

Abstract
Reactive oxygen species (ROS) generated by ischemic and pharmacological preconditioning are known to act as triggers of cardiac protection; however, the involvement of ROS in ischemic and pharmacological postconditioning (PostC) in vivo and in vitro is unknown. We tested the hypothesis that ROS are involved in PostC in the mouse heart in vivo and in the isolated adult cardiac myocyte (ACM). Mice were subjected to 30 min coronary artery occlusion followed by 2 h of reperfusion with or without ischemic or pharmacologic PostC (three cycles of 20 s reperfusion/ischemia; 1.4% isoflurane; 10 mg/kg SNC-121). Additional groups were treated with 2-mercaptopropionyl glycine (MPG), a ROS scavenger, 10 min before or after the PostC stimuli. Ischemic, isoflurane, or SNC-121 induced PostC reduced infarct size (24.1 ± 3.2, 15.7 ± 2.6, 24.9 ± 2.6%, p < 0.05, respectively) compared to the control group (43.4 ± 3.3%). These cardiac protective effects were abolished by MPG when administered before (40.0 ± 3.6, 39.3 ± 3.1, 38.5 ± 1.6%, respectively), but not after the PostC stimuli (26.6 ± 2.3, 17.0 ± 2.2, 23.9 ± 1.7%, respectively). Additionally, ACM were subjected to a simulated ischemia/reperfusion protocol with isoflurane and SNC PostC. Isoflurane- and SNC-induced PostC in vitro were abolished by prior treatment with MPG. These data indicate that ROS signaling is an essential trigger of ischemic and pharmacological PostC and this is occurring at the level of the cardiac myocyte.
Introduction
Postconditioning (PostC) is an effective, clinically relevant therapeutic strategy for cardiac protection. Zhao et al (Zhao et al., 2003) first reported ischemic PostC in which brief cycles of reperfusion and ischemia at reperfusion reduced infarct size similar to preconditioning. Importantly, PostC can be pharmacologically induced by volatile anesthetics (Chiari et al., 2005; Weihrauch et al., 2005) and opioids (Gross and Gross, 2006; Weihrauch et al., 2005), compounds shown to produce potent preconditioning effects. As prior knowledge of the onset of ischemia (as in preconditioning) is no longer a prerequisite, the therapeutic potential for PostC is significant. Signaling pathways involved in PostC include several anti-apoptotic pro-survival signaling kinases (Tsang et al., 2004; Weihrauch et al., 2005), ATP sensitive potassium channels (Krolikowski et al., 2005; Yang et al., 2004), and the mitochondrial permeability transition pore (Argaud et al., 2005; Krolikowski et al., 2005); however, no studies have investigated potential upstream triggering events involved in PostC in vivo.
Reactive oxygen species (ROS) generation has been observed within the first minutes of reperfusion and is proposed as a major mechanism of reperfusion injury (Becker, 2004). Cardiac protective stimuli such as ischemic preconditioning, volatile anesthetics, and opioids have been shown to induce a “signaling burst” of ROS that protects from subsequent injury (Cohen et al., 2001; Pain et al., 2000; Tanaka et al., 2002). Whether this is the mechanism by which they trigger PostC is not known. PostC attenuates deleterious ROS generation, and this mechanism may be an important component of the cardiac protection afforded by PostC in both in vivo (Kin et al., 2004) and in vitro (Sun et al., 2005) models. Penna et al (Penna et al., 2006) showed that ROS scavengers block infarct size reduction of PostC in an ex vivo rat heart suggesting that ROS trigger PostC; however, the ex vivo model presents certain important limitations. Therefore, in the present study, utilizing a mouse in vivo model or PostC, we investigated whether ROS are involved in the signaling cascade leading to ischemic and pharmacological PostC and determined if this mechanism was responsible for PostC in isolated ventricular cardiac myocytes.
Methods
Animals
The studies complied with the Guide for the Care and Use of Laboratory Animals (National Academy of Science); animal use protocols were approved by the VA San Diego IACUC. Male C57BL/6 mice (8–10 wk old, 24–26 g body weight) were kept in a temperature-controlled room and had ad lib access to food and water.
Experimental Design
The surgical methods were similar to those previously described (Tsutsumi et al., 2006). Mice were anesthetized with pentobarbital (80 mg/kg, ip) and mechanically ventilated with oxygen. Core temperature was maintained with a heating pad and ECG leads were placed to record heart rate. After thoracotomy and stabilization, all mice underwent a 30 min left coronary artery occlusion followed by 2 h of reperfusion. Control groups were exposed to ischemia and reperfusion only (Figure 1). Other mice were randomly assigned to receive three cycles of 20-s reperfusion/20-s ischemia started after 30 min occlusion or 1.4% isoflurane (1.0 minimum alveolar concentration for mice) or δ-opioid receptor agonist, SNC-121 (10 mg/kg), administered for 3 min before and 2 min after reperfusion (Figure 1). Other mice received the ROS scavenger, 2-mercaptopropionyl glycine (MPG; 20 mg/kg, Sigma) 10 min before or after PostC stimuli (Figure 1).
Figure 1.
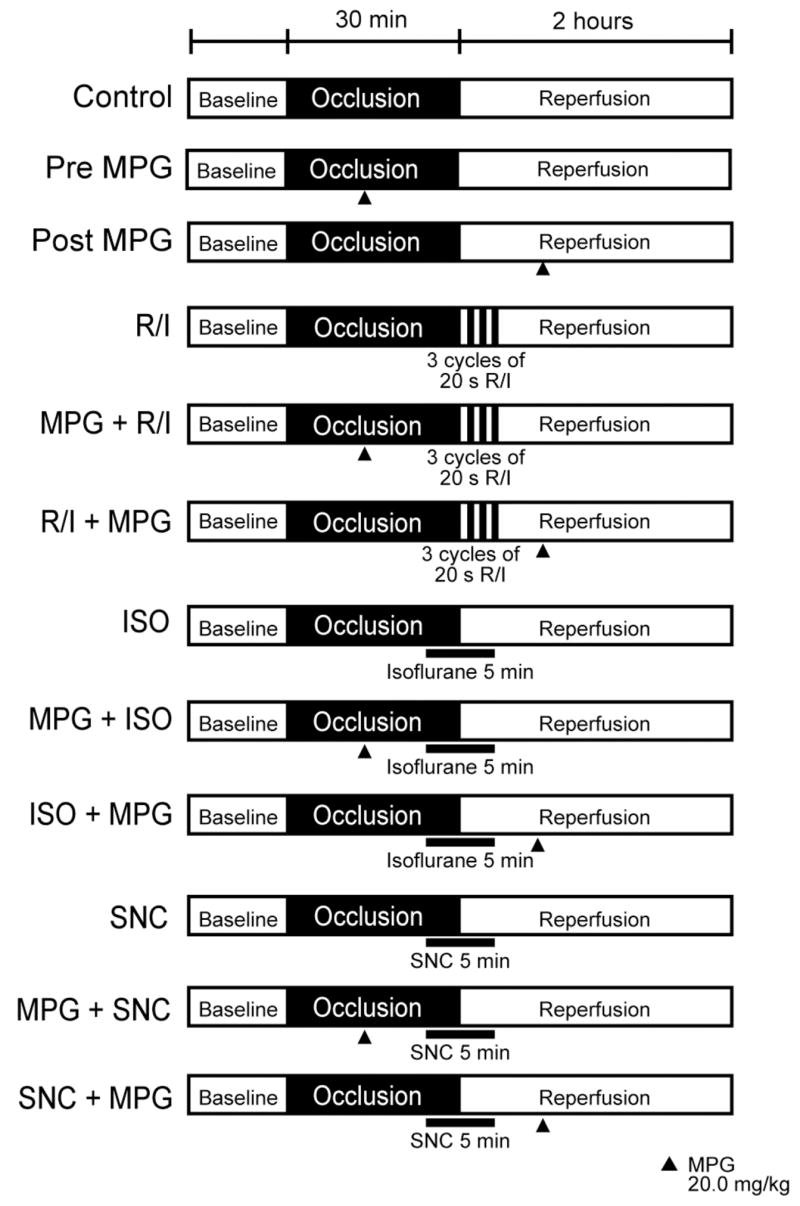
Schematic illustration of the experimental protocol. Mice were subjected to 30 min coronary artery occlusion and 2 h of reperfusion. No intervention was performed in Control animals (n = 9). Hearts underwent postconditioning (PostC) in early reperfusion periods. Ischemic PostC (R/I) was elicited by three cycles of 20 s reperfusion and ischemia before 2 h of reperfusion (n = 8). Isoflurane (ISO, n = 8) or SNC-121 (SNC, n = 8) was administered for 5 min, beginning at 3 min before and ending 2 min after reperfusion. Groups were treated with 2-mercaptopropionyl glycine (MPG) before reperfusion (Pre MPG, MPG + R/I, MPG + ISO, and MPG + SNC, n = 8), or after reperfusion (Post MPG, R/I + MPG, ISO + MPG, and SNC + MPG, n = 8).
After reperfusion the coronary artery was again occluded and area at risk (AAR) determined by staining with 1% Evans blue. The heart was cut into 1 mm slices and each slice of left ventricle was counterstained with 1% 2,3,5,-triphenyltetrazolium chloride (Sigma). After overnight storage in 10% formaldehyde, slices were weighed and visualized. The images were analyzed (Image-Pro Plus, Media Cybernetics) and infarct size was determined by planimetry. Infarct size was expressed as a percentage of the AAR.
Hemodynamic measurements were made in a separate group of animals (n=4 per group) exposed to the ischemia-reperfusion protocol. Mice were anesthetized with pentobarbital. The right carotid artery was instrumented with 1.4F Mikro-tip pressure transducer (Millar), which was connected to TCB-500 amplifier (Millar) for determination of heart rate and arterial blood pressure. Pressure signals were digitally converted (IOX version 1.8.5.11, Emka) and stored on a computer.
In Vitro Post-Conditioning
Adult cardiac myocytes were isolated as previously published (Patel et al., 2006). Hearts were retrograde perfused on a Langendorff apparatus and digested with collagenase (Worthington). Isolated cardiac myocytes were then plated in Media 199 (4% fetal bovine serum and 1% pen/strep) for one hour. Media was then replaced with maintenance Media 199 (1% bovine serum albumin/pen/strep). Myocytes were allowed to recover for 24 hours at 37°C and 5% CO2. Myocytes were then pretreated with and without 2mM MPG during one-hour lethal oxygen/glucose deprivation (metabolic chamber purged with nitrogen in zero glucose media at 37°C). Three minutes prior to reperfusion (restoration of normal oxygen and glucose containing media) 0.1mM SNC was added via an injection port and left in media during 1 hr reperfusion at 37°C and 5% CO2. For volatile anesthetic post-conditioning, cells were exposed to 1.4% isoflurane in the metabolic chamber 10 min prior to reperfusion. Immediately, at reperfusion, media was replaced with normal maintenance media with or without MPG or 1.4% isoflurane during the 1 hr reperfusion period. Myocytes were then stained with trypan blue and fixed in 4% formalin. Cells were counted (3 fields per well with 100 cells/field) using ImageJ software to determine percent cell death.
Statistical Analysis
Statistical analyses were performed by one-way ANOVA, followed by Bonferroni post-hoc test or unpaired Student’s t-test. All data are expressed as mean ± SEM. Statistical significance was defined as p < 0.05.
Results
Involvement of ROS in PostC In Vivo
There were no differences in pre-occlusion baseline hemodynamics among groups (Table 1). After 30 min of ischemia, PostC produced by repeated reperfusion-ischemia, isoflurane, or SNC-121 (R/I, ISO, or SNC, respectively) at early reperfusion maintained heart function (rate pressure product, Table 1). R/I, ISO, or SNC decreased infarct size (24.1 ± 3.2, 15.7 ± 2.6, 24.9 ± 2.6%, respectively) when compared with control hearts (43.4 ± 3.3%; Figure 2). MPG blocked the protective effects of PostC on heart function and infarct size when administered prior to the stimuli (MPG + R/I, MPG + ISO, or MPG + SNC; 40.0 ± 3.6, 39.3 ± 3.1, 38.5 ± 1.6%, respectively), but not after the stimuli (R/I + MPG, ISO + MPG, or SNC + MPG; 26.6 ± 2.3, 17.0 ± 2.2, 23.9 ± 1.7%, respectively). MPG alone (Pre MPG and Post MPG) did not affect infarct size (44.8 ± 1.6 and 41.5 ± 2.5%, respectively).
Table 1.
Hemodynamics
| Pre-occlusion | Ischemia | Reperfusion | |
|---|---|---|---|
| Heart rate, beats · min−1 | |||
| Control | 412 ± 24 | 394 ± 21 | 367 ± 11 |
| Pre-MPG | 409 ± 31 | 389 ± 15 | 316 ± 7* |
| Post-MPG | 411 ± 16 | 401 ± 15 | 380 ± 15 |
| R/I | 420 ± 31 | 400 ± 30 | 399 ± 29 |
| MPG + R/I | 399 ± 23 | 409 ± 18 | 381 ± 13 |
| R/I + MPG | 395 ± 26 | 403 ± 25 | 401 ± 26 |
| Isoflurane | 404 ± 36 | 401 ± 31 | 418 ± 33 |
| MPG + Isoflurane | 425 ± 29 | 400 ± 15 | 347 ± 12* |
| Isoflurane + MPG | 398 ± 16 | 403 ± 12 | 391 ± 26 |
| SNC-121 | 400 ± 18 | 395 ± 14 | 371 ± 14 |
| MPG + SNC-121 | 409 ± 30 | 413 ± 17 | 343 ± 14 |
| SNC + MPG-121 | 391 ± 13 | 393 ± 12 | 393 ± 15 |
| Mean arterial pressure, mmHg | |||
| Control | 71 ± 1 | 67 ± 3 | 61 ± 2* |
| Pre-MPG | 70 ± 1 | 67 ± 3 | 60 ± 2 |
| Post-MPG | 70 ± 2 | 68 ± 2 | 69 ± 2 |
| R/I | 71 ± 3 | 68 ± 2 | 69 ± 3 |
| MPG + R/I | 72 ± 2 | 67 ± 2 | 59 ± 3* |
| R/I + MPG | 69 ± 2 | 69 ± 2 | 68 ± 3 |
| Isoflurane | 70 ± 1 | 68 ± 1 | 73 ± 2† |
| MPG + Isoflurane | 70 ± 2 | 64 ± 2 | 69 ± 3* |
| SNC-121 | 70 ± 2 | 70 ± 4 | 72 ± 2† |
| MPG +SNC-121 | 71 ± 1 | 61 ± 3 | 56 ± 3* |
| SNC-121 + MPG | 69 ± 2 | 70 ± 1 | 71 ± 2 |
| Rate-Pressure Product, beats · min−1 · mmHg · 103 | |||
| Control | 29.3 ± 2.0 | 26.5 ± 2.1 | 22.4 ± 1.2* |
| Pre-MPG | 28.6 ± 1.9 | 26.2 ± 1.1 | 20.1 ± 0.6* |
| Post-MPG | 28.9 ± 0.9 | 27.2 ± 1.6 | 26.2 ± 0.4 |
| R/I | 29.9 ± 2.6 | 27.3 ± 2.2 | 27.5 ± 2.9 |
| MPG + R/I | 28.5 ± 1.7 | 27.3 ± 1.1 | 22.5 ± 0.8* |
| R/I + MPG | 27.3 ± 2.2 | 27.7 ± 1.2 | 27.0 ± 1.4 |
| Isoflurane | 28.3 ± 3.0 | 27.3 ± 1.8 | 30.6 ± 2.2† |
| MPG + Isoflurane | 29.6 ± 1.7 | 25.7 ± 1.7 | 21.0 ± 1.1* |
| Isoflurane + MPG | 26.8 ± 2.2 | 28.1 ± 0.6 | 26.8 ± 1.2 |
| SNC-121 | 28.2 ± 1.9 | 27.5 ± 1.9 | 26.8 ± 1.2 |
| MPG +SNC-121 | 29.0 ± 1.7 | 25.4 ± 1.9 | 19.3 ± 1.0* |
| SNC-121 + MPG | 26.8 ± 0.8 | 27.4 ± 0.7 | 27.9 ± 1.2 |
Data are mean ± SEM.
Significantly (p < 0.05) different from pre-occlusion (intragroup comparison).
Significantly (p < 0.05) different from Control (intergroup comparison).
Figure 2.

Myocardial infarct size expressed as percentage of the left ventricular area at risk. Data are means ± SEM. *p < 0.05 vs. Control; **p < 0.05 vs. Post MPG; #p < 0.05 vs. R/I; §p < 0.05 vs. ISO; †p < 0.05 vs. SNC.
Involvement of ROS in PostC In Vitro
Percent cell death in the presence and absence of MPG was determined in isolated ACM undergoing SNC-induced PostC. Isoflurane exposure 10 min or SNC treatment 3 min prior to the restoration of oxygen and glucose significantly reduced the number of ACM undergoing cell death compared to basal ACM undergoing only simulated ischemia/reperfusion (30.8±10 and 50.7±6 vs. 81.0±4%, respectively, p<0.01, Figure 3). ACM pretreated with MPG no longer showed isoflurane- or SNC-induced PostC and were not distinguishable from the basal group undergoing simulated ischemia/reperfusion (61.0±9 and 77.8±2 vs. 81.0±4, respectively, Figure 3).
Figure 3.

Percent cell death as determined by trypan blue staining. Data are means ± SEM. *p < 0.01 vs. Basal Ischemia.
Discussion
We show that repeated reperfusion-ischemia, isoflurane and SNC-121 induce PostC and protect mouse myocardium against ischemia/reperfusion injury in vivo. Importantly, we show that MPG, a ROS scavenger, abolishes PostC induced by these three stimuli. The PostC effects of isoflurane and SNC-121 were recapitulated in an in vitro model of cell injury and again MPG was shown to block this protection. These results suggest ROS as an important trigger in the molecular signaling involved in PostC in the whole animal that likely occurs at the level of the cardiac myocyte.
Controversy exists concerning the benefit of ROS scavenging during ischemia/reperfusion injury. ROS contribute to post-ischemic myocardial dysfunction (Bolli et al., 1989). MPG given prior to reperfusion attenuates post-ischemic myocardial dysfunction (Bolli et al., 1989; Myers et al., 1986). Conversely, ROS can trigger cardiac protection induced by ischemic (Pain et al., 2000) and pharmacological preconditioning (Cohen et al., 2001). Treatment with MPG prior to an ischemic preconditioning stimulus in the rabbit heart attenuates protection against ischemia/reperfusion injury and MPG given before the ischemic insult has no effect on infarct size (Baines et al., 1997). These studies suggest an important temporal role for ROS in preconditioning and ischemia/reperfusion injury.
The role of ROS in PostC is unclear. Recent reports show that PostC attenuates the production of ROS immediately at reperfusion in the in vivo rat heart (Kin et al., 2004). The same group demonstrated that hypoxic PostC in vitro resulted in decreased cell death, ROS production, and mitochondrial calcium loading (Sun et al., 2005). They suggest that the reduction in ROS represents an important component of cardiac protection elicited by PostC. However, Penna et al (Penna et al., 2006), show that scavenging of ROS attenuates PostC in an isolated Langendorff perfused rat heart. Our results confirm the findings of Penna et al (Penna et al., 2006). Our results suggest ROS trigger PostC in an in vivo mouse model of ischemia/reperfusion injury utilizing three different PostC stimuli. Importantly, we also show that the generation of ROS by protective stimuli in cardiac myocytes may be involved in the mechanism of PostC. The identification of the specific ROS species involved and the localized site of generation of ROS as well as the mechanism for the induction of protective signal transduction needs to be elucidated in future studies.
The current findings should be interpreted within the constraints of potential limitations. Our study utilized MPG, a sulfur containing amino acid antioxidant used by many investigators, to assess the role of ROS in ischemia/reperfusion injury. We did not directly measure ROS, the particular species of ROS, nor ROS compartmentalization. As stated above, these are important considerations in future investigations to advance the field. Additionally, we did not examine downstream activation of protein kinases in response to ROS generation that may be involved in mediating cardiac protection induced by PostC. Our data suggest a unifying mechanism for three different postconditioning stimuli in the mouse heart. SNC-121 is known to be a selective delta opioid agonist, however we cannot rule out that stimulation of other opioid receptor subtypes played a role in our findings. A more detailed pharmacological study needs to be performed to address the role of distinct opioid receptors in PostC. Importantly, we cannot rule out the possibility that our results may not be applicable to all mammalian species especially since small animal models may not adequately predict the physiological response in human pathophysiologies.
In conclusion, our results show that PostC induced by reperfusion-ischemia, isoflurane and SNC-121 protect the heart form reperfusion injury in an in vivo mouse model. Furthermore, the present findings indicate that scavenging of ROS limits the triggering of PostC by various stimuli and this may be directly involved in protective mechanisms initiated in cardiac myocytes. Our results suggest that PostC stimuli share a common triggering event (ROS) leading to downstream cardiac protection.
Acknowledgments
Supported by American Heart Association Postdoctoral Fellowship 0525190Y (YMT), Merit Award from the Department of Veterans Affairs (DMR), NIH RO1 HL081400 (DMR) and American Heart Association Scientist Development Grant 0630039N (HHP).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Argaud L, Gateau-Roesch O, Raisky O, Loufouat J, Robert D, Ovize M. Postconditioning inhibits mitochondrial permeability transition. Circulation. 2005;111(2):194–197. doi: 10.1161/01.CIR.0000151290.04952.3B. [DOI] [PubMed] [Google Scholar]
- Baines CP, Goto M, Downey JM. Oxygen radicals released during ischemic preconditioning contribute to cardioprotection in the rabbit myocardium. Journal of Molecular Cellular Cardiology. 1997;29(1):207–216. doi: 10.1006/jmcc.1996.0265. [DOI] [PubMed] [Google Scholar]
- Becker LB. New concepts in reactive oxygen species and cardiovascular reperfusion physiology. Cardiovascular Research. 2004;61(3):461–470. doi: 10.1016/j.cardiores.2003.10.025. [DOI] [PubMed] [Google Scholar]
- Bolli R, Jeroudi MO, Patel BS, Aruoma OI, Halliwell B, Lai EK, McCay PB. Marked reduction of free radical generation and contractile dysfunction by antioxidant therapy begun at the time of reperfusion. Evidence that myocardial “Stunning” Is a manifestation of reperfusion injury. Circulation Research. 1989;65(3):607–622. doi: 10.1161/01.res.65.3.607. [DOI] [PubMed] [Google Scholar]
- Bolli R, Jeroudi MO, Patel BS, DuBose CM, Lai EK, Roberts R, McCay PB. Direct evidence that oxygen-derived free radicals contribute to postischemic myocardial dysfunction in the intact dog. Proceedings of the National Academy of Sciences U S A. 1989;86(12):4695–4699. doi: 10.1073/pnas.86.12.4695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiari PC, Bienengraeber MW, Pagel PS, Krolikowski JG, Kersten JR, Warltier DC. Isoflurane protects against myocardial infarction during early reperfusion by activation of phosphatidylinositol-3-kinase signal transduction: Evidence for anesthetic-induced postconditioning in rabbits. Anesthesiology. 2005;102(1):102–109. doi: 10.1097/00000542-200501000-00018. [DOI] [PubMed] [Google Scholar]
- Cohen MV, Yang XM, Liu GS, Heusch G, Downey JM. Acetylcholine, bradykinin, opioids, and phenylephrine, but not adenosine, trigger preconditioning by generating free radicals and opening mitochondrial KATP channels. Circulation Research. 2001;89(3):273–278. doi: 10.1161/hh1501.094266. [DOI] [PubMed] [Google Scholar]
- Gross ER, Gross GJ. Ligand triggers of classical preconditioning and postconditioning. Cardiovascular Research. 2006;70(2):212–221. doi: 10.1016/j.cardiores.2005.12.019. [DOI] [PubMed] [Google Scholar]
- Kin H, Zhao ZQ, Sun HY, Wang NP, Corvera JS, Halkos ME, Kerendi F, Guyton RA, Vinten-Johansen J. Postconditioning attenuates myocardial ischemia-reperfusion injury by inhibiting events in the early minutes of reperfusion. Cardiovascular Research. 2004;62(1):74–85. doi: 10.1016/j.cardiores.2004.01.006. [DOI] [PubMed] [Google Scholar]
- Krolikowski JG, Bienengraeber M, Weihrauch D, Warltier DC, Kersten JR, Pagel PS. Inhibition of mitochondrial permeability transition enhances isoflurane-induced cardioprotection during early reperfusion: The role of mitochondrial KATP channels. Anesthesia Analgesia. 2005;101(6):1590–1596. doi: 10.1213/01.ANE.0000181288.13549.28. [DOI] [PubMed] [Google Scholar]
- Myers ML, Bolli R, Lekich RF, Hartley CJ, Roberts R. N-2-mercaptopropionylglycine improves recovery of myocardial function after reversible regional ischemia. Journal of the American College of Cardiology. 1986;8(5):1161–1168. doi: 10.1016/s0735-1097(86)80396-5. [DOI] [PubMed] [Google Scholar]
- Pain T, Yang XM, Critz SD, Yue Y, Nakano A, Liu GS, Heusch G, Cohen MV, Downey JM. Opening of mitochondrial KATP channels triggers the preconditioned state by generating free radicals. Circulation Research. 2000;87(6):460–466. doi: 10.1161/01.res.87.6.460. [DOI] [PubMed] [Google Scholar]
- Patel HH, Head BP, Petersen HN, Niesman IR, Huang D, Gross GJ, Insel PA, Roth DM. Protection of adult rat cardiac myocytes from ischemic cell death: Role of caveolar microdomains and delta-opioid receptors. American Journal of Physiology Heart and Circulatory Physiology. 2006;291(1):H344–350. doi: 10.1152/ajpheart.01100.2005. [DOI] [PubMed] [Google Scholar]
- Penna C, Rastaldo R, Mancardi D, Raimondo S, Cappello S, Gattullo D, Losano G, Pagliaro P. Post-conditioning induced cardioprotection requires signaling through a redox-sensitive mechanism, mitochondrial ATP-sensitive K+ channel and protein kinase c activation. Basic Research Cardiology. 2006;101(2):180–189. doi: 10.1007/s00395-006-0584-5. [DOI] [PubMed] [Google Scholar]
- Sun HY, Wang NP, Kerendi F, Halkos M, Kin H, Guyton RA, Vinten-Johansen J, Zhao ZQ. Hypoxic postconditioning reduces cardiomyocyte loss by inhibiting ROS generation and intracellular Ca2+ overload. American Journal of Physiology Heart and Circulatory Physiology. 2005;288(4):H1900–1908. doi: 10.1152/ajpheart.01244.2003. [DOI] [PubMed] [Google Scholar]
- Tanaka K, Weihrauch D, Kehl F, Ludwig LM, LaDisa JF, Jr, Kersten JR, Pagel PS, Warltier DC. Mechanism of preconditioning by isoflurane in rabbits: A direct role for reactive oxygen species. Anesthesiology. 2002;97(6):1485–1490. doi: 10.1097/00000542-200212000-00021. [DOI] [PubMed] [Google Scholar]
- Tsang A, Hausenloy DJ, Mocanu MM, Yellon DM. Postconditioning: A form of “Modified reperfusion” Protects the myocardium by activating the phosphatidylinositol 3-kinase-akt pathway. Circulation Research. 2004;95(3):230–232. doi: 10.1161/01.RES.0000138303.76488.fe. [DOI] [PubMed] [Google Scholar]
- Tsutsumi YM, Patel HH, Huang D, Roth DM. Role of 12-lipoxygenase in volatile anesthetic-induced delayed preconditioning in mice. American Journal of Physiology Heart and Circulatory Physiology. 2006;291(2):H979–983. doi: 10.1152/ajpheart.00266.2006. [DOI] [PubMed] [Google Scholar]
- Weihrauch D, Krolikowski JG, Bienengraeber M, Kersten JR, Warltier DC, Pagel PS. Morphine enhances isoflurane-induced postconditioning against myocardial infarction: The role of phosphatidylinositol-3-kinase and opioid receptors in rabbits. Anesthesia Analgesia. 2005;101(4):942–949. doi: 10.1213/01.ane.0000171931.08371.a2. [DOI] [PubMed] [Google Scholar]
- Yang XM, Proctor JB, Cui L, Krieg T, Downey JM, Cohen MV. Multiple, brief coronary occlusions during early reperfusion protect rabbit hearts by targeting cell signaling pathways. Journal of the American College of Cardiology. 2004;44(5):1103–1110. doi: 10.1016/j.jacc.2004.05.060. [DOI] [PubMed] [Google Scholar]
- Zhao ZQ, Corvera JS, Halkos ME, Kerendi F, Wang NP, Guyton RA, Vinten-Johansen J. Inhibition of myocardial injury by ischemic postconditioning during reperfusion: Comparison with ischemic preconditioning. American Journal of Physiology Heart and Circulatory Physiology. 2003;285(2):H579–588. doi: 10.1152/ajpheart.01064.2002. [DOI] [PubMed] [Google Scholar]