

Abstract
The production of auto-antibodies is one of the predominant characteristics of autoimmune disorders. Because IL-2 deficient mice develop autoimmunity, we asked how IL-2 deficiency might impair endogenous mechanisms of B cell tolerance. To this end, we mated BALB/c anti-dsDNA H chain knock-in mice, in which B cells producing anti-dsDNA antibodies are properly regulated, with IL-2 deficient mice and assessed the phenotype of their offspring. IL-2 deficient mice expressing the anti-dsDNA H chain knock-in allele developed anti-dsDNA antibodies of both IgM and IgG isotypes. Production of these antibodies occurred through the disruption of several mechanisms of endogenous tolerance, including deletion, maturational arrest, and follicular exclusion. In summary, our results suggest that IL-2 plays an important role in regulating B cell tolerance.
Keywords: interleukin-2, B cells, spleen, transgenic/knockout mice
Introduction
The presence of auto-antibodies against dsDNA is one of the primary diagnostic features of systemic lupus erythematosis. Several murine models have been developed to study the aberrant generation of these and other auto-antibodies. One such model is the VH3H9 “knock in” model, in which a rearranged heavy chain (VH3H9) that binds dsDNA replaces the chromosomal JH locus [1]. BALB/c mice expressing this knock-in allele exhibit several known mechanisms of B cell tolerance, and do not develop an autoimmune phenotype. Upon crossing these mice with autoimmune prone gld/gld mice, however, normal tolerance mechanisms are disrupted and high titers of anti-dsDNA antibodies are produced [2].
Mice deficient in either IL-2 or elements of its tripartite receptor also develop an autoimmune phenotype. IL-2, IL-2Rα, and IL-2Rβ deficient mice develop a severe hemolytic anemia due to the production of anti-erythrocyte antibodies [3, 4, 5]. IL-2Rβ knock out (KO) mice also exhibit low levels of anti-nuclear antibodies [5]. Production of these ntibodies in IL-2Rβ KO mice peaks at 5 weeks of age and declines by 8 weeks, likely due to the significant loss of B cells that also occurs in these mice with age [5, 6].
The presence of antibody-mediated autoimmunity in IL-2 deficiency suggests that endogenous means of B cell tolerance are impaired, however the mechanisms involved are unknown. To address this question, we generated VH3H9-KI/IL-2 KO mice and assessed their ability to generate anti-dsDNA B cells. These mice produce anti-dsDNA antibodies as a consequence of the breakdown of several endogenous mechanisms of B cell tolerance including follicular exclusion, developmental arrest, and apoptosis.
Methods
Animals
VH3H9 and VH3H9/56R knock-in mice on a BALB/c background were the kind gift of Dr. Martin Weigert, University of Chicago, Chicago, IL [1]. These mice were bred with BALB/c IL-2 KO mice expressing a TCR transgene for ovalbumin (DO11.10) [7] to eventually yield VH3H9/IL-2 KO or VH3H9/56R IL-2 KO mice. VH3H9 mice were also bred with DO11.10 IL-2 wild-type (WT) mice to generate comparable WT controls. Therefore, all mice used in this study expressing the VH3H9 transgene also express DO11.10, but are designated as VH3H9/IL-2 WT or VH3H9/IL-2 KO for simplicity. Age-matched mice between 8 and 16 weeks of age were used for all experiments. Mice were housed in specific pathogen free facilities and all experiments were in accordance with protocols approved by the University of Nebraska Institutional Animal Care and Use Committee.
Anti-nuclear antibody assay
Anti-nuclear antibodies (ANA) were detected in the sera of VH3H9/IL-2 KO or WT mice by incubation of each serially diluted sample with HEP-G2 cells (Antibodies Inc.). Homogeneous nuclear binding of IgM or IgG was detected via FITC-conjugated rat anti-mouse IgG (BD Pharmingen) or FITC-conjugated goat anti-mouse IgM antibodies Invitrogen). Serum samples were diluted on a log scale, and the ANA titer was defined as the reciprocal of the last dilution with detectable ANA staining.
Flow cytometry
Single-cell suspensions were prepared from spleens of 8- to 16-week-old VH3H9/IL-2 KO or IL-2 WT mice. Cells (1.5 × 106/ml) were stained for 30 minutes at 4°C with the fluorochrome-conjugated antibody of interest, then washed and resuspended in FACS buffer (PBS containing 2% v/v FBS and 0.1% NaN3). Nonspecific binding was blocked prior to staining using Fc block (anti-CD16/32, clone 2.4G2). Analysis was performed on a FACScan flow cytometer using CellQuest software (BD Biosciences). Anti-CD19 (clone 1D3), CD21/35 (7G6), CD16/32 (2.4G2), CD23 (B3B4), HSA (M1/69), λ1 (R11-153), CXCR5 (2G8) and Annexin V (with propidium iodide, as a kit) were purchased from BD Pharmingen.
Immunohistochemistry
Tissues were embedded in OCT (Sakura Finetechnical Co.), snap frozen in liquid nitrogen, and stored at −80°C until use. Five-micron sections were prepared, fixed in acetone, and stained with the following antibodies: FITC-labeled IgM (goat polyclonal, Invitrogen), anti-Thy 1.2 (clone 53-2.1, rat IgG2a), biotinylated anti-λ1 (clone R11-153, rat IgG1), FDC-M1 (rat IgG2c (BD Pharmingen), anti-CCL21, anti-CXCL13 (goat polyclonals, R&D Systems), MOMA-1 (rat IgG2b, Cedarlane), and ER-TR7 (rat IgG2b, BMA Biomedicals). Anti-gp38 (hamster monoclonal, 8.1.1) was the kind gift of Dr. Andrew Farr, University of Washington, Seattle. WA [8]. Antibodies to CXCL13, CCL21, and λ1 were detected via direct application of tyramide amplification (λ1) or following incubation with biotinylated anti-goat (CCL21, CXCL13) antibodies (Vector Laboratories). Tyramide amplification was performed using SA-HRP and tyramide-labeled phycoerythrin or fluorescein isothiocyanate per the manufacturer’s recommendations (BD Pharmingen). Thy 1.2, MOMA-1, ER-TR7, FDC-M1, and anti-gp38 were detected via the following fluorochrome-labeled secondaries: goat anti-rat IgG (Molecular Probes), goat anti-rat IgG (MP Biomedical), and biotinylated goat anti-hamster IgG (Kirkegaard and Perry Laboratories).
Statistics
Statistical analyses of potential differences between experimental and control groups were performed using a Fisher’s Exact Test.
Results
Identification of dsDNA B cells in VH3H9/IL-2 KO mice
To determine how IL-2 deficiency affects the generation of anti-dsDNA antibodies, BALB/c IL-2 KO mice expressing a transgenic T cell receptor for ovalbumin (DO11.10) were mated with VH3H9-KI mice to generate VH3H9/IL-2 KO mice. The BALB/c IL-2 KO mice in our colony all express a TCR transgene to improve their health and longevity. IL-2 KO mice on a BALB/c background otherwise die in utero or shortly after birth. IL-2+/−littermates from the aforementioned matings were used as phenotypic IL-2 WT controls. The frequency of T cells expressing the TCR transgene in DO11.10 heterozygotes is approximately 20%.
We first compared the frequency and total numbers of anti-dsDNA B cells in VH3H9/IL-2 KO versus VH3H9/IL-2 WT mice. Since the VH3H9 H chain typically pairs with a λ1 light chain, the anti-dsDNA B cells can be tracked using an anti-λ1 antibody [9]. As seen in Figure 1A, the frequency of λ1+ B cells was higher in VH3H9/IL-2 KO versus VH3H9/IL-2 WT mice, despite a similar frequency of λ1− B cells. Total numbers of both λ1+ and λ1− B cells were increased in VH3H9/IL-2 KO mice as compared to VH3H9/IL-2 WT mice due, at least in part, to the large numbers of splenocytes in the IL-2 KO mice (Figure 1B).
Figure 1.
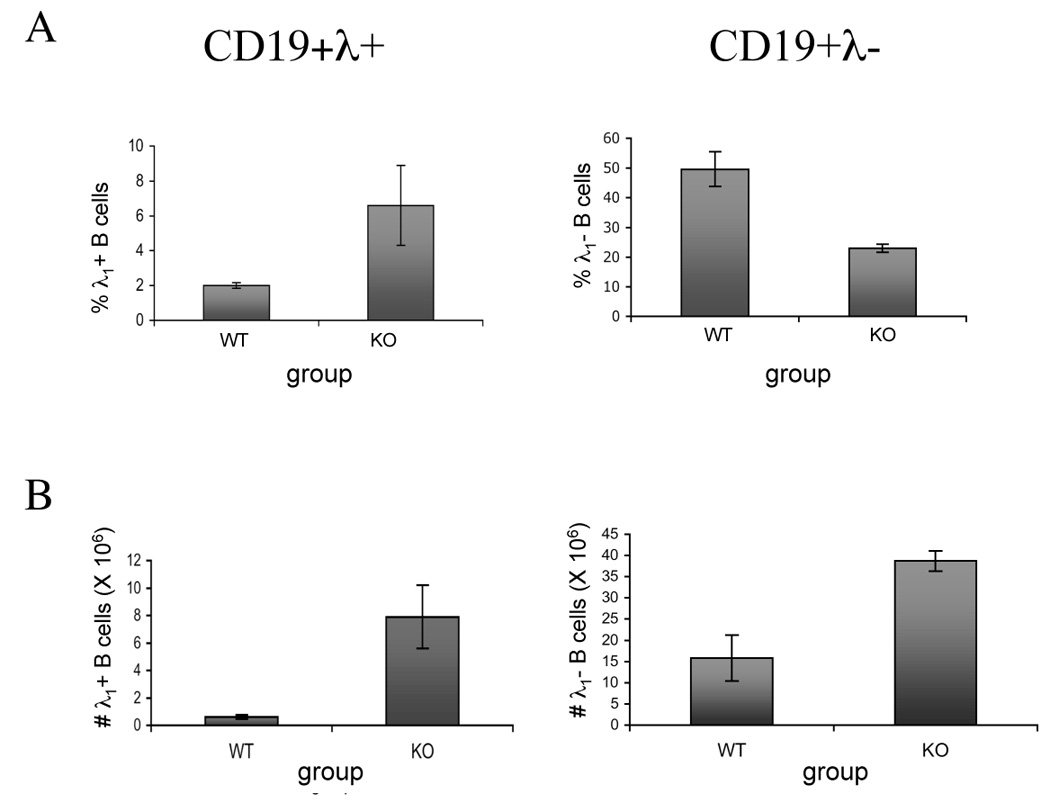
Influence of IL-2 deficiency on the frequency and number of anti-dsDNA B cells in VH3H9-KI mice. The frequency (A) and total number (B) of anti-dsDNA-producing B cells (CD19+λ1+), and all other (CD19+λ1−) B cells in spleens of VH3H9/IL-2 WT (WT) or 3H9/IL-2 KO (KO) mice were determined by flow cytometry. The frequencies shown in (A) represent the percentages of CD19+λ1+ or CD19+λ1− cells within the total splenocyte population. The results shown are the average ± SD of 3 mice per group in a single experiment, and are representative of 3 experiments in mice 10–16 weeks of age. The frequencies of T cells expressing the transgenic TCR in these mice were as follows: VH3H9/IL-2 WT 20 ± 1%; VH3H9/IL-2 KO 20 ± 4%. Statistical differences were as follows for 3 pooled experiments (n=8): %λ+ B cells, p=0.04; %λ− B cells, p=NS; #λ+ B cells, p=0.0001; #λ− B cells, p=0.04.
Influence of IL-2 deficiency on anti-dsDNA antibody production
To ascertain whether endogenous mechanisms of restricting anti-dsDNA antibody production were impaired in the absence of IL-2, we measured IgM and IgG anti-nuclear antibody (ANA) titers in the sera of VH3H9/IL-2 KO and WT mice. As seen in Figure 2, VH3H9/IL-2 KO mice developed ANA titers of IgM antibodies in the 1:100 to 1:1000 range. VH3H9/IL-2 WT mice also developed IgM antibodies, but the titers in these mice were ≤ 1:100. Regarding the production of IgG, none of the VH3H9/IL-2 WT mice produced anti-dsDNA IgG antibodies, whereas almost all VH3H9/IL-2 KO mice produced these antibodies at titers of 1:10 – 1:100. Neither DO11.10/IL-2 WT nor DO11.10/IL-2 KO mice produced detectable IgM or IgG antibodies. These data suggest that endogenous mechanisms which normally restrain autoantibody production are impaired in IL-2 deficiency.
Figure 2.
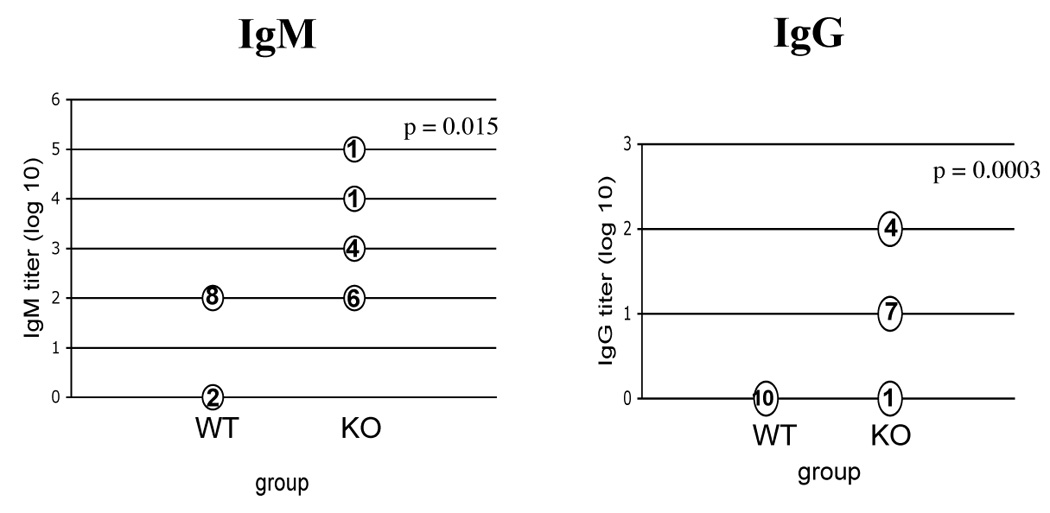
Anti-nuclear antibody production in VH3H9/IL-2 KO mice. Titers of IgM and IgG anti-nuclear antibodies were measured by ANA assay in the sera of 8- to 16-week-old VH3H9/IL-2 WT (WT) or VH3H9/IL-2 KO (KO) mice. Serum samples were serially diluted by 1:10, beginning at a dilution of 1:100 (for IgM) or 1:10 (for IgG). The ANA titer was defined as the reciprocal of the last dilution with detectable ANA staining. The numbers shown indicate the number of mice with that titer.
Apoptosis of anti-dsDNA B cells in IL-2 deficiency
In models of antibody-mediated autoimmunity, anti-dsDNA B cells have been shown to die more rapidly in mice with non-autoimmune versus autoimmune backgrounds [9]. In light of the increased frequency of anti-dsDNA B cells observed in IL-2 deficiency (see Figure 1), we first asked whether the frequency of apoptotic λ1+ B cells was altered in VH3H9/IL-2 KO mice. To address this question, we assessed cell surface expression of Annexin V in CD19+λ1+ B cells from VH3H9/IL-2 WT and KO mice. As seen in Figure 3, the frequency of apoptotic λ1+ B cells in IL-2 KO mice was approximately one third that of WT mice, suggesting that elimination of these auto-reactive cells is impaired in the absence of IL-2. Whether this defect is a direct effect of IL-2 deficiency or an indirect effect based on improper localization of these B cells (see below) remains to be determined.
Figure 3.
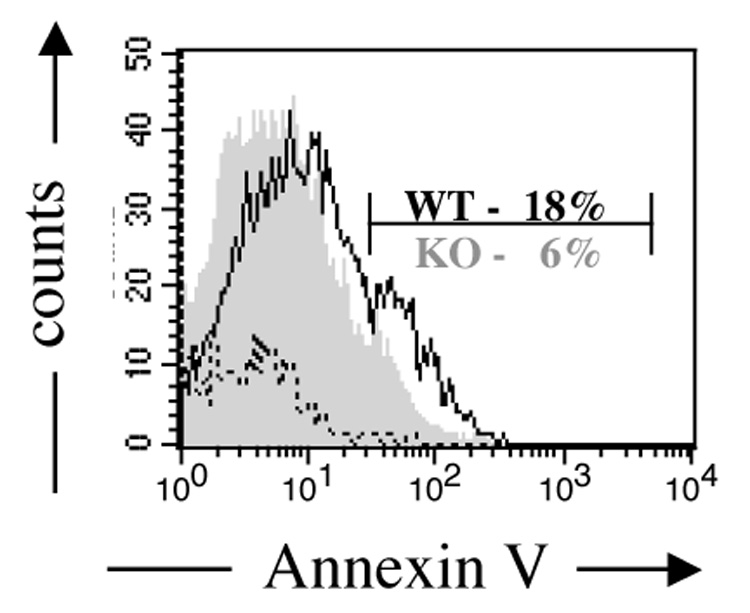
Apoptosis of anti-dsDNA B cells in VH3H9/IL-2 KO mice. The frequency of Annexin V+ cells within the propidium iodide negative population of CD19+λ1+ B cells was assessed by flow cytometry in splenocytes of VH3H9/IL-2 WT or VH3H9/IL-2 KO mice. A representative histogram is shown. Cells incubated with buffer alone (dotted line) were negative. The frequency of Annexin V+ cells was decreased in the VH3H9/IL-2 KO mice as compared to the VH3H9/IL-2 WT mice (n=8, p=0.001).
Developmental arrest and IL-2 deficiency
Given the above findings, we next asked whether additional endogenous mechanisms of tolerance were impaired in IL-2 deficiency. One potential mechanism is that of developmental arrest, in which the maturation of autoimmune B cells from the transitional stages (T1 and T2) to the mature (M) stage is inhibited. In VH3H9-lpr/lpr mice, which produce high levels of anti-dsDNA antibodies, developmental arrest is impaired [9]. In light of these findings, the developmental status of λ1+ B cells in VH3H9/IL-2 KO mice was assessed by cell surface expression of CD21 and HSA on λ1+ CD19+ B cells. As seen in Figure 4, VH3H9/IL-2 WT mice exhibited a decreased frequency of mature λ1+ B cells as compared to WT λ1− B cells (p=0.0001), suggesting that the λ1+ B cells were developmentally arrested. In VH3H9/IL-2 KO mice, there was no difference in the frequencies of T1, T2, and mature B cells between the λ1+ and λ1− groups (p=0.7). The frequency of mature λ1+ WT B cells was also decreased compared to mature λ1+ IL-2 KO B cells (p=0.0002). These results suggest that developmental arrest of anti-dsDNA B cells is impaired in the absence of IL-2. Whether these results reflect a direct effect of IL-2 or an indirect effect via modulation of factors that influence B cell maturation, such as BAFF [10], remains to be determined.
Figure 4.
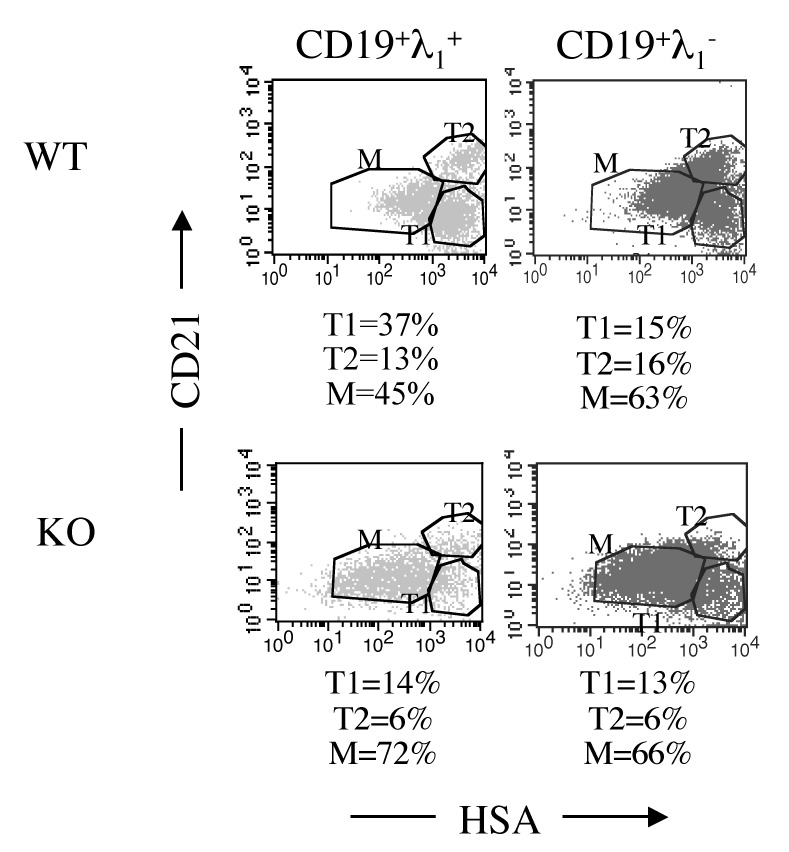
Maturational arrest of autoimmune B cells in IL-2 deficiency. The frequency of transitional (T1 – CD21loHSAhi or T2 – CD21hiHSAhi) and mature (M – CD21loHSAlo) B cells was determined by flow cytometric assessment of CD21 and HSA expression on CD19λ1+ or CD19λ1− splenocytes from VH3H9/IL-2 WT or VH3H9/IL-2 KO mice. The results shown are from a single representative experiment. Isotype controls were negative. The frequency of mature CD19λ1+ B cells was decreased in VH3H9/IL-2 WT mice as compared to VH3H9/IL-2 KO mice (n=16, p=0.0002). Mature CD191− B cells were not statistically different between these mice (n= 16, p=0.7). In addition, the frequency of mature CD19λ1+ B cells in VH3H9/IL-2 WT mice was decreased compared to mature WT λ1− B cells (n=16, p=0.0001). These latter populations were not statistically different in VH3H9/IL-2 KO mice (n=16, p=0.7).
Marginal zone B cells and IL-2 deficiency
Mice exhibiting autoimmunity, such as lupus-prone mice and NOD mice, develop an expanded marginal zone [11 – 14]. VH3H9/56R-KI mice, which are similar to VH3H9 mice but express higher affinity anti-dsDNA antibodies, exhibit a greatly expanded marginal zone when generated on a non-autoimmune background [15]. These marginal zone B cells produce antibodies expressing dual κ and λ light chains, which significantly decrease their affinity to dsDNA. It has been hypothesized that sequestration of such autoreactive B cells to the marginal zone may prevent them from entering germinal centers and developing the properties of pathogenic B cells [16]. To study localization of anti-dsDNA B cells to the marginal zone in IL-2 deficency, we generated VH3H9/56R/IL-2 KO mice and investigated their frequency of λ1+, marginal zone B cells.
As seen in Figure 5, the frequency of λ1+ marginal zone B cells was very high in VH3H9/56R mice, but was substantially decreased in VH3H9/56R/IL-2 KO mice. However, the frequency of marginal zone B cells in the λ1− B cells was also diminished relative to WT mice, suggesting that this finding is not specific for the autoimmune B cells. In VH3H9 mice, which do not exhibit a large shift of B cells to the marginal zone, the frequencies of marginal zone VH3H9/IL-2 KO λ1+ and λ1− B cells were also decreased compared to their WT counterparts (not shown). These findings suggest that mechanisms for localizing B cells to the marginal zone may be impaired in IL-2 deficiency.
Figure 5.
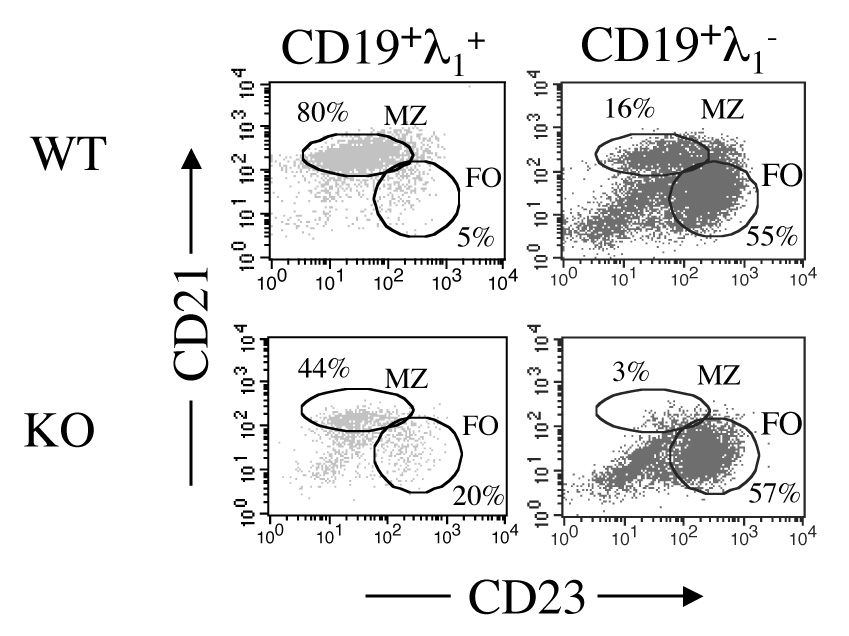
Frequency of λ1+ marginal zone B cells in VH3H9/56R-KI/IL-2 KO mice. The frequency of marginal zone (MZ – CD21hiCD23lo) and follicular (FO – CD21intCD23+)B cells was determined by flow cytometric expression of CD21 and CD23 on CD19+λ1+ or CD19+λ1− splenocytes isolated from VH3H9/56R-KI/IL-2 WT or VH3H9/56R-KI/IL-2 KO mice. Results shown are from a single representative experiment. Isotype controls were negative. The frequency of marginal zone CD19λ1+ B cells was greater in VH3H9/IL-2 KO (n=10) as compared to VH3H9/IL-2 WT (n=14) mice (p=0.0005). Marginal zone CD19λ1− B cells were also statistically different between these mice (p=0.03).
Follicular exclusion in IL-2 deficiency
Another mechanism by which autoimmune B cells are constrained is follicular exclusion. In murine models wherein self-reactive B cells are anergized, the anergic B cells do not enter the B or T cell follicles but are localized to the T/B interface [17]. To ascertain whether follicular exclusion is intact in IL-2 deficiency, localization of λ1+ B cells was assessed in spleens from VH3H9/IL-2 KO mice. As seen in Figure 6A, λ1+ B cells from VH3H9/IL-2 WT mice localized to the T/B interface, whereas B cells from VH3H9/IL-2 KO mice were spread throughout the B cell follicle. These results suggest that mechanisms responsible for follicular exclusion are impaired in IL-2 deficiency.
Figure 6.
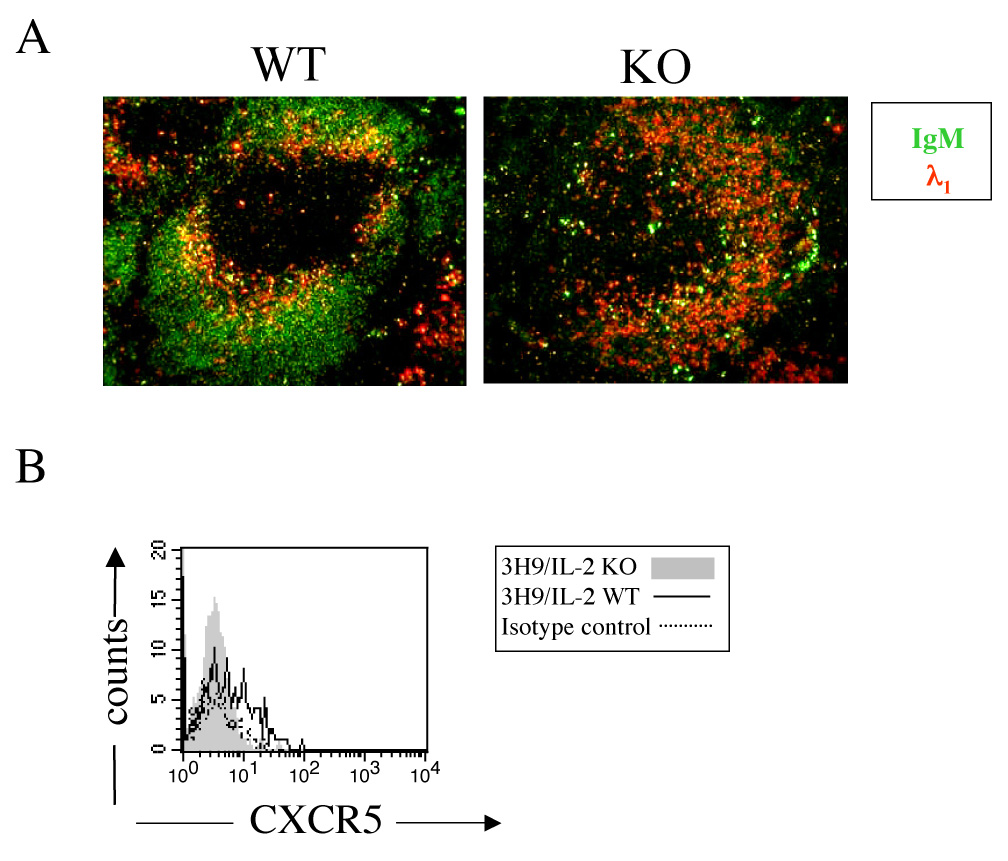
Follicular exclusion of autoimmune B cells in IL-2 deficiency. (A) The distribution of λ1+ B cells in the spleens of VH3H9/IL-2 WT or VH3H9/IL-2 KO mice was determined by immunohistochemistry, using FITC-labeled IgM and biotinylated anti-λ1 antibodies. Tyramide amplification was then applied using SA-HRP and tyramide-labeled phycoerythrin per the manufacturer’s recommendations. Original objective magnification: 10X. (B) Cell surface expression of CXCR5 was assessed by flow cytometry in CD19+λ1+ splenocytes from VH3H9/IL-2 WT or VH3H9/IL-2 IL-2 KO mice. The results shown are representative of 5 experiments. Expression of CXCR5 was decreased in VH3H9/IL-2 IL-2 KO (n=15) as compared with VH3H9/IL-2 IL-2 WT (n=14) mice (p=0.0005).
We next began to address potential mechanisms responsible for the impaired follicular exclusion observed in IL-2 deficiency. Two chemokine receptors important for proper follicular exclusion are CXCR5 and CCR7 [17]. We examined the cell surface expression of CXCR5 and CCR7 on anti-dsDNA B cells from VH3H9/IL-2 KO and WT mice. Anti-dsDNA B cells from VH3H9/IL-2 KO mice expressed lower levels of CXCR5 than VH3H9/IL-2 WT mice (Figure 6B). No clear differences in cell surface expression of CCR7 on VH3H9/IL-2 KO vs WT cells could be discerned (not shown). While interesting, these findings do not explain the lack of follicular exclusion found in VH3H9/IL-2 KO mice.
Since cell surface expression of CXCR5 and CCR7 did not explain the lack of follicular exclusion observed in IL-2 deficiency, we asked whether expression of the corresponding chemokines (CXCL13 and CCL19) was altered in the absence of IL-2. No consistent differences in the expression of either CXCL13 or CCL19, assessed by semi-quantitative RT-PCR, were found in the spleens of VH3H9/IL-2 KO and WT mice (not shown).
We have noted that the spleens of IL-2 deficient mice exhibit severely abnormal morphology over time, primarily in the areas of the marginal zone and follicle (Figure 7A and data not shown). Given these observations, we asked whether the distribution of CXCL13 or CCL21 was altered in the spleens of IL-2 deficient mice. CCL21 is a second CCR7 ligand with essentially identical function as CCL19, but higher protein levels of CCL21 are expressed in tissues [18].
Figure 7.
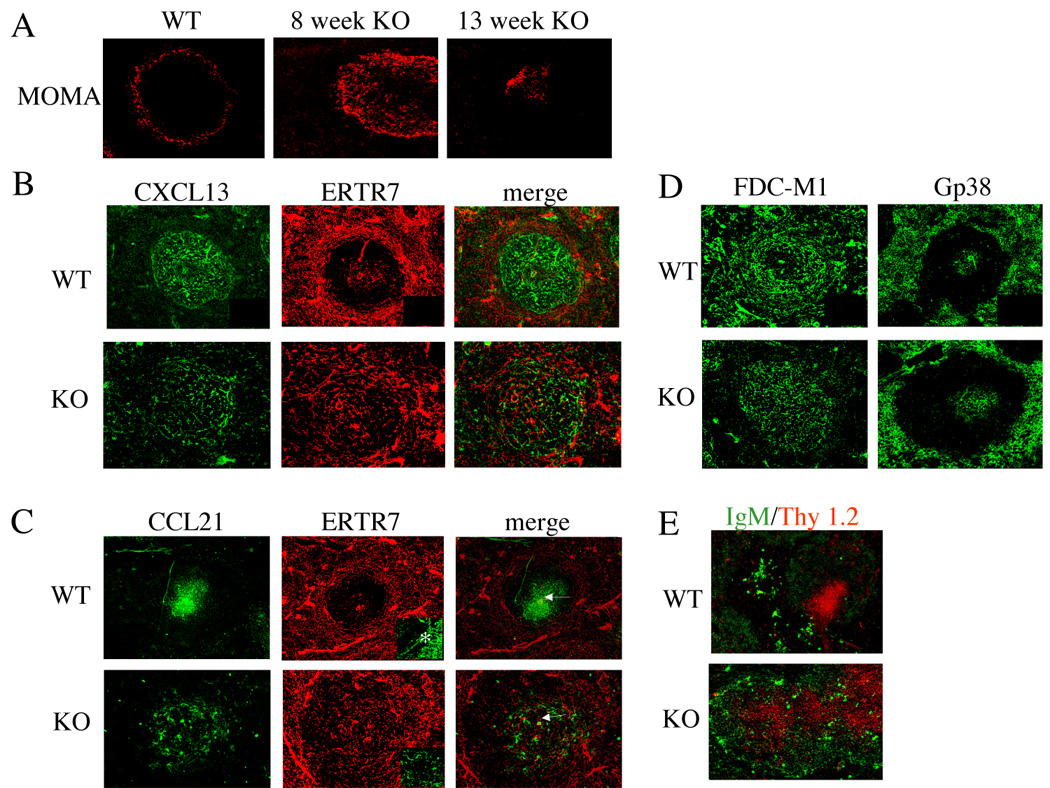
Aberrant morphology and distribution of chemokines in spleens of IL-2 deficient mice. (A) The marginal zones of VH3H9/IL-2 WT and VH3H9/IL-2 KO mice were delineated by the monoclonal antibody MOMA-1, which recognizes metallophilic macrophages present in the marginal zone. (B, C, D) The distribution of CXCL13 (B), CCL21 (C), gp38 (D), and FDC-M1 (D) in the spleens of VH3H9/IL-2 WT and KO mice was determined by immunohistochemistry, using tyramide amplification (CXCL13 and CCL21) to enhance the signal. In (C), the central arteriole is denoted by an arrow. In (B) and (C), fibroblastic reticular cells were identified by the monoclonal antibody ERTR-7. In C, the insert demonstrates the marginal sinus (*). Secondary antibody alone controls were negative (see right lower quadrant inserts, B and D). The same secondary antibody was used for CXCL13 and CCL21. (E) The distribution of B cells and T cells within splenic follicles of VH3H9/IL-2 WT and VH3H9/IL-2 KO mice was visualized via antibodies recognizing IgM and Thy 1.2. Original objective magnification for A – E: X10, C insert: X20.
In VH3H9/IL-2 WT mice, CXCL13 protein was widely distributed throughout the B cell area of the follicle, but nearly absent from the T cell area as noted by the paucity of staining around the central arteriole (Figure 7B, C). In VH3H9/IL-2 KO mice, the distribution of CXCL13 was much more disorganized, with no clear demarcation between the T and B areas of the follicle. The differences in the distribution of CCL21 were even more striking. In VH3H9/IL-2 WT mice, CCL21 was expressed in a clearly demarcated area immediately surrounding the arteriole. In the VH3H9/IL-2 KO mice, CCL21 was diffusely expressed over a larger area with a paucity of staining in the tissue immediately surrounding the arteriole (Figure 7C, central arteriole is denoted by an arrow). Accordingly, T cells in VH3H9/IL-2 WT spleens were tightly compacted in a focal area surrounding the central arteriole, whereas in VH3H9/IL-2 KO mice the T cell area was not well demarcated and the T and B cell areas overlapped considerably (Figure 7E). Interestingly, brightly staining IgM+ plasma cells were scattered throughout the follicle in VH3H9/IL-2 KO mice as compared to VH3H9/IL-2 WT mice, in which these cells are appropriately localized to the red pulp [19].
Since the distributions of both CXCL13 and CCL21 were altered in IL-2 deficiency, we next asked whether this reflected an abnormal localization of cells producing the chemokines or an abnormality in the conduit system that transports the chemokines. CXCL13 is produced by follicular dendritic cells (FDC) cells, and in tissue sections the highest expression of CXCL13 protein overlaps with these cells [18]. In VH3H9/IL-2 WT spleens, FDCs were appropriately found in the B cell area of the follicle (Figure 7D). However, in VH3H9/IL-2 KO spleens, the FDCs appeared to be spread throughout the entire follicle. Gp38+ stromal cells, which are a source of both CCL19 and CCL21 [20], were positioned appropriately in the T cell zone of the follicle in both VH3H9/IL-2 KO and VH3H9/IL-2 WT mice (Figure 7D).
Chemokines in the spleen are distributed in the white pulp by a conduit system that is surrounded by ER-TR7+ fibroblasts [21]. FDCs provide a similar reticular network in the B cell area of the follicle [21, 22]. We therefore asked whether the ERTR7+ conduit system is disrupted in IL-2 deficiency. As seen in Figure 7, the conduit system (denoted by ER-TR7+ fibroblasts) was markedly disrupted in IL-2 deficiency. The demarcation between red pulp and follicle was much less clear in VH3H9/IL-2 KO as compared to VH3H9/IL-2 WT spleens, and the marginal sinus is not seen in the IL-2 KO spleens (Figure 7C, insert). Furthermore, ER-TR7+ fibroblastic reticular cells were limited to the T cell area in VH3H9/IL-2 WT spleens but were distributed throughout the follicle in VH3H9/IL-2 KO mice. In their entirety, these findings suggest that the abnormal distributions of ER-TR7+ fibroblasts and FDCs are responsible, at least in part, for the aberrant distribution of CCL21 and CXCL13 in splenic follicles of IL-2 deficient mice. In turn, the aberrant distribution of these chemokines contributes to the altered localization of T and B cells within the follicle, and may play a role in the impaired follicular exclusion exhibited in IL-2 deficient mice.
Discussion
The development of autoimmune hemolytic anemia in mice deficient in IL-2 or its receptor suggests that IL-2 is important in the regulation of autoimmune B cells; however, the mechanisms of B cell tolerance affected by IL-2 are unknown. We now demonstrate that IL-2 influences several endogenous mechanisms of B cell tolerance including those of developmental arrest, follicular exclusion, and apoptosis.
The mechanisms of tolerance used to control autoreactive B cells of differing targets vary depending on several factors, including the affinity of the autoantigen for the B cell receptor, the concentration and form of the autoantigen, the availability of T cell help, the presence and frequency of T regulatory cells, and environmental factors [23 – 27]. Our initial studies have examined the influence of IL-2 deficiency on autoimmune B cells of moderate affinity (VH3H9) to a ubiquitous self-antigen, dsDNA. We have included, for comparison, select studies using a B cell receptor of high affinity (VH3H9/56R). Our results indicate that IL-2 deficiency impairs mechanisms of B cell tolerance used in both murine models.
Several studies by Erikson and colleagues indicate that both T helper and T regulatory cells play important roles in the regulation of autoimmune B cells [19, 28, 29]. Deletion of T cell help in MRL-lpr/lpr mice expressing the VH3H9 transgene corrected abnormalities in follicular exclusion and maturational arrest exhibited in these animals [19]. Proper follicular exclusion was mediated, at least in part, by a decrease in cell surface expression of CXCR5 on the anti-dsDNA B cells. Since the expression of CXCR5 on IL-2 deficient, anti-dsDNA B cells is very low at baseline, it is unlikely that depletion of T cell help will correct follicular exclusion by this means. Although inappropriate T cell help no doubt contributes to the generation of autoimmune B cells in IL-2 deficiency, our studies suggest that multiple factors play a role in this process.
One such factor that likely contributes to the breakdown of B cell tolerance in IL-2 deficiency is the aberrant morphology of the spleen (Figure 7). In the absence of IL-2, anti-dsDNA B cells improperly localize to B cell follicles rather than be excluded to the T/B interface. Our results suggest that this lack of follicular exclusion may be related to an altered distribution of the chemokines important for localization of T and B cells. Abnormalities in the conduit system that disseminates these chemokines is likely responsible, at least in part, for their aberrant distribution (Figure 7).
Sequestration of B cells to the marginal zone is another mechanism of B cell tolerance likely impacted by the aberrant splenic morphology observed in IL-2 deficiency. Using a model of B cell tolerance in which a large shift to the marginal zone occurs, we found that this shift was greatly diminished in IL-2 KO mice. Both autoimmune and non-autoimmune marginal zone B cells were affected, however, suggesting either a defect in the generation of marginal zone B cells, decreased expression of cell surface receptors (such as sphingosine-1-phosphate) important for localizing B cells to the marginal zone [30], or a defect in the marginal zone itself. Our finding that spleens of IL-2 deficient mice have extremely aberrant marginal zones supports the latter possibility (Figure 7).
In summary, our results demonstrate that IL-2 contributes to several mechanisms of B cell tolerance. Because patients with systemic lupus erythematosus (who produce anti-dsDNA antibodies) exhibit diminished IL-2 production [31], it is possible that this decrease contributes to autoantibody production in lupus. In fact, administration of IL-2 abrogated anti-dsDNA antibody production in MRL mice [32]. Further studies delineating the contribution of IL-2 to B cell tolerance mechanisms may lead to new therapies for lupus and other autoimmune diseases.
Acknowledgments
We thank the Cell Analysis Facility at the University of Nebraska Medical Center for performing the data acquisition portion of the flow cytometry studies. This work was supported by National Institutes of Health Grant R01 A147825.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Chen C, Prak EL, Weigert M. Editing disease-associated autoantibodies. Immunity. 1997;6:97–105. doi: 10.1016/s1074-7613(00)80673-1. [DOI] [PubMed] [Google Scholar]
- 2.Fields ML, Hondowicz BD, Wharton GN, Adair BS, Metzgar MH, Alexander ST, Caton AJ, Erikson J. The regulation and activation of lupus-associated B cells. Immunol Rev. 2005;204:165–183. doi: 10.1111/j.0105-2896.2005.00238.x. [DOI] [PubMed] [Google Scholar]
- 3.Willerford DM, Chen J, Ferry JA, Davidson L, Ma A, Alt FW. Interleukin-2 receptor alpha chain regulates the size and content of the peripheral lymphoid compartment. Immunity. 1995;3:521–530. doi: 10.1016/1074-7613(95)90180-9. [DOI] [PubMed] [Google Scholar]
- 4.Schorle H, Holtschke T, Hunig T, Schimpl A, Horak I. Development and function of T cells in mice rendered interleukin-2 deficient by gene targeting. Nature. 1991;352:621–624. doi: 10.1038/352621a0. [DOI] [PubMed] [Google Scholar]
- 5.Suzuki H, Kundig TM, Furlonger C, Wakeham A, Timmes E, Matsuyama T, Schmits R, Simard JJ, Ohashi PS, Griesser H, Taniguchi T, Paige CJ, Mak TW. Deregulated T cell activation and autoimmunity in mice lacking interleukin-2 receptor beta. Science. 1995;268:1472–1476. doi: 10.1126/science.7770771. [DOI] [PubMed] [Google Scholar]
- 6.Schultz M, Clarke SH, Arnold LW, Sartor RB, Tonkonogy SL. Disrupted B-lymphocyte development and survival in interleukin-2-deficient mice. Immunology. 2001;104:127–134. doi: 10.1046/j.0019-2805.2001.01308.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Murphy KM, Heimsberger AB, Loh DY. Induction by antigen of intrathymic apoptosis of CD4+CD8+TCRlo thymocytes in vivo. Science. 1990;250:1720–1723. doi: 10.1126/science.2125367. [DOI] [PubMed] [Google Scholar]
- 8.Farr AG, Berry ML, Kim A, Nelson AJ, Welch MP, Aruffo A. Characterization and cloning of a novel glycoprotein expressed by stromal cells in T-dependent areas of peripheral lymphoid tissues. J Exp Med. 1992;176:1477–1482. doi: 10.1084/jem.176.5.1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mandik-Nayak L, Seo S, Eaton-Bassiri A, Allman D, Hardy RR, Erikson J. Functional consequences of the developmental arrest and follicular exclusion of anti-double-stranded DNA B cells. J Immunol. 2000;164:1161–1168. doi: 10.4049/jimmunol.164.3.1161. [DOI] [PubMed] [Google Scholar]
- 10.Mackay F, Silveira PA, Brink R. B cells and the BAFF/APRIL axis: fast-forward on autoimmunity and signaling. Curr Opin Immunol. 2007 Jun;:327–336. doi: 10.1016/j.coi.2007.04.008. [DOI] [PubMed] [Google Scholar]
- 11.Duan B, Morel L. Role of B-1a cells in autoimmunity. Autoimmunity Reviews. 2006;5:403–408. doi: 10.1016/j.autrev.2005.10.007. [DOI] [PubMed] [Google Scholar]
- 12.Qian Y, Conway KL, Lu X, Seitz HM, Matsushima GK, Clarke SH. Autoreactive MZ and B-1 B cell activation by Faslpr is coincident with an increased frequency of apoptotic lymphocytes and a defect in macrophage clearance. Blood. 2006;108:974–982. doi: 10.1182/blood-2005-12-006858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Grimaldi CM, Michael DJ, Diamond B. Cutting edge: expansion and activation of a population of autoreactive marginal zone B cells in a model of estrogen-induced lupus. J Immunol. 2001;167:1886–1890. doi: 10.4049/jimmunol.167.4.1886. [DOI] [PubMed] [Google Scholar]
- 14.Rolf J, Motta V, Duarte N, Lundholm M, Berntman E, Bergman ML, Sorokin L, Cardell SL, Holmberg D. The enlarged population of marginal zone/CD1d (high) B lymphocytes in nonobese diabetic mice maps to diabetes susceptibility region Idd11. J Immunol. 2005;174:4821–4827. doi: 10.4049/jimmunol.174.8.4821. [DOI] [PubMed] [Google Scholar]
- 15.Li Y, Li H, Weigert M. Autoreactive B cells in the marginal zone that express dual receptors. J Exp Med. 2002;195:181–188. doi: 10.1084/jem.20011453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li Y, Li H, Ni D, Weigert M. Anti-DNA B cells in MRL/lpr mice show altered differentiation and editing pattern. J Exp Med. 2002;196:1543–1552. doi: 10.1084/jem.20021560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ekland EH, Forster V, Lipp V, Cyster V. Requirements for follicular exclusion and competitive elimination of autoantigen-binding B cells. J Immunol. 2004;172:4700–4708. doi: 10.4049/jimmunol.172.8.4700. [DOI] [PubMed] [Google Scholar]
- 18.Cyster JG. Chemokines, sphingosine-1-phosphate, and cell migration in secondary lymphoid organs. Annu Rev Immunol. 2005;23:127–159. doi: 10.1146/annurev.immunol.23.021704.115628. [DOI] [PubMed] [Google Scholar]
- 19.Seo SJ, Fields V, Buckler V, Reed V, Mandik-Nayak V, Nish V, Noelle V, Turka V, Finkelman V, Caton V, Erikson V. The impact of T helper and T regulatory cells on the regulation of anti-double-stranded DNA B cells. Immunity. 2002;16:535–546. doi: 10.1016/s1074-7613(02)00298-4. [DOI] [PubMed] [Google Scholar]
- 20.Luther SA, Tang V, Hyman V, Farr V, Cyster V. Coexpression of the chemokines ELC and SLC by T zone stromal cells and deletion of the ELC gene in the plt/plt mouse. Proc Natl Acad Sci USA. 2000;97:12694–12699. doi: 10.1073/pnas.97.23.12694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nolte MA, Belien V, Schadee-Eestermans V, Jansen V, Unger V, van Rooijen V, Kraal V, Mebius V. A conduit system distributes chemokines and small blood-borne molecules through the splenic white pulp. J Exp Med. 2003;198:505–512. doi: 10.1084/jem.20021801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bajenoff M, Egen V, Koo V, Laugier V, Brau V, Glaichenhaus V, Germain V. Stromal cell networks regulate lymphocyte entry, migration, and territoriality in lymph nodes. Immunity. 2006;25:989–1001. doi: 10.1016/j.immuni.2006.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Borrero M, Clarke SH. Low-affinity anti-Smith antigen B cells are regulated by anergy as opposed to developmental arrest or differentiation to B-1. J Immunol. 2002;168:13–21. doi: 10.4049/jimmunol.168.1.13. [DOI] [PubMed] [Google Scholar]
- 24.Aoki CA, Roifman CM, Lian ZX, Bowlus CL, Norman GL, Shoenfeld Y, Mackay IR, Gershwin ME. IL-2 receptor alpha deficiency and features of primary biliary cirrhosis. J Autoimmun. 2006;27:50–53. doi: 10.1016/j.jaut.2006.04.005. [DOI] [PubMed] [Google Scholar]
- 25.Youinou P. B cell conducts the lymphocyte orchestra. J Autoimmun. 2007;28:143–151. doi: 10.1016/j.jaut.2007.02.011. [DOI] [PubMed] [Google Scholar]
- 26.Sharma R, Zheng L, Guo X, Fu SM, Ju ST, Jarjour WN. Novel animal models for Sjogren's syndrome: Expression and transfer of salivary gland dysfunction from regulatory T cell-deficient mice. J Autoimmun. 2006;27:289–296. doi: 10.1016/j.jaut.2006.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ansari AA, Pereira LE, Mayne AE, Onlamoon N, Pattanapanyasat K, Mori K, Villinger F. The role of disease stage, plasma viral load and regulatory T cells (Tregs) on autoantibody production in SIV-infected non-human primates. J Autoimmun. 2007;28:152–159. doi: 10.1016/j.jaut.2007.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fields ML, Hondowicz BD, Metzgar MH, Nish SA, Wharton GN, Picca CC, Caton AJ, Erikson J. CD4+ CD25+ regulatory T cells inhibit the maturation but not the initiation of an autoantibody response. J Immunol. 2005;175:4255–4264. doi: 10.4049/jimmunol.175.7.4255. [DOI] [PubMed] [Google Scholar]
- 29.Fields ML, Nish SA, Hondowicz BD, Metzgar MH, Wharton GN, Caton AJ, Erikson J. The influence of effector T cells and Fas ligand on lupus-associated B cells. J Immunol. 2005;175:104–111. doi: 10.4049/jimmunol.175.1.104. [DOI] [PubMed] [Google Scholar]
- 30.Cinamon G, Matloubian M, Lesneski MJ, Xu Y, Low C, Lu T, Proia RL, Cyster JG. Sphingosine 1-phosphate receptor 1 promotes B cell localization in the splenic marginal zone. Nat Immunol. 2004;5:713–720. doi: 10.1038/ni1083. [DOI] [PubMed] [Google Scholar]
- 31.Katsiari CG, Tsokos GC. Transcriptional repression of interleukin-2 in human systemic lupus erythematosus. Autoimmun Rev. 2006;5:118–121. doi: 10.1016/j.autrev.2005.08.009. [DOI] [PubMed] [Google Scholar]
- 32.Huggins ML, Huang FP, Xu D, Lindop G, Stott DI. Modulation of autoimmune disease in the MRL-lpr/lpr mouse by IL-2 and TGF-beta1 gene therapy using attenuated Salmonella typhimurium as gene carrier. Lupus. 1999;8:29–38. doi: 10.1191/096120399678847308. [DOI] [PubMed] [Google Scholar]