms obtained for immobilized heme-HRP with increasing peroxide concentration.
Fig. 3.
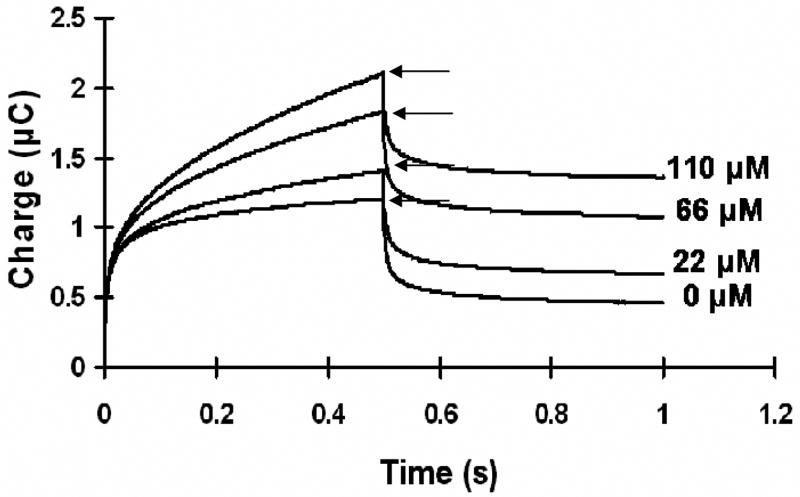
The chronocoulometric response (charge versus time response to an applied potential step) obtained for the immobilized heme-HRP system with successive addition of hydrogen peroxide to the solution. The arrows point towards the charge value corresponding to each addition of peroxide.
The net change in reduction charge from the chronocoulograms is plotted in Fig. 4 against the concentration of peroxide for five de novo proteins. In addition, HRP and ‘bare heme’ (immobilized on the electrode but without added protein) were measured as positive controls, while BSA and ‘No Heme’ served as negative controls. The response of the surface with no heme was measured, and found to have negligible activity. This indicates that the surface is densely packed with self-assembled monolayers (SAM) of thiols and has very few defects (exposed gold surface).
Fig. 4.

The net electrocatalytic reduction charge (after background subtraction of charge at 0 μM of peroxide) is plotted against the concentration of peroxide at heme modified electrodes coated with different proteins. The potential is stepped from 0 mV to −500 mV and the pulse width is 500 ms. The mediator used for the HRP and the de novo proteins is hydroxyquinone. Table 1 lists the net electrocatalytic charge (ΔQ) due to peroxidase activity of the proteins at 200 μM concentration of peroxide.
Comparison of the peroxidase activity of protein n86, heme, BSA and HRP
Among the immobilized de novo proteins, n86 (from the 74-residue first generation library) had the highest level of activity – significantly higher than BSA-heme or bare heme. Previously, it had been shown that in solution, the peroxidase activity of n86 was ~1/3 that of HRP [19]. The data shown in Fig. 3 and table 1 indicate that in the immobilized state, n86 is ~1/2 as active as HRP (assayed at a peroxide concentration of 200 μM).
Table 1.
The net charge (ΔQ) due to peroxidase activity of the protein at 200 μM peroxide. The ratio of the net charge of the protein and the net charge of HRP at the same concentration is also listed.
| Protein Immobilized on Heme modified electrode | ΔQ200μM | |
|---|---|---|
| HRP | 2.10 | 1 |
| Protein n86 | 1.06 | 0.51 |
| Protein s-23 | 0.67 | 0.32 |
| Protein s-824 | 0.55 | 0.26 |
| Protein s-836 | 0.58 | 0.28 |
| Bare Heme | 0.52 | 0.25 |
| Protein s-213 | 0.27 | 0.13 |
| Protein s-285 | 0.24 | 0.12 |
| BSA | 0.19 | 0.09 |
As shown in Fig. 4 and table 1, bare heme (without protein) shows peroxidase activity around 1/4 that of immobilized HRP. This stands in contrast to a ratio of ~1/430 using heme free in solution [19, 29]. This finding can be explained by the following: Heme immobilized on a surface is: (a) less prone to aggregation and subsequent precipitation; (b) has improved access for the peroxide; and (c) provides an increase in electron-transfer rate from the electrode to the heme. Indeed, a related study of heme immobilized on gold surface by a short organic linker showed that the peroxidase activity of immobilized heme was similar to that of microperoxidase – even though bare heme lacks the imidazole ring coordinating the central iron atom [30].
Fig. 4 shows that the peroxidase activity of immobilized heme decreases by 62% on binding BSA. BSA is known to inhibit heme peroxidase activity in solution by 50 to 60% [31, 32]. This BSA control shows that protein binding per se is not reponsible for the observed peroxidase activity. Thus, binding of BSA reduces the peroxidase activity of heme, whereas binding of protein n86 enhances peroxidase activity.
Comparison of peroxidase activity of second generation de novo proteins with protein n86 (first generation protein)
The peroxidase activities of the second generation proteins (marked with the ‘S’ prefix) were compared to protein n86. The second generation proteins were designed from n86 by elongating the four alpha helices [16]. The second generation apo-proteins are structurally and thermodynamically superior to protein n86; all of them are considerably more stable, and several of them (S-824 and S-836) have native-like 3-dimensional structures as determined by NMR [16, 17]. Earlier studies of the CO binding affinity showed that although the second generation apo-proteins are very different from protein n86 in terms of packing and rigidity, they probably have similarly exposed heme binding sites [20]. Here we show that although similar heme binding sites in these proteins produced similar CO binding affinities, these sites do not yield similar peroxidase activities.
Among the de novo heme proteins studied thus far, n86 from the first generation library shows the highest peroxidase activity. The second generation proteins show considerable variation of peroxidase activity: Protein S-23 shows the highest activity – approximately 1/3 the activity of HRP. Proteins S-824 and S-836 show activity in the range of immobilized heme, and proteins S-213 and S-285 show poor peroxidase activity – similar to heme-BSA. Fig. 4 shows that the peroxidase activity of HRP and n86 are directly proportional to the increasing concentration of peroxide in the range of concentration studied. It is expected that the activity saturates at a higher concentation of peroxide (data not shown). However, the activity plots of the other proteins (and bare heme) approach saturation even in the low concentration range studied.
2.3 Peroxidase activity of the de novo proteins in solution
The peroxidase activities of the de novo proteins were also measured in solution. These solution studies were motivated by two considerations:
First, the possiblity that immobilization of the heme proteins leads to a reduction in activity had not been ruled out. Therefore activity assays in solution were performed to confirm that the trends of activity among the different proteins immobilized on the electrode mirrored the corresponding trends in solution (Fig. 5).
Fig. 5.

Peroxidase activity of the de novo proteins in solution. Purified protein was used in the assay. The typical reaction mixture contained 50 μM purified protein, 4 μM heme chloride, 10 μM TMB (3,3,5,5′ tetramethyl benzidine), and 0.00075% H2O2. Absorbance at 450 nm was recorded and was normalized by dividing it with concentration of protein determined from absorbance at 280 using extinction coefficient of 6990 cm−1.
Second, one might have hypothesized that the flexible proteins n86 and S-23 would bind to the surface more easily than the well-folded proteins S-824, S-836, and that this enhanced binding would be the sole reason for the higher activity of n86 and S-23. To address these concerns, peroxidase activity was also studied in solution.
Previously, we had shown that the peroxidase activity of protesin n86 in solution is approximately 1/3 that of HRP [19]. Here, the levels of peroxidase activity in solution for protein n86 and the second generation heme proteins were compared using UV spectroscopy. As shown in Fig. 5, n86 shows the highest peroxidase activity followed by the other proteins. The trends of relative activities of the de novo proteins in solution are similar to the trends of relative activities of the same proteins immobilized on a surface. The specific activities differ in the two environments, presumably because the activities of the immobilized proteins are dictated by the kinetics of electron transfer from surface to the heme center.
3. Discussion
From the results described above, we conclude the following:
In the immobilized state, protein n86 from the first generation library is ~1/2 as active as HRP, and twice as active as heme.
In the solution state, protein n86 is 1/3 as active as HRP, and 120-fold more active than heme [19].
The second generation proteins (derived from n86 by elongating the alpha helices) show lower peroxidase activity than protein n86.
Among the second-generation proteins, de novo protein S-23 shows the highest peroxidase activity, which is ~1/6 that of HRP. Thus, the structurally less ordered de novo proteins n86 and S-23 show higher peroxidase activity than well-folded proteins S-824 and S-836.
The control, bare heme, shows a 20-fold increase in peroxidase activity when immobilized relative to heme free in solution.
Immobilized BSA-heme shows 62% lower peroxidase activity than immobilized heme.
Immobilized de novo proteins S-285 and S-213 show lower peroxidase activity than heme alone.
Taken together, these results demonstrate that proteins binding to a heme modified electrode can exert a substantial effect (increase or decrease) on the heme dependent peroxidase activity.
We speculate that the peroxidase activity shown by the de novo proteins is catalysed by a five coordinate, histidine ligated, Fe(III) heme with partial access to solvent. In the following discussion, we summarize several features consistent with this proposal.
The de novo proteins devised by the binary code strategy were not explicitly designed to bind heme. As per the binary code strategy, histidine residues (polar) were designed to be on the surface of the proteins. Upon heme binding, structural reorganization presumably moves at least one histidine side chain into a position consistent with binding to a partially buried heme. Indeed, previous results have demonstrated that although the heme sites in the de novo proteins are less buried than in myoglobin, they are more buried than in microperoxidase [20, 22]. Access of the heme to water presumably accounts for the relatively low reduction potential of the proteins: −174 mV compared to free heme, which is −220 mV (both vs NHE) [21].
The presence of a histidine (rather than methionine) ligand is supported by the observed reduction potentials. (Cysteine can be excluded because it does not occur in the sequences of de novo proteins.) In absence of structural data on the de novo heme proteins, we have no reason to expect a histidine side chain in the sixth axial position of the heme in the de novo heme protein. Indeed, our previous work demonstrated that the sixth ligand in the de novo heme proteins is easily displacable by small molecules, such as pyridine and imidazole derivatives [22]. Thus the sixth axial ligation site of the heme is likely to by occupied by an aquo ligand.
Hence, similar to horseradish peroxidase, the peroxidase activity of the de novo proteins is probably catalysed by a five coordinate Fe(III) resting state with a single histidine ligation.
Previous structural and thermodynmaic studies of the de novo proteins in their apo forms showed that proteins S-23 and n86 are less ordered (more dynamic) than proteins S-824, S-836, S-213 and S-285 [16]. Yet the results described herein demonstrate that proteins S-23 and n86 have higher levels of peroxidase activity than the other de novo proteins. Perhaps, these more flexible apo-proteins become more ordered upon binding heme, and thereby form an active site that is more efficient for peroxidase activity. In contrast, the ordered structures of apo-proteins S-824, S-836, S-213 and S-285 may become less ordered upon binding heme and may therefore be less efficient peroxidases. Indeed, formation of a five coordinate heme active site may occur more easily for the less ordered structures of n86 and S-23 compared to well ordered apo-proteins S-824, S-836, S-213 and S-285.
The difference in peroxidase activity among the various de novo proteins stands in marked contrast to their nearly uniform abilities to bind carbon monoxide [20]. The similar rates of CO association kinetics (kon) of the de novo proteins indicated that heme exposure in all of the de novo proteins were similar [20]. Despite this similarity in heme exposure, the results described in this study show that their peroxidase activities differ. These findings demonstrate that in addition to heme accessibility, other factors contribute to the relative levels of peroxidase activity of these de novo proteins.
Future work could involve stabilizing protein n86 and protein S-23 using directed mutagenesis and screening for both increased peroxidase activity and structural stability. Also, different immobilization strategies could be explored to ensure that the surface is stable at high concentrations of peroxide. For example, future studies will be done with more stable surfaces, such as silicon or ITO (Indium tin oxide). Finally, we note that modified versions of these de novo heme proteins may be useful as components in biosensors to detect peroxides.
4. Materials and Methods
4.1. Chemicals
Horse radish peroxidase (HRP; E.C.1.11.1.7) 150 units/mg, 1,12-diaminododecane and 3,3′-dithiopropionic acid di-(N-succinimidyl ester), 3-carboxypropyl disulphide, 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDC), heme (H-5533), hydroquinone, and butanone, were obtained from Aldrich-Sigma-Fluka. The buffer salts (KH2PO4.3H2O and KH2PO4) were analytical grade and DMSO was obtained from Fisher Scientific. 30 % hydrogen peroxide was obtained from Baker (Dveneter, Netherlands). All the electrodes were from Bioanalytical systems Inc. (West Lafayette, IN).
4.2. De novo Protein Expression and Purification
De novo proteins were expressed in E.Coli strain BL21 (DE3) [33] and grown in 2xYT. The proteins were extracted from the cells using a freeze-thaw protocol and then solubilized in 100 mM MgCl2 [34] Cellular contaminants were removed by acid precipitation in 50 mM sodium acetate buffer (pH 4.2) [16]. The resulting supernatant was loaded onto a POROS HS cation exchange column (PerSeptive Biosystems), and eluted using a gradient of NaCl from 0 M to 1.5 M. The buffers used for HPLC were degassed and purged with helium before use. Purified proteins were concentrated, and buffer exchanged using Centricon Plus-20 filters (Millipore). Protein expression was evaluated by SDS-PAGE. Concentrations were measured by UV-VIS spectroscopy using a Hewlett Packard 8452A diode array spectrophotometer and a molar extinction coefficient of 6990 M−1 cm −1.
4.3. Preparation of apo-HRP
Apo-HRP was purified from HRP using published methods [35, 36]. HRP was dissolved in cold 100mM sodium acetate buffer pH 2.6 to yield a final concentration of 1mg/mL. This solution was added to a separatory funnel containing ice-cold butanone (1mL). After mixing, the phases were allowed to separate, resulting in the bottom aqueous phase containing the apo-HRP and the top butanone phase containing heme [35, 36]. The aqueous phase was collected and then buffer exchanged to PBS buffer pH 7.4 using Centricon Plus-20 filters (Millipore catalog. No. UFV4BCC25). The apo-HRP was diluted with PBS buffer to a final concentration of 0.1 mg/mL. The UV spectrum of the apo-HRP was recorded to ensure there was no heme. Concentration of apo-HRP was determined using molar extinction coefficient of 20000 M−1 cm−1 at 280 nm [36]. The concentration of peroxide was determined by titration with KMnO4. The working stock solution concentration was adjusted by diluting the major stock.
4.4. Electrode preparation
Gold electrodes with a diameter of 1.6 mm were purchased from BAS (bioanalytical systems MF-2014). The electrodes were polished (bioanalytical polishing kit MF-2060) followed by electrochemical cleaning by cycling from − 3 V to + 3 V in 0.1 M H2SO4 [37]. Cyclic voltammogram (CV) recorded in 0.5 M H2SO4 was used to determine the purity of the electrode surface just before modification. The real electrode surface area of the bare gold was determined from chronocoulometry of 0.1 mM K3Fe(CN)6 in 0.1M KCl as a supporting electrolyte. From this the area of the electrode was estimated to be 0.015 cm2.
The electrodes were immobilized with heme using published methods with minor modifications [23]. The cleaned electrodes were immersed into a 100 mM solution of thiols in DMSO. The ratio of the activated to the unactivated thiols was kept constant at 1:5. After incubation for 90 minutes at room temperature, the electrodes were thoroughly rinsed with water and then immersed into 20 mM 1,12-diaminododecane. The C12 spacer (1,12 diaminodecane) was used for all proteins. It was previously shown that the distance form heme to the surface of the protein in HRP is at least 10 Å. The C12 spacer (16 Å) allows the heme to reach the active site of HRP. After 4 hours, the electrodes were thoroughly washed with water and transferred to heme coupling solution. The heme coupling solution was made as follows: heme was dissolved in DMSO for a final concentration of 1 mM and then diluted ten times in 10 mM HEPES buffer (pH 7.8) containing 150 mM EDC (1-ethyl-3-(3-dimethylaminopropyl) carbodiimide) coupling agent. The heme coupling was done at 4°C overnight. The electrodes were rinsed with distilled water followed by brief rinsing with 100 mM sodium perchlorate in DMSO to remove adsorbed heme on the surface of the electrode. Finally, the electrodes were incubated in 400 μM protein solutions for eight hours at 4°C.
4.5 Electrochemical measurements
Cyclic voltammetry and chronocoulometry experiments were recorded using a BAS 100B/W potentiostat. All measurements were carried out at ambient temperature (~22°C) in a conventional three-compartment electrochemical cell consisting of the chemically modified gold electrode as a working electrode, a platinum auxiliary electrode and a saturated Ag/AgCl/KCl electrode as reference. 50 mM sodium phosphate buffer pH 7.4 was used as the electrolyte. During the experiments inert atmosphere was maintained by providing an argon blanket at all times.
For cyclic voltammetry, the following parameters were used: the system was scanned from potential −600 to 0 mV and the scan rates were varied. The parameters used for differential pulse voltammetry are: scan rate, 20 mV/s; pulse amplitude, 50 mV; pulse period, 200 ms; pulse width, 50 ms; sample width 17 ms. All potentials are reported with respect to the Ag/AgCl/KCl reference electrode.
Mediator based electron transfer
Direct electron transfer between redox enzyme and the electrode is precluded by steric hindrance and slow electron transfer from electrode to the redox center in the protein [13, 14]. Therefore, a mediator is required to shuttle electrons between the electrode and the heme reaction center of the protein. Usually millimolar concentrations of mediator are needed when measuring micromolar concentration of peroxide in order to overcome the dependence of the signal on the mediator concentration [38]. Therefore, to measure the peroxidase activity of the proteins, 2 mM hydroquinone was added to the electrolyte solution as a mediator.
Peroxidase activity measurements
Chronocoulometry was used to determine the charge of the system due to electrocatalytic reduction of peroxide. The steady state charge after successive addition of aliquots of peroxide solution was plotted against peroxide concentration. In all experiments, high saturating concentrations of peroxide were not used. Parameters used for double step constant potential chronocoulometry were: potential, −500 mV and pulse width, 500 ms.
Acknowledgments
This work was supported by the NIH Grant R01 GM062869 (M.H.H.). A.D acknowledges FMC Graduate fellowship. The authors are grateful to Dr. Scott A. Trammell and Prof. Andrew B. Bocarsly for helpful discussions. We further thank Prof. John T. Groves for the use of the potentiostat.
Footnotes
This article is dedicated to the memory of Ed Stiefel. As his student and his colleague, the authors were educated by his intellect and inspired by his exuberance.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Arnold F. Nature. 2001;409:253–257. doi: 10.1038/35051731. [DOI] [PubMed] [Google Scholar]
- 2.Benson DE, Michael SW, Hellinga HW. Proc Nat Acad Sci USA. 2000;97:6292–6297. doi: 10.1073/pnas.97.12.6292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Moffet DA, Hecht MH. Chemical Reviews. 2001;101:3191–3204. doi: 10.1021/cr000051e. [DOI] [PubMed] [Google Scholar]
- 4.Hecht MH, Das A, Go A, Bradley LH, Wei Y. Protein Science. 2004;13:1711–1723. doi: 10.1110/ps.04690804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Baltzer L, Nilsson J. Curr Opin Biotechnol. 2001;12:355–360. doi: 10.1016/s0958-1669(00)00227-5. [DOI] [PubMed] [Google Scholar]
- 6.Bolon DN, Mayo SL. Proc Nat Acad Sci USA. 2001;98:14274–14279. doi: 10.1073/pnas.251555398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wei Y, Hecht MH. Protein Engineering Design and Selection. 2004;17:67–75. doi: 10.1093/protein/gzh007. [DOI] [PubMed] [Google Scholar]
- 8.Robertson DE, Farid RS, Moser CC, Urbauer JL, Mulholland SE, Pidikiti R, Lear JD, Wand AJ, DeGrado WF, Dutton PL. Nature. 1994;368:425–432. doi: 10.1038/368425a0. [DOI] [PubMed] [Google Scholar]
- 9.Tommos C, Skalicky JJ, Pilloud DL, Wand AJ, Dutton PL. Biochemistry. 1999;38:9495–9507. doi: 10.1021/bi990609g. [DOI] [PubMed] [Google Scholar]
- 10.Kaplan J, DeGrado WF. Proc Nat Acad Sci USA. 2004;101:11566–11570. doi: 10.1073/pnas.0404387101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Korkegian A, Black ME, Baker D, Stoddard BL. Science. 2005;308:857–860. doi: 10.1126/science.1107387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Veitch NC. Phytochemistry. 2004;65:249–259. doi: 10.1016/j.phytochem.2003.10.022. [DOI] [PubMed] [Google Scholar]
- 13.Limoges B, Saveant JM, Yazidi D. J Am Chem Soc. 2003;125:9192–203. doi: 10.1021/ja0354263. [DOI] [PubMed] [Google Scholar]
- 14.Dequaire M, Limoges B, Moiroux J, Saveant JM. J Am Chem Soc. 2002;124:240–253. doi: 10.1021/ja0170706. [DOI] [PubMed] [Google Scholar]
- 15.Kamtekar S, Schiffer JM, Xiong H, Babik JM, Hecht MH. Science. 1993;262:1680–1685. doi: 10.1126/science.8259512. [DOI] [PubMed] [Google Scholar]
- 16.Wei Y, Liu T, Sazinsky SL, Moffet DA, Pelczer I, Hecht MH. Protein Sci. 2003;12:92–102. doi: 10.1110/ps.0228003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wei Y, Kim S, Fela D, Baum J, Hecht MH. Proc Natl Acad Sci USA. 2003;100:13270–13273. doi: 10.1073/pnas.1835644100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rojas NRL, Kamtekar S, Simons CT, McLean JE, Vogel KM, Spiro TG, Farid RS, Hecht MH. Protein Science. 1997;6:2512–2524. doi: 10.1002/pro.5560061204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Moffet DA, Certain LK, Smith AJ, Kessel AJ, Beckwith KA, Hecht MH. J Am Chem Soc. 2000;122:7612–7613. [Google Scholar]
- 20.Moffet DA, Case MA, House JC, Vogel K, Williams RD, Spiro TG, McLendon GL, Hecht MH. J Am Chem Soc. 2001;123:2109–2115. doi: 10.1021/ja0036007. [DOI] [PubMed] [Google Scholar]
- 21.Moffet DA, Foley J, Hecht MH. Biophysical Chemistry. 2003;105:231–239. doi: 10.1016/s0301-4622(03)00072-3. [DOI] [PubMed] [Google Scholar]
- 22.Das A, Hecht MH. Biophysical Chemistry. 2006;123:102–112. doi: 10.1016/j.bpc.2006.04.011. [DOI] [PubMed] [Google Scholar]
- 23.Zimmermann H, Lindgren A, Schuhmann W, Gorton L. Chemistry. 2000;6:592–599. doi: 10.1002/(sici)1521-3765(20000218)6:4<592::aid-chem592>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- 24.Riklin A, Katz E, Willner I, Stocker A, Buckmann AF. Nature. 1995;376:672–675. doi: 10.1038/376672a0. [DOI] [PubMed] [Google Scholar]
- 25.Zayats M, Katz E, Willner I. J Am Chem Soc. 2002;124:2120–2121. doi: 10.1021/ja025503e. [DOI] [PubMed] [Google Scholar]
- 26.Dunford HB. In: Peroxidases in Chemistry and Biology. Everse J, Everse KE, Grisham MB, editors. II. CRC press; Boca Raton, FL: 1991. pp. 1–24. [Google Scholar]
- 27.Bott AW, Heineman WR. Current Separations. 2004;20:121–126. [Google Scholar]
- 28.Kolpin CF, Swofford HS., Jr Analytical Chemistry. 1978;50:916–920. doi: 10.1021/ac50029a024. [DOI] [PubMed] [Google Scholar]
- 29.Cochran AG, Schultz PG. J Am Chem Soc. 1990;112:9414–9415. [Google Scholar]
- 30.Lotzbeyer T, Schuhmann W, Schmidt HL. Journal of Electroanalytical Chemistry. 1995;395:341–344. [Google Scholar]
- 31.Beaven GH, Chen S, D’Albis A, Gratm WB. Eur J Biochem. 1974;41:539–546. doi: 10.1111/j.1432-1033.1974.tb03295.x. [DOI] [PubMed] [Google Scholar]
- 32.Grinberg LN, O’Brien PJ, Hrkal Z. Free Radic Biol Med. 1999;27:214–219. doi: 10.1016/s0891-5849(99)00082-9. [DOI] [PubMed] [Google Scholar]
- 33.Studier FW, Rosenberg AH, Dunn JJ, Dubendorff JW. Methods in Enzymology. 1990;185:60–89. doi: 10.1016/0076-6879(90)85008-c. [DOI] [PubMed] [Google Scholar]
- 34.Johnson BH, Hecht MH. Biotechnology. 1994;12:1357–1360. doi: 10.1038/nbt1294-1357. [DOI] [PubMed] [Google Scholar]
- 35.Teale FWJ. Biochim Biophys Acta. 1959;35:543. doi: 10.1016/0006-3002(59)90407-x. [DOI] [PubMed] [Google Scholar]
- 36.Tamura M, Asakura T, Yonetani T. Biochim Biophys Acta. 1972;268:292–304. doi: 10.1016/0005-2744(72)90324-5. [DOI] [PubMed] [Google Scholar]
- 37.Katz EY, Solov’ev AA. Journal of Electroanalytical Chemistry. 1990;291:171–186. [Google Scholar]
- 38.Ruzgas T, Csoeregi E, Emneus J, Gorton L, Marko-Varga G. Anal Chim Acta. 1996;330:123–138. [Google Scholar]