

Summary
The acquisition of drug resistance by Plasmodium falciparum has severely curtailed global efforts to control malaria. Our ability to define resistance has been greatly enhanced by recent advances in Plasmodium genetics and genomics. Sequencing and microarray studies have identified thousands of polymorphisms in the P. falciparum genome, and linkage disequilibrium analyses have exploited these to rapidly identify known and novel loci that influence parasite susceptibility to antimalarials such as chloroquine, quinine, and sulfadoxine-pyrimethamine. Genetic approaches have also been designed to predict determinants of in vivo resistance to new antimalarials such as the artemisinins. Transfection methodologies have defined the role of determinants including pfcrt, pfmdr1 and dhfr. This knowledge can be leveraged to develop more efficient methods of surveillance and treatment.
Introduction
Malaria devastates the lives of millions of people each year. Eradication efforts based on the use of chloroquine (CQ) faltered in the 1960s, following the development of drug-resistant parasites [1]. Other antimalarial drug regimens, such as sulfadoxine-pyrimethamine (SP), have also selected for resistant parasites [2]. Recent genetics and genomics advances have paved the way for discoveries into the origins and spread of antimalarial drug resistance and the underlying molecular mechanisms. Researchers can now use data from genome sequencing projects to identify genetic regions linked to resistance phenotypes. The development of transfection and integration techniques permits researchers to test candidate genes for their contribution to resistance under controlled laboratory conditions. Genetic markers can also now be readily tracked in natural populations. These innovations can be used to predict drug efficacy in the field, with implications for public health policy. Here, we review how these new methodologies can expand and accelerate research into antimalarial drug resistance.
Genomic Studies
Using Polymorphisms to Identify Resistance Loci
The sequencing and annotation of the 23 Mb P. falciparum genome in 2002 provided a superb resource for localizing and identifying gene candidates within a particular locus [3]. Linking a specific locus with a given phenotype such as drug resistance, however, requires the ability to compare the genotypes of resistant and sensitive parasites. Rather than sequencing the entire genome of each resistant or sensitive clone, recent advances have exploited the presence of conserved polymorphisms in the genome as surrogate markers for the resistance determinant(s). Polymorphisms can include microsatellites (consisting of repeats of a short nucleotide sequence), single nucleotide polymorphisms (SNPs), or small insertions or deletions (indels).
A trio of papers, published in Nature Genetics in early 2007, moved the field substantially closer to a comprehensive polymorphism map for the P. falciparum genome [4–6]. These papers describe the sequencing of the entire genome, or selected regions, from multiple P. falciparum strains. The authors estimate the number of SNPs in the P. falciparum genome as ranging from about 25,000 to 50,000, corresponding to one SNP every 400 to 800 base pairs. In P. falciparum, as in humans, these SNPs do not segregate randomly. Instead they tend to cluster in “blocks,” called haplotype blocks, delimited by recombination hotspots. Association studies thus need only to track a signature set of SNP tags that identify a particular haplotype block. Studies indicate that recombination rates vary substantially between different strains of P. falciparum, with ones in Africa demonstrating the highest rates [7]. The number of polymorphisms varies for different gene classes and for different regions within chromosomes. This presumably reflects the influence of diversifying selection exerted on genes by factors such as host immunity and drug selection. [4–8]. High rates of recombination, such as that observed among African P. falciparum strains [7], will tend to obscure the linkage between ancestral traits. The phenomenon of drug resistance, however, is a relatively recent evolutionary event. Consequently, the use of linkage disequilibrium (LD) is ideally suited for tracking the spread of a resistance gene throughout a population.
Roper et al. [9] analyzed microsatellites surrounding alleles of the dihydrofolate reductase (dhfr) gene that confer resistance to pyrimethamine. They concluded that the most resistant form of dhfr commonly found in Africa, characterized by three point mutations, was associated with a set of related haplotypes that originated in Southeast Asia (Figure 1). Data collected subsequently by McCollum et al. [10], suggests that triple mutant parasites in Africa may have had additional independent origins. The findings of Roper et al. echoed the work of Wootton et al. [11] who suggested that CQ resistance spread in a selective sweep from Asia into Africa. That conclusion was based on the extensive LD among microsatellite markers surrounding the previously identified [12] Plasmodium falciparum chloroquine resistance transporter (pfcrt).
Figure 1. Identification of a selective sweep of mutant dhfr conferring pyrimethamine resistance from Asia to Africa.

(a) Genotype data are shown for 12 Thai isolates with dhfr alleles that harbor 2–4 resistance mutations, 24 African isolates with triple-mutant alleles, and 18 African parasites with sensitive dhfr alleles. The four-letter codes designate amino acids* present at positions 51, 59, 108, and 164 in the predicted DHFR protein. Amino acids conferring resistance are underlined, and dhfr alleles are shaded yellow, orange, red, and black in order of increasing resistance. The sensitive allele is shaded blue. Fragment lengths are shown for eight microsatellites positioned at –0.1, –4.4, –5.3, –10, and –20 kb upstream and +0.5, +6, +10 kb and +20 kb downstream of dhfr. Dots and yellow shading indicate microsatellite sizes that are identical to the predominant resistant haplotype (shown on rightmost column). Figure reproduced from Roper et al. [9], reprinted with permission from AAAS. (b) Selective sweep of dhfr triple mutants. Resistance to pyrimethamine originated and spread in Southeast Asia in the early 1970s. Triple mutant resistant parasites arrived circa 1978 in Africa, by unknown routes, and spread in a selective sweep. Data from McCollum et al. [10] suggest an independent origin for South American dhfr resistance alleles. Orange shading denotes malaria endemic regions. Stars represent approximate origins of identified pyrimethamine resistance sweeps. Figure adapted from Tim Anderson with the author’s kind permission. *Single-letter abbreviations are: C, Cys; I, Ile; L, Leu; N, Asn; R, Arg; S, Ser.
More recently, Volkman et al. [6] used SNPs, identified in their extensive sequencing project, to analyze 12 culture-adapted parasite lines with differing drug response profiles. They detected several selective sweeps associated with CQ resistance, including the previously described region on chromosome 7 containing pfcrt, as well as loci on chromosomes 5 (harboring the multidrug resistance gene homolog pfmdr1) and 11. Focusing on pyrimethamine resistant clones, they were able to detect two candidate selective sweeps on chromosomes 13 and 14, which were of particular interest because they demonstrated a stronger signal than the previously identified sweep at the dhfr locus on chromosome 4 [9, 13, 14].
Rapid Identification of Resistance Loci
A promising technique for exploiting polymorphisms was described by Kidgell et al. [15], who used a P. falciparum microarray to analyze genome variability. This array contained 25-mer probes covering approximately 50 percent of all coding regions. Polymorphisms were identified by measuring the loss of hybridization signal associated with mismatches between genomic DNA and the 25-mer probes. Gene amplifications were identified via their increased hybridization intensity. Using 14 cloned P. falciparum lines, a total of 23,653 single feature polymorphisms were identified, which included both SNPs and indels. This data set revealed a region on chromosome 7, encompassing pfcrt, that demonstrated extensive LD in the CQ resistant clones. They also identified numerous clones with a gene amplification of GTP-cyclohydrolase, an enzyme in the folate biosynthesis pathway (Figure 2). The authors hypothesized that this amplification might represent a novel mechanism of antifolate resistance. The power of this system, as noted by the authors, is that “tens of thousands of genetic markers can be both discovered and typed in as little as one day in any parasite isolate, potentially using only a few milliliters of infected patient blood.” The potential for rapidly identifying resistance loci from a sampling of clinical isolates stands in marked contrast to the classical approach of crossing a CQ resistant and sensitive clone, which was first reported in 1990 and culminated in the identification of pfcrt a full ten years later [12].
Figure 2. Identification of a genetic locus of variable copy number, postulated to alter parasite susceptibility to SP.
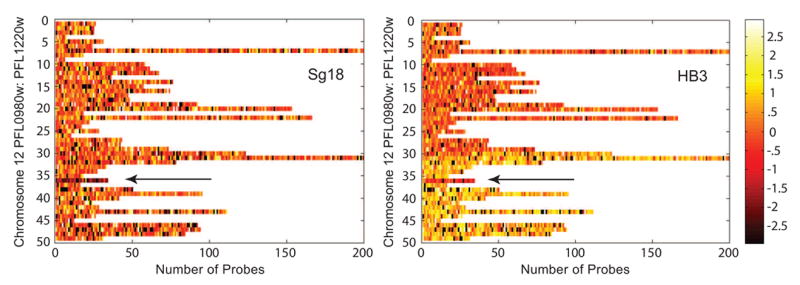
This heat map, generated from a high-density microarray analysis, shows the base 2 logarithm of the ratio of the normalized, background-subtracted probe signal for the listed lines (Sg18 and HB3), relative to the sequenced line 3D7 [15]. All probes on the array were selected to be unique within 3D7. Each horizontal bar represents a single gene on the right arm of chromosome 12. Dark probes, with low signal intensities, do not match any region in the genome of the listed line, while light probes contain matches to multiple regions of the genome. Based on comparisons of multiple clones, the authors detected heterogeneity in probe signal intensities at and around the GTP-cyclohydrolase gene (PFL1155w), indicated by the arrow. Further investigations demonstrated that the 3D7 reference line for the microarray had multiple copies of PFL1155w. Thus some lines, such as Sg18, demonstrate a reduced hybridization signal (dark probes) at this locus relative to 3D7. Others, such as HB3, demonstrate neutral or even increased hybridization signals, implying that they harbor amplifications of the PFL1155w gene. Amplification of DNA on either side of PFL1155w is also apparent in HB3 compared to 3D7. The authors postulate that the PFL1155w amplification may affect SP sensitivity. Figure reproduced from Kidgell et al. [15] with kind permission from Elizabeth Winzeler and PLoS Pathogens.
Identifying Multiple Contributing Loci
While CQ sensitivity is primarily determined by pfcrt, for other drugs the situation is not always as clear. Multiple genes located at different loci may each contribute additively, or depend on pairwise interactions, to produce a resistant phenotype. Resistance to quinine, a drug used for over 350 years, exhibits such a phenotype. To explore this, Ferdig et al. [16] assessed 35 independent progeny, derived from the cross of a low level quinine resistant and a quinine sensitive clone, for their degree of quinine sensitivity. They then statistically analyzed microsatellite markers from each progeny to map the quantitative trait loci (QTL). This revealed five distinct regions with either additive or pairwise effects on resistance. Two peaks, including pfcrt and a locus on chromosome 13, dominated the sensitivity phenotype. After subtracting out the effect of these two loci, additional regions became apparent, including the region encompassing pfmdr1 (figure 3). Both pfcrt and pfmdr1 had previously been demonstrated by allelic exchange to contribute to quinine resistance [17,18], thereby confirming the authors’ approach. The genes at the three additional QTL remained indeterminate. The authors however, predicted that the pfnhe1 gene, located on chromosome 13 and encoding a putative sodium/hydrogen exchanger, might contribute to quinine resistance. Physiological studies support the idea that variant pfnhe1, in concert with other parasite determinants, contributes to quinine resistance [19].
Figure 3. Identification of multiple loci associated with quinine resistance using QTL mapping.
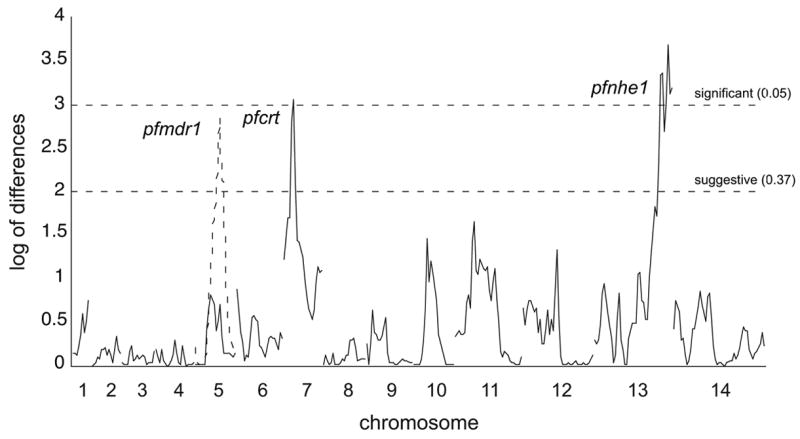
Ferdig et al. [16] mapped the QTL associated with quinine resistance using 35 independent progeny from the cross of quinine low level resistant and sensitive clones. Markers from linkage groups on each of the 14 P. falciparum chromosomes are distributed on the horizontal axis. Log of Difference scores are plotted on the vertical axis as a function of genome location. Horizontal dashed lines indicate threshold values from 1,000 permutations. Peaks at chromosomes 7 and 13 (colocalizing with pfcrt and pfnhe1 respectively) indicate QTL associated with elevated quinine 90% inhibitory concentrations. The peak at chromosome 5 (colocalizing with pfmdr1 and shown as a dashed line) was identified in a secondary scan after removing the effects from the major QTL defined by pfcrt and pfnhe1. Adapted from [16] with kind permission from Michael Ferdig.
Identifying Emerging Resistance Loci
Population studies that include drug resistant and sensitive parasites provide a powerful tool for identifying resistance loci. However, some circumstances will require alternative approaches to their identification, notably when predicting resistance to drugs used in new antimalarial regimens. Sidhu et al. [20] generated P. falciparum lines resistant to azithromycin in vitro. By taking a candidate gene approach, they were able to identify a mutation in the apicoplast-encoded ribosomal protein L4, which on the basis of structural models and literature on azithromycin-resistant bacteria was predicted to confer resistance.
Other groups have used rodent models to select for resistant parasites. While resistance to CQ arose through pfcrt independent mechanisms in the rodent parasite P. chabaudi [21], human and rodent Plasmodia do share in common their mode of resistance to atovaquone, mefloquine, and pyrimethamine (reviewed in [22]). Thus, on the balance, rodent malaria models have provided informative data on mechanisms of resistance. Using a process of gradual selection, Afonso et al. [23] recently generated artemisinin resistant lines in P. chabaudi (Figure 4a). This is the first confirmed in vivo report of Plasmodium resistance to artemisinin. Like Sidhu et al. [20] they used a candidate gene approach to identify possible mechanisms of artemisinin resistance, but failed to detect any alterations. An approach that is particularly well suited to identifying resistance genes in this case is Linkage Group Selection (LGS), which adapts a classical genetics approach related to “bulked segregant analysis” [24]. With both approaches, a resistant clone and a susceptible clone are intercrossed to generate progeny with mixed genotypes. With LGS, rather than cloning out individual progeny and testing them for their drug resistance phenotype, researchers subject the progeny to drug selection and then test the genotypes of the surviving population en masse. Genetic markers linked to the susceptible genotype are selectively lost from the population, creating a “selection valley” around the determinant (Figure 4b). Because researchers assess the genotype of the progeny in bulk, they must use techniques that allow them to assess the proportion of each haplotype within the sample (reviewed in [25]). Culleton et al. validated this technique as a method for identifying loci of resistance by crossing pyrimethamine resistant and sensitive lines of P. chabaudi and using LGS to identify a resistance locus including dhfr [26]. Applying LGS to the artemisinin pressured P. chabaudi line, Hunt et al. [27] identified a locus on chromosome 2 harboring a de-ubiquitinating enzyme that is currently a candidate.
Figure 4. Selection of artemisinin-resistant P. chabaudi and the LGS approach to identifying an in vivo determinant of resistance.
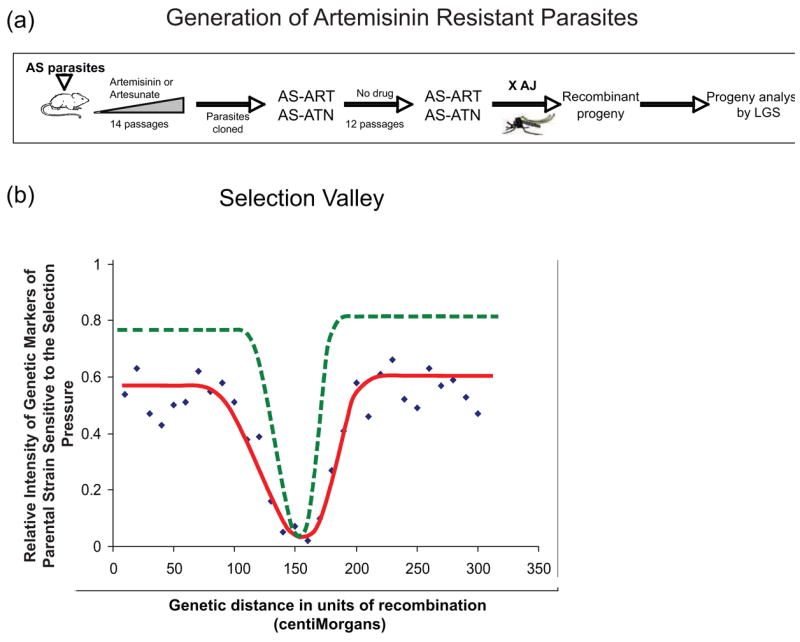
a) Artemisinin-sensitive P. chabaudi parasites of the AS lineage were exposed to increasing concentrations of artemisinin (ART) or artesunate (ATN) over 14 passages in the mouse. This produced the AS-ART and AS-ATN resistant lines that were respectively 15-fold and 6-fold more resistant to their selecting agents compared to the parental AS line, displayed cross-resistance, and were genetically stable [23]. The AS-ART clone was then crossed with AJ and the progeny subjected to LGS. Ongoing studies associate resistance with a locus on chromosome 2 [27]. (b) Simulated results of a LGS analysis. Resistant and sensitive parasite clones are crossed and the progeny are subjected to a specific selection pressure. The relative intensities of quantitative markers of the sensitive parental line compared to the drug-pressured line are plotted against the genetic distance of each marker along a parasite chromosome (data points and line of best fit are represented as purple diamonds and red line). Data are plotted and analyzed for every chromosome. This graph illustrates a “selection valley” that has formed in a region spanning about 100 centiMorgans of genetic distance. Markers at the lowest point in the selection valley are predicted to be closest to the gene that determines resistance to the applied selection pressure. Backcrossing a selected progeny with the sensitive parent allows LGS to be repeated iteratively, thereby producing a steeper selection valley (green dashed line). Figure 4b was reproduced from [25], copyright Elsevier Press, with kind permission from Richard Carter.
Manipulating the P. falciparum Genome
Allelic Exchange
Recent advances in genomic analyses have enormously aided our ability to localize drug resistance loci. However, the regions identified with these techniques generally span several hundred Kb and may contain dozens to hundreds of predicted genes. The literature contains many examples of candidate genes that were predicted to account for a given phenotype but which proved wrong upon more extensive analysis. The gold standard for confirming the identity of a resistance gene involves allelic exchange. If a gene truly confers resistance, then replacing the sensitive allele with the putative resistant allele, on the sensitive background, should confer resistance. The stable transfection of P. falciparum parasites, first described in 1995, paved the way for gene integration and allelic exchange studies (reviewed in [28]). In an early application, Triglia and coworkers [29] demonstrated that dihydropteroate synthase (dhps) mutants conferred sulfadoxine resistance. Later, Sidhu et al. [17] definitively showed that pfcrt conferred resistance to CQ by replacing the pfcrt allele of a sensitive line with the pfcrt alleles of resistant lines from South America, Asia, and Africa. Reed et al. [18] also employed allelic exchange to demonstrate that allelic variants of pfmdr1 could modulate the degree of parasite susceptibility to mefloquine, quinine, halofantrine, CQ, and artemisinin. In some instances, such as for pfmdr1, in vitro resistance and clinical treatment failure have been attributed to gene amplification events [30]. Sidhu et al. [31] recently engineered the targeted disruption of one copy of pfmdr1 in a clone with duplicate copies. Their findings confirmed that pfmdr1 amplifications decrease sensitivity to mefloquine, lumefantrine, halofantrine, quinine, and artemisinin.
Gene Integration
Technical difficulties have hampered efforts to perform the reciprocal experiment, i.e. inserting extra copies of a putative resistance gene into the genome of sensitive parasites. Genomic integration happens inefficiently in P. falciparum. Researchers therefore have tended to rely on episomally replicating plasmids in order to express transgenes. This technique suffers however from the plasmids having low and variable numbers of copies in the transfected parasites. Balu et al. [32] described a technique for stably transfecting P. falciparum using transposable elements. While they report high transfection efficiencies, the transposable elements insert randomly at TTAA sites throughout the genome, making the system more suitable for mutagenesis studies than for generating stable integrants [32]. Another technique described recently by Nkrumah et al. [33], employs a mycobacterial integrase to transfect P. falciparum. This integrase catalyzes recombination between an attP sequence motif located on a transfected plasmid and an attB site located in the genome. This site has been introduced into three P. falciparum lines and additional lines can be generated using a classical homologous recombination strategy. While there are several applications for this site-specific integration technique, it should prove particularly useful for rapidly generating phenotypically and genetically homogeneous transgenic parasites that express putative drug resistance alleles. It also allows for the introduction of additional copies of genes that appear to confer resistance via copy number amplification.
Tracking Known Resistance Mutations
Allele Identification
Several papers have introduced interesting methods for evaluating the frequency of drug resistant genotypes within the context of heterogeneous pathogen populations [26, 34, 35]. Most techniques employ PCR-based amplification of SNP markers surrounding the resistance locus. The PCR product is then either sequenced using a quantitative sequencing technique or subjected to an oligonucleotide ligation assay. While not yet validated for Plasmodium, SNP microarrays have been used in other systems to determine the frequency of different alleles within mixed populations [36].
Field Applications for Molecular Markers of Resistance
McCollum et al. [37] assayed for the presence of dhfr and dhps mutations associated with SP resistance in Venezuela. They found that the mutations continue to persist in the population despite the fact that SP usage was discontinued in the region in 1998. Their results suggest that this drug combination may remain ineffective indefinitely within this region. More promising news was reported from Malawi where researchers found that the resistant form of pfcrt essentially disappeared less than a decade after CQ was replaced by SP as the first line of therapy [38]. A clinical study recently concurred that CQ sensitivity has returned to Malawi [39], confirming the predictive value of the genetic screening techniques. The presence of resistant lines in surrounding countries precludes the immediate return to CQ monotherapy in Malawi. However, the data suggests that CQ could potentially be used again in the future as part of a rotating arsenal of antimalarials with a rotation period of mere decades.
Conclusions
The development of CQ resistance has had a devastating effect on our ability to control malaria. No subsequent antimalarial regimen has contained malaria as successfully and cost effectively. As researchers develop and introduce new antimalarial drugs there is a dire need to ensure that we preserve their effectiveness for as long as possible. Clinical reports of treatment failure provide one estimate of resistance. Clinical studies, however, are generally costly, suffer from confounding factors such as poor compliance, and tend to focus on the predominant drugs utilized within a given country. Molecular studies tracking the presence of drug resistant determinants in the malarial population can thus provide critical data complementing clinical observations. New genetic tools give us an unprecedented ability to track new mutations as they arise, confirm their importance and mode of action in the laboratory, and measure their prevalence in the population. Public policy decisions should benefit from the development of these new tools to ensure that malarial eradication programs are as effective as possible.
Acknowledgments
The research of David A. Fidock, Ph.D., is supported in part by the Investigators in Pathogenesis of Infectious Disease Award from the Burroughs Wellcome Fund and by the NIH (R01 AI50234). Eric H. Ekland, Ph.D., is a Hoffman-LaRoche Fellow of the Life Sciences Research Foundation. We extend our gracious thanks to Tim Anderson, Elizabeth Winzeler, Michael Ferdig, Paul Hunt and Richard Carter for providing figures that were adapted for this review.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and recommended reading
Papers of particular interest, published within the period of review, have been highlighted as:
● of special interest
● ● of outstanding interest
- 1.Wellems TE, Plowe CV. Chloroquine-resistant malaria. J Infect Dis. 2001;184:770–776. doi: 10.1086/322858. [DOI] [PubMed] [Google Scholar]
- 2.Woodrow CJ, Krishna S. Antimalarial drugs: recent advances in molecular determinants of resistance and their clinical significance. Cell Mol Life Sci. 2006;63:1586–1596. doi: 10.1007/s00018-006-6071-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gardner MJ, Hall N, Fung E, White O, Berriman M, Hyman RW, Carlton JM, Pain A, Nelson KE, Bowman S, et al. Genome sequence of the human malaria parasite Plasmodium falciparum. Nature. 2002;419:498–511. doi: 10.1038/nature01097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jeffares DC, Pain A, Berry A, Cox AV, Stalker J, Ingle CE, Thomas A, Quail MA, Siebenthall K, Uhlemann AC, et al. Genome variation and evolution of the malaria parasite Plasmodium falciparum. Nat Genet. 2007;39:120–125. doi: 10.1038/ng1931. ●The authors describe the genome wide shotgun sequencing of a Ghanaian clinical isolate, the IT laboratory strain, and the related chimpanzee parasite P. reichenowi. They identify 27,000 nonredundant SNPs and a similar number of indels. The paper includes a comparison of protein evolutionary rates relative to their expression levels, developmental stage of expression and cellular localization. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mu J, Awadalla P, Duan J, McGee KM, Keebler J, Seydel K, McVean GA, Su XZ. Genome-wide variation and identification of vaccine targets in the Plasmodium falciparum genome. Nat Genet. 2007;39:126–130. doi: 10.1038/ng1924. ● These authors sequenced 3,539 genes (or about 65% of the total predicted genes in the P. falciparum genome) from the clones Dd2, Hb3, D10 and 7G8. They identify specific genes and genomic regions under diversifying selective pressure. [DOI] [PubMed] [Google Scholar]
- 6.Volkman SK, Sabeti PC, DeCaprio D, Neafsey DE, Schaffner SF, Milner DA, Jr, Daily JP, Sarr O, Ndiaye D, Ndir O, et al. A genome-wide map of diversity in Plasmodium falciparum. Nat Genet. 2007;39:113–119. doi: 10.1038/ng1930. ● ● The authors generated high quality draft genome sequences for the clones HB3 and Dd2 and supplement this effort with extensive sequencing from additional clones and clinical isolates. They identify 47,937 SNPs and an additional 37,0039 indels. Of particular interest, in drug resistant parasites they discovered multiple regions with signatures of selective sweeps and propose a novel pyrimethamine resistance locus. [DOI] [PubMed] [Google Scholar]
- 7.Mu J, Awadalla P, Duan J, McGee KM, Joy DA, McVean GA, Su XZ. Recombination hotspots and population structure in Plasmodium falciparum. PLoS Biol. 2005;3:e335. doi: 10.1371/journal.pbio.0030335. ● In this paper the authors conduct a thorough investigation of chromosome 3, generating SNP haplotype and population recombination maps from 99 global isolates. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Volkman SK, Lozovsky E, Barry AE, Bedford T, Bethke L, Myrick A, Day KP, Hartl DL, Wirth DF, Sawyer SA. Genomic heterogeneity in the density of noncoding single-nucleotide and microsatellite polymorphisms in Plasmodium falciparum. Gene. 2007;387:1–6. doi: 10.1016/j.gene.2006.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Roper C, Pearce R, Nair S, Sharp B, Nosten F, Anderson T. Intercontinental spread of pyrimethamine-resistant malaria. Science. 2004;305:1124. doi: 10.1126/science.1098876. [DOI] [PubMed] [Google Scholar]
- 10.McCollum AM, Poe AC, Hamel M, Huber C, Zhou Z, Shi YP, Ouma P, Vulule J, Bloland P, Slutsker L, et al. Antifolate resistance in Plasmodium falciparum: multiple origins and identification of novel dhfr alleles. J Infect Dis. 2006;194:189–197. doi: 10.1086/504687. [DOI] [PubMed] [Google Scholar]
- 11.Wootton JC, Feng X, Ferdig MT, Cooper RA, Mu J, Baruch DI, Magill AJ, Su XZ. Genetic diversity and chloroquine selective sweeps in Plasmodium falciparum. Nature. 2002;418:320–323. doi: 10.1038/nature00813. [DOI] [PubMed] [Google Scholar]
- 12.Fidock DA, Nomura T, Talley AK, Cooper RA, Dzekunov SM, Ferdig MT, Ursos LM, Sidhu AB, Naude B, Deitsch KW, et al. Mutations in the P. falciparum digestive vacuole transmembrane protein PfCRT and evidence for their role in chloroquine resistance. Mol Cell. 2000;6:861–871. doi: 10.1016/s1097-2765(05)00077-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Roper C, Pearce R, Bredenkamp B, Gumede J, Drakeley C, Mosha F, Chandramohan D, Sharp B. Antifolate antimalarial resistance in southeast Africa: a population-based analysis. Lancet. 2003;361:1174–1181. doi: 10.1016/S0140-6736(03)12951-0. [DOI] [PubMed] [Google Scholar]
- 14.Nair S, Williams JT, Brockman A, Paiphun L, Mayxay M, Newton PN, Guthmann JP, Smithuis FM, Hien TT, White NJ, et al. A selective sweep driven by pyrimethamine treatment in southeast asian malaria parasites. Mol Biol Evol. 2003;20:1526–1536. doi: 10.1093/molbev/msg162. [DOI] [PubMed] [Google Scholar]
- 15.Kidgell C, Volkman SK, Daily J, Borevitz JO, Plouffe D, Zhou Y, Johnson JR, Le Roch K, Sarr O, Ndir O, et al. A systematic map of genetic variation in Plasmodium falciparum. PLoS Pathog. 2006;2:e57. doi: 10.1371/journal.ppat.0020057. ● ● Using a microarray that covers approximately 55% of the malarial genome, the authors survey 14 different clones and identify 23,653 single feature polymorphisms. The strength of the technique lies in its ability to rapidly screen whole genomes for single feature polymorphisms. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ferdig MT, Cooper RA, Mu J, Deng B, Joy DA, Su XZ, Wellems TE. Dissecting the loci of low-level quinine resistance in malaria parasites. Mol Microbiol. 2004;52:985–997. doi: 10.1111/j.1365-2958.2004.04035.x. [DOI] [PubMed] [Google Scholar]
- 17.Sidhu AB, Verdier-Pinard D, Fidock DA. Chloroquine resistance in Plasmodium falciparum malaria parasites conferred by pfcrt mutations. Science. 2002;298:210–213. doi: 10.1126/science.1074045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Reed MB, Saliba KJ, Caruana SR, Kirk K, Cowman AF. Pgh1 modulates sensitivity and resistance to multiple antimalarials in Plasmodium falciparum. Nature. 2000;403:906–909. doi: 10.1038/35002615. [DOI] [PubMed] [Google Scholar]
- 19.Bennett TN, Patel J, Ferdig MT, Roepe PD. Plasmodium falciparum Na(+)/H(+) exchanger activity and quinine resistance. Mol Biochem Parasitol. 2007;153:48–58. doi: 10.1016/j.molbiopara.2007.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sidhu AB, Sun Q, Nkrumah LJ, Dunne MW, Sacchettini JC, Fidock DA. In vitro efficacy, resistance selection, and structural modeling studies implicate the malarial parasite apicoplast as the target of azithromycin. J Biol Chem. 2007;282:2494–2504. doi: 10.1074/jbc.M608615200. [DOI] [PubMed] [Google Scholar]
- 21.Hunt P, Cravo PV, Donleavy P, Carlton JM, Walliker D. Chloroquine resistance in Plasmodium chabaudi: are chloroquine-resistance transporter (crt) and multi-drug resistance (mdr1) orthologues involved? Mol Biochem Parasitol. 2004;133:27–35. doi: 10.1016/j.molbiopara.2003.08.010. [DOI] [PubMed] [Google Scholar]
- 22.Carlton JM, Hayton K, Cravo PV, Walliker D. Of mice and malaria mutants: unravelling the genetics of drug resistance using rodent malaria models. Trends Parasitol. 2001;17:236–242. doi: 10.1016/s1471-4922(01)01899-2. [DOI] [PubMed] [Google Scholar]
- 23.Afonso A, Hunt P, Cheesman S, Alves AC, Cunha CV, do Rosario V, Cravo P. Malaria parasites can develop stable resistance to artemisinin but lack mutations in candidate genes atp6 (encoding the sarcoplasmic and endoplasmic reticulum Ca2+ ATPase), tctp, mdr1, and cg10. Antimicrob Agents Chemother. 2006;50:480–489. doi: 10.1128/AAC.50.2.480-489.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Michelmore RW, Paran I, Kesseli RV. Identification of markers linked to disease-resistance genes by bulked segregant analysis: a rapid method to detect markers in specific genomic regions by using segregating populations. Proc Natl Acad Sci USA. 1991;88:9828–9832. doi: 10.1073/pnas.88.21.9828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Carter R, Hunt P, Cheesman S. Linkage Group Selection--a fast approach to the genetic analysis of malaria parasites. Int J Parasitol. 2007;37:285–293. doi: 10.1016/j.ijpara.2006.11.013. [DOI] [PubMed] [Google Scholar]
- 26.Culleton R, Martinelli A, Hunt P, Carter R. Linkage group selection: rapid gene discovery in malaria parasites. Genome Res. 2005;15:92–97. doi: 10.1101/gr.2866205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hunt P, Afonso A, Creasey A, Culleton R, Sidhu ABS, Logan J, Valderramos S, McNae I, Cheesman S, do Rosario V, et al. Gene encoding a de-ubiquitinating enzyme is mutated in artemisinin- and chloroquine-resistant rodent malaria parasites. Mol Microbiol. 2007 doi: 10.1111/j.1365-2958.2007.05753.x. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Carvalho TG, Menard R. Manipulating the Plasmodium genome. Curr Issues Mol Biol. 2005;7:39–55. [PubMed] [Google Scholar]
- 29.Triglia T, Wang P, Sims PF, Hyde JE, Cowman AF. Allelic exchange at the endogenous genomic locus in Plasmodium falciparum proves the role of dihydropteroate synthase in sulfadoxine-resistant malaria. Embo J. 1998;17:3807–3815. doi: 10.1093/emboj/17.14.3807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Price RN, Uhlemann AC, Brockman A, McGready R, Ashley E, Phaipun L, Patel R, Laing K, Looareesuwan S, White NJ, et al. Mefloquine resistance in Plasmodium falciparum and increased pfmdr1 gene copy number. Lancet. 2004;364:438–447. doi: 10.1016/S0140-6736(04)16767-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sidhu AB, Valderramos SG, Fidock DA. pfmdr1 mutations contribute to quinine resistance and enhance mefloquine and artemisinin sensitivity in Plasmodium falciparum. Mol Microbiol. 2005;57:913–926. doi: 10.1111/j.1365-2958.2005.04729.x. [DOI] [PubMed] [Google Scholar]
- 32.Balu B, Shoue DA, Fraser MJ, Jr, Adams JH. High-efficiency transformation of Plasmodium falciparum by the lepidopteran transposable element piggyBac. Proc Natl Acad Sci USA. 2005;102:16391–16396. doi: 10.1073/pnas.0504679102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nkrumah LJ, Muhle RA, Moura PA, Ghosh P, Hatfull GF, Jacobs WR, Jr, Fidock DA. Efficient site-specific integration in Plasmodium falciparum chromosomes mediated by mycobacteriophage Bxb1 integrase. Nat Methods. 2006;3:615–621. doi: 10.1038/nmeth904. ● The techniques published in this paper promise to dramatically reduce the amount of time required to generate stable transgene integrants. This will prove valuable for characterizing resistance determinants in phenotypically and genetically homogeneous parasite cultures. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhou Z, Poe AC, Limor J, Grady KK, Goldman I, McCollum AM, Escalante AA, Barnwell JW, Udhayakumar V. Pyrosequencing, a high-throughput method for detecting single nucleotide polymorphisms in the dihydrofolate reductase and dihydropteroate synthetase genes of Plasmodium falciparum. J Clin Microbiol. 2006;44:3900–3910. doi: 10.1128/JCM.01209-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cole-Tobian JL, Zimmerman PA, King CL. High-throughput identification of the predominant malaria parasite clone in complex blood stage infections using a multi-SNP molecular haplotyping assay. Am J Trop Med Hyg. 2007;76:12–19. [PMC free article] [PubMed] [Google Scholar]
- 36.Meaburn E, Butcher LM, Liu L, Fernandes C, Hansen V, Al-Chalabi A, Plomin R, Craig I, Schalkwyk LC. Genotyping DNA pools on microarrays: tackling the QTL problem of large samples and large numbers of SNPs. BMC Genomics. 2005;6:52. doi: 10.1186/1471-2164-6-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.McCollum AM, Mueller K, Villegas L, Udhayakumar V, Escalante AA. Common origin and fixation of Plasmodium falciparum dhfr and dhps mutations associated with sulphadoxine-pyrimethamine resistance in a low transmission area in South America. Antimicrob Agents Chemother. 2007 doi: 10.1128/AAC.01228-06. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kublin JG, Cortese JF, Njunju EM, Mukadam RA, Wirima JJ, Kazembe PN, Djimde AA, Kouriba B, Taylor TE, Plowe CV. Reemergence of chloroquine-sensitive Plasmodium falciparum malaria after cessation of chloroquine use in Malawi. J Infect Dis. 2003;187:1870–1875. doi: 10.1086/375419. [DOI] [PubMed] [Google Scholar]
- 39.Laufer MK, Thesing PC, Eddington ND, Masonga R, Dzinjalamala FK, Takala SL, Taylor TE, Plowe CV. Return of chloroquine antimalarial efficacy in Malawi. N Engl J Med. 2006;355:1959–1966. doi: 10.1056/NEJMoa062032. ● ● Following up on their molecular studies, the authors conduct a clinical trial indicating that susceptibility to CQ has returned to Malawi. One of the implications is that the resistant allele of pfcrt is selectively disadvantageous during the natural life cycle. Another is that CQ may be reintroduced at a future date in combination with other antimalarials. [DOI] [PubMed] [Google Scholar]