

Abstract
Enabled/vasodilator-stimulated phosphoprotein (Ena/VASP) proteins are key actin regulators that localize at regions of dynamic actin remodeling, including cellular protrusions and cell–cell and cell–matrix junctions. Several studies have suggested that Ena/VASP proteins are involved in the formation and function of cellular junctions. Here, we establish the importance of Ena/VASP in endothelial junctions in vivo by analysis of Ena/VASP-deficient animals. In the absence of Ena/VASP, the vasculature exhibits patterning defects and lacks structural integrity, leading to edema, hemorrhaging, and late stage embryonic lethality. In endothelial cells, we find that Ena/VASP activity is required for normal F-actin content, actomyosin contractility, and proper response to shear stress. These findings demonstrate that Ena/VASP is critical for actin cytoskeleton remodeling events involved in the maintenance of functional endothelia.
Introduction
The vascular system is lined with endothelial cells that serve as a barrier separating blood from underlying tissues. Maintenance and modulation of endothelial barrier function is necessary to control the movement of nutrients, fluids, and immune cells between the intravascular and extravascular compartments. Barrier function requires cell–cell interactions, mediated in large part by adherens and tight junctions, which act to restrict the passage of material between cells in the endothelial lining (Corada et al., 1999; Hordijk et al., 1999). Loss of cell–cell junctional integrity in the endothelial lining results in increased permeability and edema (Wysolmerski et al., 1984; Wysolmerski and Lagunoff, 1985; Rotrosen and Gallin, 1986).
Both adherens and tight junctions use transmembrane proteins that interact with homotypic receptors on neighboring cells. Ligation of these receptors stimulates the cytoplasmic recruitment of multiprotein complexes that are involved in signal transduction and interaction with the cytoskeleton. Endothelial adherens junctions are formed by the homophilic interaction of transmembrane VE-cadherin molecules (Dejana et al., 1999). The intracellular domain of vascular endothelial–cadherin (VE-cadherin) interacts with β-catenin and recruits molecules such as plakoglobin, α-catenin, zyxin, p120 catenin, and vinculin (Aberle et al., 1996; Pokutta and Weis, 2002; Bazzoni and Dejana, 2004). Tight junctions contain several transmembrane components, including occludin, claudins, and junctional adhesion molecule-A (Martin-Padura et al., 1998; Mitic and Anderson, 1998). These interact with intracellular components such as the zonula occludens (ZO) proteins (ZO-1, ZO-2, and ZO-3) and AF-6/Afadin.
Functionally, cell–matrix associations, such as focal adhesions, are similar to cell–cell junctions in that they link the cytoskeleton with external contact points and are important for barrier function (Wu, 2005). In focal adhesions, transmembrane integrin receptors bind extracellular matrix and recruit intracellular multiprotein complexes that interact with the cytoskeleton. Many of the proteins found in focal adhesions are also present in cell–cell junctions.
Members of the Enabled/vasodilator-stimulated phosphoprotein (Ena/VASP) family of actin regulatory proteins are found at both focal adhesions and cell–cell junctions, where they interact and colocalize with components of adherens and tight junctions (Reinhard et al., 1992; Lawrence et al., 2002; Tanoue and Takeichi, 2004). In addition to localization to sites involved in barrier function, Ena/VASP is a well-established substrate for PKA and PKG (Waldmann et al., 1987; Halbrugge and Walter, 1989; Haffner et al., 1995). PKA and PKG signaling pathways are involved in barrier function regulation, which raises the possibility that Ena/VASP could be a downstream effector of these pathways.
At the leading edge of lamellipodia and the tips of filopodia, Ena/VASP localizes to regions with dynamic actin reorganization (Reinhard et al., 1992; Gertler et al., 1996; Lanier et al., 1999; Rottner et al., 1999; Lambrechts et al., 2000), where it has been proposed to promote actin polymerization via an anticapping mechanism (Bear et al., 2002; Barzik et al., 2005). Dynamic actin is also found at focal adhesions and cell–cell junctions, and ligation of cell–cell receptors, such as cadherins, results in reorganization of the underlying actin cytoskeleton (Yonemura et al., 1995; Adams and Nelson, 1998). Disruption of Ena/VASP has been shown to reduce the formation of perijunctional actin filaments (Scott et al., 2006) and perturbs the dynamics of epithelial sheet sealing in Drosophila melanogaster (Gates et al., 2007). Although these data suggest that Ena/VASP regulates actin dynamics at cell–cell and cell–matrix junctions, the functional implications of this are unclear.
In mammals, the Ena/VASP family consists of three members, mammalian Ena (Mena), VASP, and Ena/Vasp-like (Reinhard et al., 1992; Gertler et al., 1996). Because these members have overlapping function and expression patterns, knocking out individual family members results in relatively minor phenotypes in mouse models (Aszodi et al., 1999; Hauser et al., 1999; Lanier et al., 1999). To determine the biological function of this family of proteins, we have generated an Ena/VASP triple null (mmvvee) mouse. mmvvee mice exhibit neuronal defects, including exencephaly, cobblestone cortex, and lack of cortical fiber tract formation, which have previously been characterized elsewhere (Kwiatkowski et al., 2007). In addition to these phenotypes, mmvvee mice display severe vascular defects that are described here.
Results
mmvvee mice exhibit edema
Gross examination of mmvvee embryos revealed profound edema, with 82% of embryos (14 out of 17) at embryonic day (E) 14.5 appearing edematous to the naked eye (Fig. 1 A). The edema was most pronounced in the torso, where transverse sectioning showed that it was largely restricted to the subdermal region (Fig. 1 B). Edema did not result from exencephaly because we observed edematous mmvvee embryos that were not exencephalic (unpublished data).
Figure 1.

Edema phenotype of mmvvee embryos. (A) mmvvee E14.5 embryos exhibit edema (arrow) as compared with littermate control. (B) Hematoxylin and eosin staining of transverse histological sections from E14.5 embryos. Subdermal region is greatly expanded in mmvvee embryos (arrows).
Edema is a common endpoint for a variety of physiological defects. Alteration of either hydrostatic or oncotic pressure can result from impaired kidney function leading to alterations in salt and water handling and/or protein wasting. However, the onset of edema in mmvvee embryos was apparent from histological sectioning as early as E12.5 (unpublished data). At this stage in development the embryonic kidney is not functional, ruling out kidney dysfunction as the cause of edema.
Embryonic edema can result from cardiac defects (Komatsu et al., 2002; Fan et al., 2005; Li et al., 2006). Congenital heart defects typically manifest with cardiac dilation and congestive heart failure, resulting in increases in the hydrostatic pressure within the vascular system, which drive fluid into the interstitial tissue. To determine if heart defects could explain the edema observed in mmvvee embryos, we analyzed the anatomy of the mmvvee embryonic heart. Histologically, mmvvee hearts appeared structurally normal (Fig. 2 A) with no sign of dilation. Ena/VASP has been implicated in Sema6D signaling events necessary for proper cardiac chamber formation and trabeculation (Toyofuku et al., 2004); however, trabeculation in the mmvvee heart was normal (Fig. 2 A), and major structural features of the mmvvee heart, including septum, valves, and associated veins and arteries, displayed no defects (not depicted). Closer analysis by electron microscopy showed that mmvvee cardiac myocytes formed Z lines and intercalated discs that appeared similar to those of littermate controls (Fig. 2, B and C). Although there is no evidence of structural or functional heart abnormalities that could explain the edema phenotype, we did notice that mmvvee endocardia displayed elongated cell membranes and were less closely associated with the cardiac myocytes than endocardia in the controls (Fig. 2 D). Because the endocardium is the endothelial lining of the heart, this defect suggested that endothelial dysfunction might be at the root of the edema caused by loss of Ena/VASP.
Figure 2.

mmvvee embryonic heart has a thin endocardium. (A) mmvvee embryonic hearts are not hypertrophic. Cardiac chambers are not dilated and have normal patterns of trabeculation as shown by hematoxylin and eosin staining of histological sections. Electron microscopy demonstrates formation of Z lines (B, arrows) and intercalated discs (C) in mmvvee embryos. Magnified images of intercalated discs identified by red rectangles are shown under C. (D) The endocardial lining (arrowheads) of mmvvee embryonic hearts are thinner than littermate controls and are less closely associated with the underlying cardiac myocytes (arrows).
Vessel patterning and integrity are compromised in the absence of Ena/VASP
The loss of junctional proteins VE-cadherin, β-catenin, and FAK in the endothelia of mice results in edema and the regression of blood vessels during development (Carmeliet et al., 1999; Cattelino et al., 2003; Shen et al., 2005; Braren et al., 2006). To determine the patterning and integrity of blood vessels in mmvvee mice, whole mount embryos were stained for the endothelial-specific marker platelet/endothelial cell adhesion molecule (PECAM) 1, and vessels were found to be grossly intact at E10.5 (Fig. 3 A). Although major vessels appeared normal in mmvvee embryos, there were differences in small vessels evident at E10.5, including disruption in connectivity between intersomitic vessels in the mid-torso region (Fig. 3 B) and irregular patterning in the vascular plexus of the tail (Fig. 3 C). These subtle differences in the small vessels of mmvvee embryos are relatively minor in comparison to the vessel disruption observed in mice that were null for endothelial junctional proteins such as β-catenin (Cattelino et al., 2003). Importantly, these patterning defects are not the beginning of widespread vascular regression because vessels visualized under a dissection microscope were intact in the amniotic sac and body as late as E16.5 (Fig. 3, D and E). Vessels in the body and amnion of E16.5 mmvvee mice did not exhibit any differences in patterning or vascular density.
Figure 3.

Vascular formation and patterning in mmvvee embryos. (A) E10.5 embryos after whole-mount immunohistochemistry with anti-PECAM antibodies. Major blood vessels are present and intact in the mmvvee embryo. (B) Intersomitic vessels appear normal in mmvvee embryos (arrowheads) but there is a reduction in connectivity between them (arrows). (C) The vascular plexus in the mmvvee tail (indicated by brackets and magnified under C) exhibits irregular patterning. Images were gamma adjusted. Major vessels in the amniotic sac (D) and body (E) of mmvvee embryos appear normal in formation and patterning at E16.5. Note the increased volume and turbidity of the mmvvee amniotic fluid.
Disruption in endothelial barrier function allows the leakage of plasma proteins into the interstitial space. This alters the oncotic pressure gradient that normally functions to draw fluid back into the vasculature, resulting in the accumulation of interstitial fluid. A more profound disruption in barrier function allows the hemorrhaging of red blood cells. Consistent with a role for Ena/VASP in promoting barrier function, we observed sporadic hemorrhaging in mmvvee embryos (Fig. 4 A). The earliest observations of hemorrhage were at E14.5 and increased throughout development. Hemorrhaging was also evident by an accumulation of blood in the amnion (Fig. 4 A). Similar to the pattern seen in the embryo of edema followed by hemorrhage, analysis of embryos at different stages revealed that the amnion first accumulated excess amniotic fluid (Fig. 3 D) followed by hemorrhage. By E18.5, 70% of mmvvee amnions (60 out of 86) exhibited an accumulation of blood. Careful examination of recently harvested mmvvee embryos failed to identify an obvious source of hemorrhage that would account for the accumulation of amniotic blood. Although it is likely that the blood in the amnion is caused by hemorrhaging of the amniotic vasculature, it is possible that it comes from the embryo itself.
Figure 4.

Venule discontinuity leads to hemorrhages in mmvvee embryos at later embryonic stages. (A) Sporadic hemorrhages (arrows) in the trunk of mmvvee embryos are first visible around E16.5 and become progressively more prominent (top left). At E18.5 the amnion is filled with blood and can be identified during dissection (mmvvee embryo held with forceps). Bottom shows littermate control and mmvvee embryos in yolk sac with placenta attached. (B) Hematoxylin and eosin staining of histological sections show numerous small vessels in mmvvee embryos that contain red blood cells and appear to be discontinuous as early as E14.5 (arrowhead). Bar, 10 μm. (C) Electron micrographs of blood vessels indicate that the endothelial junctions in arterioles of mmvvee are intact (open arrowheads), but numerous ruptures are present in venules (closed arrowheads) with evidence of escaping red blood cells (asterisk). The endothelial cells of these venules displayed extensive cellular lamellipodia protruding in the vascular lumen or extending in the subendothelial space (small arrows).
Histological analysis indicated a loss of integrity of small blood vessels in mmvvee subdermal tissue (Fig. 4 B). This was confirmed by transmission electron microscopy, which found that 60% of blood vessels (6 out of 10) >10 μm in diameter were discontinuous, meaning there was an opening in the endothelial lining and escaping red blood cells (Fig. 4 C). These discontinuous vessels were determined to be venules, based on size and their lack of surrounding adventitia. In addition to discontinuity, mmvvee venules also displayed endothelial cell protrusions into the vascular lumen and subendothelial space (Fig. 4 C), further suggesting a defect in cell–cell contacts. In contrast, no examples of discontinuous capillaries/small arterioles or larger veins and arteries were evident in mmvvee embryos. Very small vessels (<10 μm), presumed to be capillaries, appeared to be consistently intact in both groups. These findings suggest that hemorrhaging in mmvvee embryos results specifically from open venules.
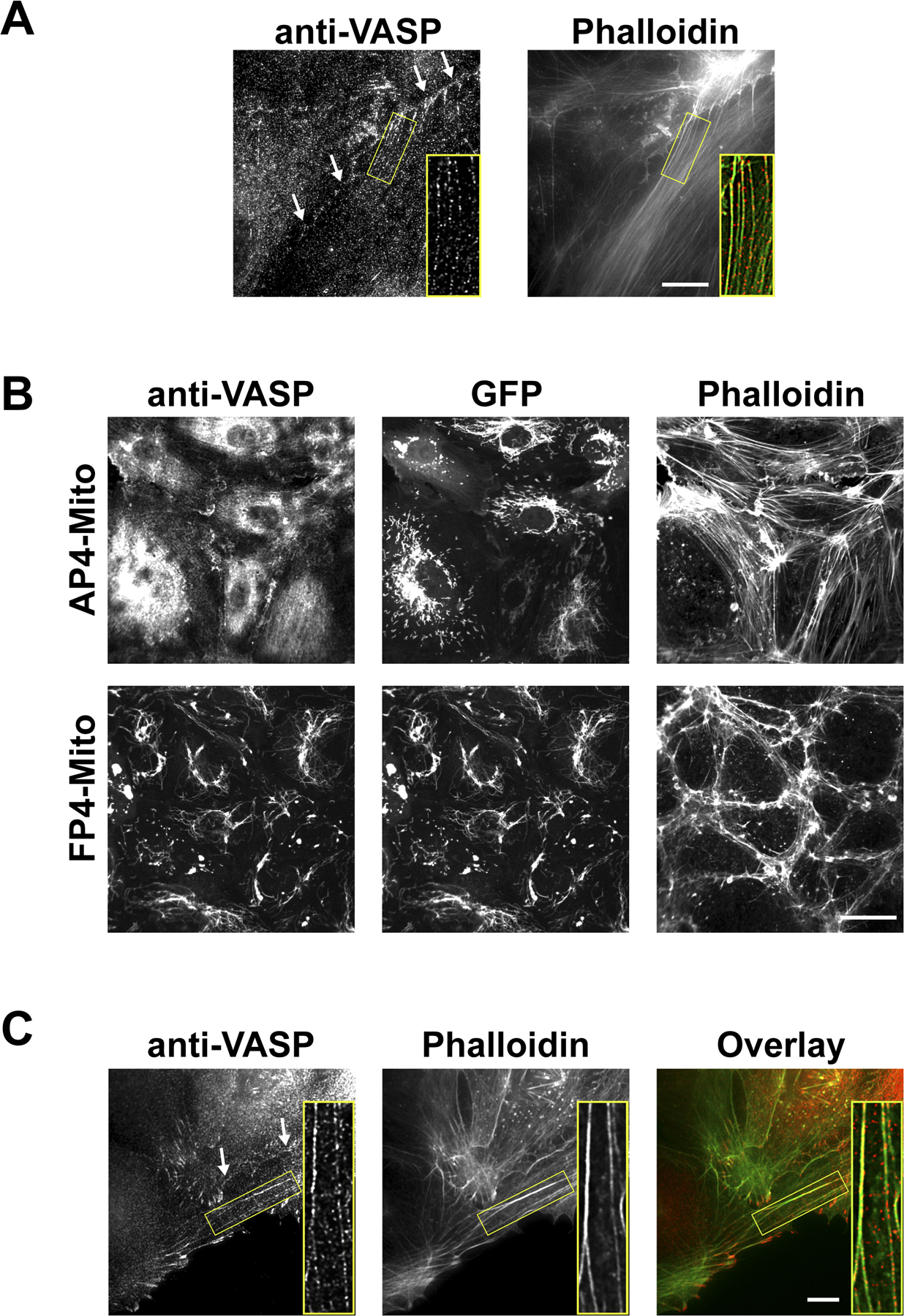
Ena/VASP activity is required for proper barrier function
To measure the effect of Ena/VASP on endothelial barrier function, we used a cell-based assay to test the ability of fluorescently tagged dextran to cross a confluent monolayer of endothelial cells. The dextran molecule had a molecular weight comparable to albumin, the chief protein component of blood plasma. The isolation and culture of sufficient quantities of primary endothelial cells from mmvvee embryonic mice was not possible, so we used human umbilical vein endothelial cells (HUVECs) for the barrier function assays. Consistent with localization in other cell types, Ena/VASP proteins exhibited localization at focal adhesions, stress fibers, and cell–cell contacts in HUVECs (Fig. S1 A, available at http://www.jcb.org/cgi/content/full/jcb.200705002/DC1). Previous studies of the role of Ena/VASP on barrier function focused on VASP and relied on the overexpression of VASP fragments to interfere with VASP function (Vasioukhin et al., 2000; Comerford et al., 2002; Lawrence et al., 2002) or used siRNA to knock down VASP (Rosenberger et al., 2007), which did not eliminate the function of other Ena/VASP proteins expressed in endothelial cells (unpublished data). To overcome these potential limitations, we used a strategy proven to block the activity of all Ena/VASP family members. This approach involves expression of the EGFP-FP4-Mito construct that sequesters all Ena/VASP proteins to the surface of the mitochondria (Fig. S1 B) and mimics loss of function (Bear et al., 2000, 2002; Lebrand et al., 2004). EGFP-FP4-Mito–expressing HUVECs were grown to confluence and used in barrier function assays. When Ena/VASP activity was inhibited, basal barrier function was significantly reduced as compared with control HUVECs expressing EGFP or EGFP-AP4-Mito, a construct that targets mitochondria but does not sequester Ena/VASP (Fig. 5 A). Inhibition of Ena/VASP activity resulted in a reduction of barrier function similar to that attained through the addition of VEGF, a known permeability enhancing agent, to control monolayers (Fig. 5 B). The high permeability of Ena/VASP-inactivated monolayers could be further augmented with VEGF treatment, indicating that Ena/VASP is not required for response to VEGF. Rather, Ena/VASP activity appears to be important in the maintenance of basal barrier function. In support of this, overexpression of EGFP-VASP improved barrier function (Fig. 5 B). Collectively, these data are consistent with a model in which the edema and hemorrhage observed in mmvvee embryos result from defects in endothelial barrier function.
Figure 5.

Barrier function is reduced in endothelial cells lacking Ena/VASP activity. (A) HUVECs expressing FP4-Mito, a construct that inactivates Ena/VASP proteins, exhibit an increase in the movement of Texas red–dextran across confluent monolayers compared with control cells expressing GFP or AP4-Mito, a mutated version of the sequestration construct that targets the mitochondria but is incapable of binding Ena/VASP proteins (*, P < 0.05; n = 6). Error bars represent SD. (B) Stimulation of HUVEC monolayers with VEGF increases the permeability of control monolayers to levels similar to FP4-Mito monolayers without VEGF treatment. Overexpression of VASP decreases permeability compared with GFP control. VEGF treatment further reduces barrier function of FP4-Mito monolayers as compared with FP4-Mito without VEGF treatment (*, P < 0.05; n = 3). Error bars represent SD.
The assembly of cell–cell junction protein complexes is unperturbed by the loss of Ena/VASP
The defective vascular integrity of mmvvee endothelium and reduced barrier function described in the previous sections could result from a failure to recruit the normal complement of cell–cell junction proteins. In fact, overexpression of a fragment of Ena/VASP thought to act as a dominant negative has been shown to block E-cadherin recruitment in keratinocytes (Vasioukhin et al., 2000). Furthermore, it has been suggested that phosphorylation of VASP by PKA is important for ZO-1 recruitment to cell–cell junctions (Kohler et al., 2004). To determine if the genetic loss of Ena/VASP affects the recruitment of junctional proteins, we isolated primary PECAM-positive endothelial cells from the aortae of control and mmvvee embryos and stained for junctional components. The resulting small clusters of primary endothelial cells showed that localization of PECAM, VE-cadherin, β-catenin, α-catenin, and ZO-1 to cell–cell junctions were unaffected by the absence of Ena/VASP (Fig. 6 A). Immunohistochemistry on tissue sections also showed a normal distribution of cell junctional markers in the mmvvee vasculature (unpublished data). Similarly, inactivation of Ena/VASP in HUVECs had no effect on the recruitment of the same junctional proteins to established cell–cell junctions (Fig. 6 B) despite the observed barrier defects described in the previous section. The recruitment of junctional proteins was also unperturbed by VASP overexpression (Fig. 6 B).
Figure 6.

Ena/VASP is not required for proper localization of junctional proteins. (A) Junctional proteins, as indicated, localize to cell junctions in primary endothelial cells isolated from mmvvee and control embryonic aortae. No obvious changes in intensity or localization of junctional proteins are evident, although there is a decrease in stress fibers in mmvvee cells. Bar, 10 μm. (B) Localization of junctional proteins VE-cadherin, ZO-1, and β-catenin is unaffected by Ena/VASP inactivation in HUVECs, although the morphology of cell–cell junctions is altered. Inactivation of Ena/VASP results in a smoother appearing junction, whereas junctions are highly striated in HUVECs overexpressing Ena/VASP. Bar, 50 μm. (C) VE-cadherin recovery to cell junctions after calcium switch is unaffected by Ena/VASP. HUVECs were grown to confluence and treated with 5 mM EGTA for 30 min to disrupt adherens junctions. EGTA was washed out and cells were allowed to recover for the indicated times, followed by fixation and immunofluorescence for VE-cadherin. Recovery rates of VE-cadherin were indistinguishable between control HUVECs and HUVECs with altered Ena/VASP activity. Bar, 50 μm.
To test if the kinetics of cadherin-dependent contact formation was perturbed in the absence of Ena/VASP, we performed calcium switch assays on HUVEC cultures. Chelation of calcium disrupts the ligation and localization of VE-cadherin at cell junctions. After the reintroduction of calcium, the recovery rate of VE-cadherin to junctions of Ena/VASP-inactivated cells was identical to controls, with localization first detected after 30 min and full recovery after 4 h (Fig. 6 C). These results indicate that neither cadherin-mediated adhesion nor the recruitment of junctional proteins to cell–cell contacts are affected by the absence of Ena/VASP, which suggests that mislocalization or delayed recruitment of junctional proteins does not cause the edema observed in mmvvee embryos.
Ena/VASP is important for shear stress response
Loss of Ena/VASP activity in primary endothelial cells (Fig. 6 A) and HUVEC cultures (Fig. 7 A), either by genetic knockout or protein inactivation, respectively, led to a visible reduction in F-actin content. This reduction in F-actin was measured with microscopy by quantitating the signal intensity of fluorescently tagged phalloidin (Fig. 7 B). Although overexpression of Ena/VASP in HUVEC monolayers did not cause a substantial increase in F-actin content, it did result in a striking rearrangement of actin into parallel ventral stress fibers that aligned with each other across multiple cell lengths (Fig. 7 A).
Figure 7.

Stress fiber formation is affected by Ena/VASP activity. (A) Inactivation of Ena/VASP in confluent HUVEC monolayers decreases stress fiber formation, whereas overexpression of Ena/VASP results in the formation of parallel ventral stress fibers as compared with controls. Bar, 50 μm. Stress fibers at regions of cell–cell junctions colocalize with the junctional marker β-catenin (inset). (B) Box-and-Whisker plots of the decrease of F-actin in Ena/VASP-inactivated HUVECs and mmvvee primary aorta endothelial cells (*, P < 0.05; n = 20). Despite changes in actin architecture, total F-actin content is not altered in HUVECs overexpressing VASP. The middle line of the box indicates median, the top of the box indicates 75th quartile, the bottom of the box indicates 25th quartile, and whiskers extend to the highest and lowest values. (C) HUVECs expressing FP4-Mito fail to form stress fibers in response to shear stress (10 dyne/cm2, 16–20 h). Magnified images identified by red rectangles are shown (bottom). Flow direction is indicated by arrow. Images are gamma adjusted. Bar, 50 μm. (D) Confocal micrographs of phalloidin- and anti-PECAM–labeled dorsal aortae show that cells in the endothelial lining of mmvvee aortae fail to form stress fibers and elongate in the direction of flow. Bar, 10 μm.
The actin reorganization caused by Ena/VASP overexpression is similar to that induced by shear stress. Under physiological conditions, flowing blood subjects endothelial cells to shear stress, which has been shown to modulate numerous endothelial cell functions, particularly the strengthening of cell–cell junctions and elongation of cell shape in the direction of flow (Seebach et al., 2000; Shikata et al., 2005). To determine if the cytoskeletal changes that facilitate response to shear stress are impaired in the absence of Ena/VASP activity, we cultured Ena/VASP-inactivated HUVECs in flow chambers. Confluent monolayers of control HUVECs subjected to shear stress overnight responded as expected by forming parallel stress fibers but, strikingly, Ena/VASP-inactivated HUVECs failed to form stress fibers (Fig. 7 C). Instead, shear stress appeared to result in a slight decrease in the amount of stress fibers as compared with cells cultured without flow. In contrast, cortical actin staining appeared unaffected by inhibition of Ena/VASP. To determine if shear stress response was also impaired in mmvvee embryos, we looked at actin organization and endothelial cell shape in aortae. Control aortae had robust stress fibers that ran in the direction of blood flow, whereas the stress fibers of mmvvee embryos were less organized. Immunostaining for PECAM in control aortae revealed a strong linear pattern consistent with cells that had elongated in the direction of blood flow, but PECAM staining of endothelial cells in the lining of mmvvee aortae was visibly disorganized (Fig. 7 D). These data indicate that Ena/VASP is critical for the reorganization of the cytoskeleton and changes in cell morphology in response to shear stress.
Actin incorporation and actomyosin contractility are affected by Ena/VASP
Biochemical experiments suggest Ena/VASP binds at, or near, the barbed ends of F-actin and permits continued incorporation of monomeric actin in the presence of capping protein by antagonizing capping activity. We hypothesized that the observed changes in F-actin content were attributable to changes in actin incorporation at barbed ends. To test this possibility, we pulsed permeabilized HUVECs with fluorescently labeled monomeric actin and measured its incorporation at free barbed ends by quantitative microscopy. G-actin incorporation into barbed ends was reduced in cells in which Ena/VASP was inhibited and increased upon the overexpression of VASP (Fig. 8, A and B). The sites of the greatest barbed end labeling were at the leading edges of lamellipodia, focal adhesions, and along stress fibers in discrete puncta known as dense bodies, which is consistent with previously published results using nonendothelial cell types (Kreis et al., 1979). To determine the effect of Ena/VASP activity on these different structures, we independently measured barbed end labeling at the cell periphery (leading edge), cell–cell junctions, and internal regions of the cell, where the signal was largely caused by incorporation into stress fibers. Barbed end labeling at all of these sites was found to be sensitive to Ena/VASP activity, although the most profound effect was observed along stress fibers, with signal being frequently undetectable in cells expressing FP4-Mito (Fig. 8 A). This same pattern was found using dissociated primary endothelial cells, with barbed end incorporation reduced in cells from mmvvee aorta as compared with littermate controls (Fig. 8 C). Ena/VASP has previously been described to decorate stress fibers in discrete puncta (Reinhard et al., 1992; Boukhelifa et al., 2004), a localization we confirmed in both HUVECs and primary endothelial cells (Fig. S1, A and C). Higher resolution analysis of stress fibers showed that GFP-VASP localizes in puncta that overlap with, or are immediately adjacent to, regions of barbed end labeling (Fig. 8 D). Puncta were spaced at a mean of 0.852 ± 0.285 μm from each other, a distribution that was unaffected by Ena/VASP activity. The colocalization of Ena/VASP and sites of G-actin incorporation along stress fibers, and the reduction of G-actin incorporation in stress fibers upon the inactivation of Ena/VASP, indicates that Ena/VASP promotes actin monomer incorporation along stress fibers.
Figure 8.

G-actin incorporation is reduced in cells lacking Ena/VASP activity. (A) Fluorescent images showing F-actin (phalloidin) and sites of recent G-actin incorporation at barbed ends. G-actin incorporation at barbed ends is highest at the cell periphery and stress fibers and is visibly reduced in stress fibers by Ena/VASP inactivation. Overexpression of Ena/VASP proteins increases G-actin incorporation. Incorporation at barbed ends in HUVECs expressing AP4-Mito was comparable to the EGFP control. Bar, 50 μm. (B) Box-and-Whisker plots of total cellular F-actin content and G-actin incorporation in HUVECs (n > 23). Peripheral measurements were taken from the first 5 μm, starting at the cell edge of subconfluent cells. F- and G-actin content in cell junctions under confluent conditions were measured by linescan (n = 40). (C) Box-and-Whisker plots of F-actin content and G-actin incorporation in internal regions and cell–cell junctions of primary endothelial cells. Actin incorporation was reduced in cells derived from mmvvee embryos as compared with littermate controls (n > 17). The middle line of the box indicates median, the top of the box indicates 75th quartile, the bottom of the box indicates 25th quartile, and whiskers extend to the highest and lowest values. (D) Higher resolution images of stress fibers (blue) show that G-actin (red) incorporation occurs in discrete punta that colocalize with EGFP-VASP (green). Bar, 25 μm. *, P < 0.05.
Stress fibers are associated with the generation of contractile forces within the cell. Other proteins known to localize in puncta along stress fibers include α-actinin, filamin, tropomyosin, and myosin, all of which are involved in actomyosin contraction (Lazarides and Burridge, 1975; Gordon, 1978; Kreis and Birchmeier, 1980; Byers et al., 1984; Drenckhahn and Wagner, 1986; Svitkina et al., 1989). Because the actin stress fiber rearrangements that accompany Ena/VASP overexpression are similar to those observed in highly contractile cells, we measured the expression of myosin light chain (MLC) in cells with altered Ena/VASP activity. Ena/VASP overexpression increased levels of MLC over that in controls, whereas inactivation of Ena/VASP reduced MLC expression (Fig. 9, A and B). These changes in the level of MLC expression were accompanied by changes in MLC phosphorylation that have been shown to be indicative of enhanced actomyosin contraction (Sobieszek, 1977).
Figure 9.

Ena/VASP activity affects actomyosin contractility. (A) Western blots with antibodies to total or phosphorylated MLC show that inactivation of Ena/VASP in HUVECs reduces the level of MLC expression, whereas cells overexpressing Ena/VASP have higher levels of MLC expression. (B) Quantitation of total and phospho MLC (*, P < 0.05; n = 5). Error bars represent SD. (C) HUVEC cells with inactivated Ena/VASP exert less force, as measured by contraction of collagen matrices, whereas overexpression of VASP increases force generation compared with GFP control (*, P < 0.05; n = 10). Contraction of HUVECs expressing AP4-Mito was statistically indistinguishable from GFP control (not depicted).
To determine if changes in MLC expression and phosphorylation state had the predicted effect on actomyosin contraction, we seeded endothelial cells into three-dimensional collagen matrices and used matrix compaction as an indication of the force exerted upon the collagen by the cells. Consistent with activation of MLC, HUVECs overexpressing Ena/VASP exerted more force on the collagen, whereas Ena/VASP-inactivated cells were less contractile than controls (Fig. 9 C).
Ena/VASP activity alters junction morphology
Cell–cell and cell–matrix contacts attach to the actin cytoskeleton and are influenced by force exerted on them through actomyosin contraction. Although the recruitment of adherens and tight junction proteins to cell–cell contacts was not altered by Ena/VASP activity, the gross morphology of the junction was affected. In less contractile Ena/VASP-inactivated HUVECS, staining for junctional marker proteins VE-cadherin, ZO-1, and β-catenin revealed junctions that were slightly smoother than in controls, whereas Ena/VASP overexpression resulted in a striated appearance of junctions, which is indicative of increased force generation (Figs. 6 B and 7 A). These results show that the shape of cell–cell junctions is altered by Ena/VASP activity, presumably through an increase in actomyosin contraction. The application of tension upon cell junctions by the cytoskeleton is necessary for the formation and maintenance of junctions in a process known as force strengthening. Thus, reduction of actomyosin contraction in the absence of Ena/VASP may interfere with force strengthening, resulting in the observed barrier function defects.
Discussion
Mice lacking Ena/VASP proteins develop edema and hemorrhage, which lead to embryonic lethality. Edema and hemorrhage can both result from disruption in endothelial barrier function, which we show is decreased upon Ena/VASP inactivation in a cell-based barrier function assay. Barrier function depends on the formation and maintenance of cell–cell contacts, which interact with one another and the underlying cytoskeleton. Loss of Ena/VASP activity reduces stress fiber formation, impairs shear stress response, and inhibits actomyosin contraction. Relaxation of the actin cytoskeleton in the absence of Ena/VASP activity may result in weak cell–cell junctions that do not strengthen appropriately, culminating in the barrier function defects observed in mmvvee mice.
Ena/VASP and cardiac function
Edema can arise from several conditions, including late stage heart failure. A previous study has shown that displacement of Ena/VASP by cardiac-specific expression of the VASP-EVH1 domain causes dilated cardiomyopathy, bradycardia, and myocyte hypertrophy (Eigenthaler et al., 2003). Furthermore, the hearts of Mena-null mice develop structural and electrical conductivity abnormalities postnatally (unpublished data). In contrast, hearts from mmvvee embryos appear to form normal Z lines and intercalated disks and are not enlarged, suggesting that a heart defect is not the primary cause of the edema. The discrepancy between our work and other studies may arise because of off-target effects of the dominant-negative systems used to inhibit Ena/VASP function, or the phenotypes may be restricted to postnatal animals. Mena-dependent heart defects that develop later in life may result from failure of cardiac myocytes to adapt to normal postnatal cardiac stresses. The only defect we observed in the mmvvee embryonic heart was a thinning and detachment of the endocardium. This phenotype is strikingly similar to that seen in the β-catenin conditional knockout mouse (Cattelino et al., 2003), suggesting a defect in cell–cell contacts.
Barrier function and cell–cell junctions
Although knockout mice lacking individual members of the Ena/VASP family do not exhibit barrier defects, our work shows that simultaneous knockout of all three Ena/VASP family members results in severe endothelial barrier disruption. These in vivo findings are consistent with earlier studies that suggested a link between VASP and barrier function (Sporbert et al., 1999; Comerford et al., 2002; Lawrence et al., 2002). Several signaling pathways that reduce barrier function result in the phosphorylation of VASP by PKG, raising the possibility that Ena/VASP is an important downstream target in barrier function regulation. VEGF is one agent that activates nitric oxide/PKG signaling and results in VASP phosphorylation. However, Ena/VASP-inactivated cells have an intact response to VEGF, which suggests that, at least in the case of VEGF signaling, phosphorylation of VASP by PKG is not required for barrier function modulation.
A recent study has shown that VASP is down-regulated in response to hypoxic conditions, providing another mechanism for Ena/VASP family members to regulate barrier function (Rosenberger et al., 2007). Targeting of VASP by siRNA reduced barrier function in human microvascular endothelial cells, whereas overexpression of VASP increased basal barrier function and protected cells from hypoxia-induced barrier disruption. The VASP-null mouse, however, does not exhibit signs of barrier disruption such as edema, likely because of the presence of the highly related Mena and Ena/Vasp-like proteins in the mouse vasculature.
Although Ena/VASP has been implicated in the regulation of barrier function, its mechanism of action is poorly understood. It has been proposed that Ena/VASP is essential for proper recruitment of cell junction proteins, potentially explaining its importance for barrier function (Vasioukhin et al., 2000; Lawrence et al., 2002; Kohler et al., 2004). Indeed, genetic ablation of several proteins involved in cell–matrix (FAK) or cell–cell contacts (β-catenin and VE-cadherin) results in edematous hemorrhagic phenotypes that are similar to those in mmvvee mice (Carmeliet et al., 1999; Cattelino et al., 2003; Shen et al., 2005; Braren et al., 2006). Nevertheless, we see no evidence of a failure to recruit junctional proteins in either primary endothelial cells lacking Ena/VASP proteins or Ena/VASP-inactivated HUVEC cultures, suggesting that barrier disruption caused by loss of Ena/VASP does not impair junction formation.
Underlying cause of barrier function defect
Dynamic actin has been implicated in the maintenance of barrier function, particularly peripheral cortical actin, which is proximal to cell junctions. As actin regulators, Ena/VASP proteins have been proposed to affect barrier function through modulation of actin dynamics. Using a G-actin barbed end incorporation assay, we found that loss of Ena/VASP activity leads to only a modest reduction in incorporation at the cell periphery, including regions of cell–cell junctions. This suggests that changes in cortical actin dynamics do not explain the role of Ena/VASP on barrier function. We did find that G-actin incorporation along barbed ends within stress fibers was dramatically reduced in the absence of Ena/VASP activity, whereas cells overexpressing Ena/VASP had increased barbed end labeling and formed large ventral stress fibers. Our results provide a mechanistic explanation for the previous report that VASP overexpression in bovine aortic endothelial cells increased stress fibers (Price and Brindle, 2000). Ena/VASP-induced parallel ventral stress fibers mimic actin cytoskeletal rearrangements that occur in response to shear stress and actomyosin contraction. Indeed, VASP overexpression increased actomyosin contraction, whereas cells lacking Ena/VASP activity were less contractile than control cells. Ena/VASP-deficient cells also failed to reorganize their stress fibers or alter cell morphology in response to shear stress, a defect we confirmed in vivo in the endothelial lining of aorta from mmvvee mice. These findings are consistent with the hypothesis that Ena/VASP may be a downstream component of the zyxin-dependant mechanosensory mechanism (Yoshigi et al., 2005). Endothelial shear stress response increases barrier function (Shikata et al., 2005) and affects signaling pathways, including the activation of integrins (Orr et al., 2006). Thus, although we have documented the importance of Ena/VASP for barrier function under static conditions, impaired response to shear stress may exacerbate the barrier defect in mmvvee embryos. VASP has been shown to become transiently phosphorylated in cells subjected to shear stress before undergoing actin rearrangement (Wei et al., 2003), suggesting that Ena/VASP response to shear stress may be phosphoregulated.
Stress fiber formation is regulated by the small GTPase Rho (Ridley and Hall, 1992). Rho is involved in the regulation of numerous processes, including cell–cell contact function and barrier function maintenance (Nusrat et al., 1995; Braga et al., 1997, 1999; Carbajal and Schaeffer, 1999), actomyosin contractility (van Nieuw Amerongen and van Hinsbergh, 2001), and actin rearrangement in response to shear stress (Li et al., 1999). The overlap of Rho-dependent processes with those affected by Ena/VASP suggests that Ena/VASP and Rho may act in the same signaling pathway. Ena/VASP did not alter Rho activity as measured by Rho pulldown assays (unpublished data), indicating that if Ena/VASP is in a common signaling pathway with Rho, it is probably downstream. Interestingly, other Rho family proteins have been proposed to signal through Ena/VASP, particularly Rac1, which has been shown to lead to a translocation of VASP to cell junctions and to increase barrier function (Waschke et al., 2006). Similar to our findings concerning Rho activation, we detected no effect on Rac1 activity by Ena/VASP (unpublished data).
Ena/VASP and force strengthening
In addition to increasing F-actin levels, Ena/VASP activity may increase actomyosin contractility through up-regulation of MLC. MLC expression is regulated by the transcription factor serum response factor (Papadopoulos and Crow, 1993; Latinkic et al., 2004), which is activated under conditions that reduce G-actin pools, including Ena/VASP overexpression (Sotiropoulos et al., 1999; Grosse et al., 2003). In the absence of Ena/VASP, conditions that would further compromise actomyosin contractility, stress fibers, and F-actin content are reduced.
Actomyosin contractility exerts force on focal adhesions and enhances the recruitment of proteins to the adhesion site in a process known as force strengthening (Geiger and Bershadsky, 2001). Recent work extends the importance of force generation to the formation and functioning of adherens and tight junctions (Miyake et al., 2006). Response to shear stress can also be thought of as a form of force strengthening and has been shown to enhance the strength of cell–cell contacts and thus barrier function. Ena/VASP alters the morphology of cell–cell junctions in a manner consistent with changes in actomyosin contractility. Actomyosin contraction is generally thought to reduce endothelial barrier function by favoring the formation of paracellular pores. However, many permeability enhancing agents not only stimulate actomyosin contraction but also destabilize cell–cell junctions. For example, p21-activated kinase triggers actomyosin contraction by phosphorylating MLC (Stockton et al., 2004) and destabilizes cell–cell junctions through the phosphorylation and internalization of VE-cadherin (Gavard and Gutkind, 2006). Furthermore, activation of MLC has been shown to mediate enhanced endothelial barrier function in response to treatment with sphingosine 1–phosphate (Dudek et al., 2004) and overexpression of an inactive MLC mutant reduces epithelial barrier function (Gandhi et al., 1997). These observations suggest that the relationship between actomyosin contraction and barrier function is complex. We speculate that loss of Ena/VASP results in a chronic relaxation of force exerted on cell contacts by the actin cytoskeleton, leading to a failure of the cell contacts to properly strengthen. Impaired cell contacts may first manifest in the vascular system because this is a tissue that exists under strong physical stress, resulting in the fluid loss and vessel discontinuity observed in the mmvvee mouse. Intriguingly, vessel rupture appeared to be limited to venules. This subset of the vascular bed is responsible for the bulk of transendothelial movement of circulating immune cells and plasma. Postcapillary venules are particularly sensitive to the barrier function antagonists histamine and bradykinin (Bazzoni and Dejana, 2004), a fact potentially explained by the poor organization of tight junctions in this endothelial tissue or the relative lack of supporting cells around the venule endothelium. Similarly, the disorganization of tight junctions or lack of supporting cells may render venules particularly sensitive to a reduction in barrier function caused by the loss of Ena/VASP.
The ability to modulate endothelial barrier function is an important clinical objective because barrier dysfunction underlies the morbidity of numerous disease processes, including inflammation, sepsis, and noncardiogenic pulmonary edema. In these processes, an inflammatory milieu of cytokines acts through several downstream pathways to enhance vascular permeability by disruption of cellular junctions and alteration of the actin cytoskeleton. Another disease state that exhibits abnormally low endothelial barrier function involves pathological vasculature induced by solid tumors. In tumors, barrier dysfunction leads to an increase in interstitial fluid pressure in the tumor microenvironment, which may interfere with the delivery of anticancer drugs to their intended sites. The efficacy of anti-VEGF therapies in combination with chemotherapy has been postulated to result from vascular normalization, including an increase in tumor barrier function, which enhances delivery of chemotherapeutic agents to the tumor. In this work, we demonstrate that Ena/VASP is critical for barrier function in vivo and in vitro. Previous work has identified Ena/VASP as an end effector for several converging signaling pathways. This suggests that Ena/VASP may provide an attractive therapeutic target to modify the morbidity associated with the increased permeability observed in a variety of clinical conditions.
Materials and methods
Mice
The generation of Ena/VASP-null (mmvvee) mice has been previously described (Kwiatkowski et al., 2007). All animal work was approved by the Massachussetts Institute of Technology Committee on Animal Care. Because a single allele of Mena was sufficient to prevent the described vascular defects, breeding pairs were selected to generate MMvvee or Mmvvee littermate controls.
Morphological and histological analysis
Bouin's fixed tissues were embedded in paraffin, sectioned, and stained with hematoxylin and eosin using standard techniques. Sections were imaged with a microscope (Eclipse TE300; Nikon) equipped with a digital camera (Spot Flex; Diagnostic Instruments, Inc.). Whole mount staining was performed using a standard protocol. In brief, E10.5 embryos were fixed in paraformaldehyde, permeabilized in methanol with DMSO, and blocked with hydrogen peroxide. Anti-PECAM1 was used at a 1:20 dilution (BD Biosciences) and visualized using horseradish peroxidase (Vectastain Elite ABC kit; Vector Laboratories). Stained embryos were imaged using a stereoscopic zoom microscope (SMZ-U; Nikon).
Electron microscopy
Tissues were fixed with 2.5% glutaraldehyde and 2.5% formaldehyde in 0.1 M sodium cacodylate–HCl, pH 7.2, for 60–90 min at 4°C, postfixed with 2% osmium tetroxide for 60 min at 4°C, dehydrated in up to 70% graded alcohol, and en bloc stained with 0.2% uranyl acetate in 70% alcohol for 60 min at 4°C. The samples were then processed for epon embedding. Thin epon sections (400 nm) were cut with a diamond knife, poststained with uranyl acetate and Reynold's lead citrate, and viewed with an electron microscope (G2 Spirit BioTWIN; Tecnai) operated at 80 kV. Digital images were taken with a camera (2k CCD; Advanced Microscopy Techniques; provided by the Harvard Medical School Electron Microscopy Facility).
Cell isolation and culture
Primary endothelial cells were isolated from the aorta dissected from E14.5 embryos. Aorta were rinsed in HBSS without calcium and magnesium and treated with 1 mg/ml collagenase (Sigma-Aldrich) in primary endothelial media for 15 min at 37°C. Collagenase was removed with a PBS rinse and aorta were treated with 0.25% trypsin-EDTA (Invitrogen) for 15 min at 37°C. Aorta were then triterated and passed through a 70-μm cell strainer (BD Biosciences), and dissociated cells were plated directly on collagen I–coated coverslips. Primary endothelial cells were cultured at 37°C in RPMI supplemented with 10% fetal calf serum, penicillin/streptomycin, 1 μg/ml hydrocortisone, 10 U/ml heparin, 1 μg/ml DMSO, and 0.1 mg/ml endothelial cell growth supplement (Sigma-Aldrich) and used for experiments within 48 h. HUVECs (Cascade Biologics) were maintained on collagen I– coated dishes in endothelial basal media–2 (Clonetics). Cells expressing EGFP control, EGFP-Ena/VASP proteins, EGFP-FP4-Mito, and EGFP-AP4-Mito constructs were generated using vesicular stomatitis virus G–pseudotyped retroviruses as previously described (Bear et al., 2000; Furman et al., 2002). Infected HUVECs were FACS sorted for equivalent levels of expression and used in cell-based assays between passages five and eight.
Immunofluorescence
Cells were plated onto collagen I–coated coverslips and fixed with warmed 4% paraformaldehye/0.25% gluteraldehyde in Krebs-Ringer solution with 0.4 M sucrose. Cells were blocked in 10% BSA/PBS, extracted with 0.2% Triton X-100 for 3 min, and stained with indicated primary antibodies. Alexa- phalloidin (Invitrogen) was used at 1:50 dilution and Alexa-conjugated secondary antibodies were used at 1:1,000 (Invitrogen).
Anti–VE-cadherin and anti-PECAM (Becton Dickinson), anti-ZO1 (Chemicon), anti–α-catenin (Invitrogen), and anti–β-catenin (Sigma-Aldrich) were used for staining primary endothelial cells. Anti–VE-cadherin antibody (Chemicon) was used for HUVEC cultures. Images were acquired with a microscope (Deltavision [Carl Zeiss, Inc.]; Eclipse TE300 [Nikon]) and deconvolved using processing software (Softworx; Applied Precision) or a digital camera (Orca-ER; Hamamatsu). Descending aorta were dissected from E14.5 embryos, fixed with 4% paraformaldehyde in PBS for 2 h, immunostained with anti-PECAM antibody, and imaged with a microscope (TE2000; Nikon) equipped with a spinning disk confocal head (Yokagawa), a Lambda 10–2 multiposition filter wheel (Sutter Instrument Co.), and a camera (Orca-ER cooled CCD; Hammamatsu). This system was coupled to a multi-band laser (Innova 70C 2.5W; Coherent). Wavelengths were selected with separate excitation and emission filters (Chroma Technology Corp.) in the filter wheels. All hardware was controlled by Metamorph software (Invitrogen). Aorta images are maximum projections of five consecutive 1-μm planes, using a Plan Apo objective (60×/1.40 DIC; Nikon).
Shear stress
HUVECs were seeded at confluence in fibronectin-coated Ibidi μ-slide VI flow through chambers (Integrated BioDiagnostics). 10 dynes/cm2 of laminar shear stress was applied to cells for 16 to 20 h by flowing media through a loop consisting of a media reservoir, peristaltic pump, and compliance chamber. Cells were fixed in warm 4% paraformaldehyde and stained with Alexa-phalloidin. Images were captured with a microscope (Eclipse TE300) using a Plan Fluor objective (10×/0.3; Nikon).
G-actin incorporation assay
HUVECs and primary mouse endothelial cells were plated on collagen-coated coverslips, and the rate of G-actin incorporation at barbed ends was determined using a previously described technique (Symons and Mitchison, 1991). In brief, cells were permeabilized with 0.125 mg/ml saponin in the presence of 0.5 mM Alexa 568–conjugated G-actin (Invitrogen). After a 2-min labeling period, samples were fixed in 0.5% glutaraldehyde, permeabilized with 0.5% Triton X-100, and blocked in the presence of Alexa 350–phalloidin (Invitrogen). Images were acquired with a microscope (Eclipse TE300) using a Plan Flour objective (40×/1.30 DIC; Nikon). Cellular F- and G-actin content was quantitated using Metamorph software.
MLC quantitation
HUVEC cultures were maintained on collagen-coated 10-cm2 plates at confluence for 3 d, lysed in SDS sample buffer, sonicated for 10 s, and subjected to SDS-PAGE. Western blot analysis was performed using antibodies to total and Serine19 phosphorylated MLC 2 (Cell Signaling Technology). Blots were probed with anti-RhoA (Cytoskeleton, Inc.) and anti-actin (Chemicon) antibodies to ensure equivalent loading.
Contraction assay
106 cells/ml of HUVECs were suspended in 2 mg/ml of type I collagen and cast in 200-μl discs as previously described (Sieminski et al., 2004). Projected areas of collagen gel discs were measured immediately after release from molds and after 2 d of culture in endothelial basal media supplemented with 50 ng/ml VEGF (R&D Systems), basic fibroblast growth factor (R&D Systems), and phorbol myristate acetate (Sigma-Aldrich) for 2 d.
Permeability assay
HUVECs were seeded at confluence on polycarbonate transwell membrane inserts (Corning 3402) and cultured for 3 d. 70 kD of Texas red–dextran (Invitrogen) was added to the top chamber at 2 mg/ml, and its movement into the bottom chamber was monitored over 4 h by spectrophotometer.
Statistical analysis
Statistical differences between two conditions were determined using t test. For multiple conditions, means were compared by analysis of variance. All data found to be significant (P < 0.05) by analysis of variance were compared with Tukey's honestly significant difference post hoc test to reveal statistically different groups.
Online supplemental material
Fig. S1 shows the localization of endogenous VASP at cell–cell junctions and stress fibers in HUVEC and primary endothelial cell cultures. Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.200705002/DC1.
Supplemental Material
Acknowledgments
We would like to thank Erik Dent and Rob Makar for assistance in embryo manipulation, microdissection, and substantial intellectual involvement, as well as Roger Kamm for generous support of this work. We also thank Anne Ridley, Jenny McKenzie, and Donald Senger for helpful advice. We thank Maria Ericsson and Elizabeth Benecchi from the Electron Microscopy Facility at Harvard Medical School for their excellent help with the tissue ultramicrotomy. Melanie Barzik, Frauke Drees, and Irina Shapiro provided critical reading of the manuscript and helpful comments.
This work was supported by National Institutes of Health grant GM58801 to F.B. Gertler and funds from the Max Planck Institute to R. Fässler.
C. Furman's present address is Pfizer Research Technology Center, Cambridge, MA 02139.
A.V. Kwiatkowski's present address is Dept. of Biological Sciences, Stanford University, Stanford, CA 94305.
Abbreviations used in this paper: E, embryonic day; Ena/VASP, Enabled/vasodilator-stimulated phosphoprotein; HUVEC, human umbilical vein endothelial cell; Mena, mammalian Ena; MLC, myosin light chain; PECAM, platelet/endothelial cell adhesion molecule; VE-cadherin, vascular endothelial–cadherin; ZO, zonula occludens.
References
- Aberle, H., H. Schwartz, and R. Kemler. 1996. Cadherin-catenin complex: protein interactions and their implications for cadherin function. J. Cell. Biochem. 61:514–523. [DOI] [PubMed] [Google Scholar]
- Adams, C.L., and W.J. Nelson. 1998. Cytomechanics of cadherin-mediated cell-cell adhesion. Curr. Opin. Cell Biol. 10:572–577. [DOI] [PubMed] [Google Scholar]
- Aszodi, A., A. Pfeifer, M. Ahmad, M. Glauner, X.H. Zhou, L. Ny, K.E. Andersson, B. Kehrel, S. Offermanns, and R. Fassler. 1999. The vasodilator-stimulated phosphoprotein (VASP) is involved in cGMP- and cAMP-mediated inhibition of agonist-induced platelet aggregation, but is dispensable for smooth muscle function. EMBO J. 18:37–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barzik, M., T.I. Kotova, H.N. Higgs, L. Hazelwood, D. Hanein, F.B. Gertler, and D.A. Schafer. 2005. Ena/VASP proteins enhance actin polymerization in the presence of barbed end capping proteins. J. Biol. Chem. 280:28653–28662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bazzoni, G., and E. Dejana. 2004. Endothelial cell-to-cell junctions: molecular organization and role in vascular homeostasis. Physiol. Rev. 84:869–901. [DOI] [PubMed] [Google Scholar]
- Bear, J.E., J.J. Loureiro, I. Libova, R. Fassler, J. Wehland, and F.B. Gertler. 2000. Negative regulation of fibroblast motility by Ena/VASP proteins. Cell. 101:717–728. [DOI] [PubMed] [Google Scholar]
- Bear, J.E., T.M. Svitkina, M. Krause, D.A. Schafer, J.J. Loureiro, G.A. Strasser, I.V. Maly, O.Y. Chaga, J.A. Cooper, G.G. Borisy, and F.B. Gertler. 2002. Antagonism between Ena/VASP proteins and actin filament capping regulates fibroblast motility. Cell. 109:509–521. [DOI] [PubMed] [Google Scholar]
- Boukhelifa, M., M.M. Parast, J.E. Bear, F.B. Gertler, and C.A. Otey. 2004. Palladin is a novel binding partner for Ena/VASP family members. Cell Motil. Cytoskeleton. 58:17–29. [DOI] [PubMed] [Google Scholar]
- Braga, V.M., L.M. Machesky, A. Hall, and N.A. Hotchin. 1997. The small GTPases Rho and Rac are required for the establishment of cadherin-dependent cell–cell contacts. J. Cell Biol. 137:1421–1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braga, V.M., A. Del Maschio, L. Machesky, and E. Dejana. 1999. Regulation of cadherin function by Rho and Rac: modulation by junction maturation and cellular context. Mol. Biol. Cell. 10:9–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braren, R., H. Hu, Y.H. Kim, H.E. Beggs, L.F. Reichardt, and R. Wang. 2006. Endothelial FAK is essential for vascular network stability, cell survival, and lamellipodial formation. J. Cell Biol. 172:151–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byers, H.R., G.E. White, and K. Fujiwara. 1984. Organization and function of stress fibers in cells in vitro and in situ. A review. Cell Muscle Motil. 5:83–137. [DOI] [PubMed] [Google Scholar]
- Carbajal, J.M., and R.C. Schaeffer Jr. 1999. RhoA inactivation enhances endothelial barrier function. Am. J. Physiol. 277:C955–C964. [DOI] [PubMed] [Google Scholar]
- Carmeliet, P., M.G. Lampugnani, L. Moons, F. Breviario, V. Compernolle, F. Bono, G. Balconi, R. Spagnuolo, B. Oostuyse, M. Dewerchin, et al. 1999. Targeted deficiency or cytosolic truncation of the VE-cadherin gene in mice impairs VEGF-mediated endothelial survival and angiogenesis. Cell. 98:147–157. [DOI] [PubMed] [Google Scholar]
- Cattelino, A., S. Liebner, R. Gallini, A. Zanetti, G. Balconi, A. Corsi, P. Bianco, H. Wolburg, R. Moore, B. Oreda, et al. 2003. The conditional inactivation of the β-catenin gene in endothelial cells causes a defective vascular pattern and increased vascular fragility. J. Cell Biol. 162:1111–1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comerford, K.M., D.W. Lawrence, K. Synnestvedt, B.P. Levi, and S.P. Colgan. 2002. Role of vasodilator-stimulated phosphoprotein in PKA-induced changes in endothelial junctional permeability. FASEB J. 16:583–585. [DOI] [PubMed] [Google Scholar]
- Corada, M., M. Mariotti, G. Thurston, K. Smith, R. Kunkel, M. Brockhaus, M.G. Lampugnani, I. Martin-Padura, A. Stoppacciaro, L. Ruco, et al. 1999. Vascular endothelial-cadherin is an important determinant of microvascular integrity in vivo. Proc. Natl. Acad. Sci. USA. 96:9815–9820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dejana, E., G. Bazzoni, and M.G. Lampugnani. 1999. Vascular endothelial (VE)-cadherin: only an intercellular glue? Exp. Cell Res. 252:13–19. [DOI] [PubMed] [Google Scholar]
- Drenckhahn, D., and J. Wagner. 1986. Stress fibers in the splenic sinus endothelium in situ: molecular structure, relationship to the extracellular matrix, and contractility. J. Cell Biol. 102:1738–1747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudek, S.M., J.R. Jacobson, E.T. Chiang, K.G. Birukov, P. Wang, X. Zhan, and J.G. Garcia. 2004. Pulmonary endothelial cell barrier enhancement by sphingosine 1-phosphate: roles for cortactin and myosin light chain kinase. J. Biol. Chem. 279:24692–24700. [DOI] [PubMed] [Google Scholar]
- Eigenthaler, M., S. Engelhardt, B. Schinke, A. Kobsar, E. Schmitteckert, S. Gambaryan, C.M. Engelhardt, V. Krenn, M. Eliava, T. Jarchau, et al. 2003. Disruption of cardiac Ena-VASP protein localization in intercalated disks causes dilated cardiomyopathy. Am. J. Physiol. Heart Circ. Physiol. 285:H2471–H2481. [DOI] [PubMed] [Google Scholar]
- Fan, G., Y.P. Jiang, Z. Lu, D.W. Martin, D.J. Kelly, J.M. Zuckerman, L.M. Ballou, I.S. Cohen, and R.Z. Lin. 2005. A transgenic mouse model of heart failure using inducible Galpha q. J. Biol. Chem. 280:40337–40346. [DOI] [PubMed] [Google Scholar]
- Furman, C., S.M. Short, R.R. Subramanian, B.R. Zetter, and T.M. Roberts. 2002. DEF-1/ASAP1 is a GTPase-activating protein (GAP) for ARF1 that enhances cell motility through a GAP-dependent mechanism. J. Biol. Chem. 277:7962–7969. [DOI] [PubMed] [Google Scholar]
- Gandhi, S., D.D. Lorimer, and P. de Lanerolle. 1997. Expression of a mutant myosin light chain that cannot be phosphorylated increases paracellular permeability. Am. J. Physiol. 272:F214–F221. [DOI] [PubMed] [Google Scholar]
- Gates, J., J.P. Mahaffrey, S.L. Rogers, M. Emerson, E.M. Rogers, S.L. Sottile, D. Van Vactor, F.B. Gertler, and M. Peifer. 2007. Enabled plays key roles in embryonic epithelial morphogenesis in Drosophila. Development. 134:2027–2039. [DOI] [PubMed] [Google Scholar]
- Gavard, J., and J.S. Gutkind. 2006. VEGF controls endothelial-cell permeability by promoting the beta-arrestin-dependent endocytosis of VE-cadherin. Nat. Cell Biol. 8:1223–1234. [DOI] [PubMed] [Google Scholar]
- Geiger, B., and A. Bershadsky. 2001. Assembly and mechanosensory function of focal contacts. Curr. Opin. Cell Biol. 13:584–592. [DOI] [PubMed] [Google Scholar]
- Gertler, F.B., K. Niebuhr, M. Reinhard, J. Wehland, and P. Soriano. 1996. Mena, a relative of VASP and Drosophila Enabled, is implicated in the control of microfilament dynamics. Cell. 87:227–239. [DOI] [PubMed] [Google Scholar]
- Gordon, W.E., III. 1978. Immunofluorescent and ultrastructural studies of “sarcomeric” units in stress fibers of cultured non-muscle cells. Exp. Cell Res. 117:253–260. [DOI] [PubMed] [Google Scholar]
- Grosse, R., J.W. Copeland, T.P. Newsome, M. Way, and R. Treisman. 2003. A role for VASP in RhoA-Diaphanous signalling to actin dynamics and SRF activity. EMBO J. 22:3050–3061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haffner, C., T. Jarchau, M. Reinhard, J. Hoppe, S.M. Lohmann, and U. Walter. 1995. Molecular cloning, structural analysis and functional expression of the proline-rich focal adhesion and microfilament-associated protein VASP. EMBO J. 14:19–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halbrugge, M., and U. Walter. 1989. Purification of a vasodilator-regulated phosphoprotein from human platelets. Eur. J. Biochem. 185:41–50. [DOI] [PubMed] [Google Scholar]
- Hauser, W., K.P. Knobeloch, M. Eigenthaler, S. Gambaryan, V. Krenn, J. Geiger, M. Glazova, E. Rohde, I. Horak, U. Walter, and M. Zimmer. 1999. Megakaryocyte hyperplasia and enhanced agonist-induced platelet activation in vasodilator-stimulated phosphoprotein knockout mice. Proc. Natl. Acad. Sci. USA. 96:8120–8125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hordijk, P.L., E. Anthony, F.P. Mul, R. Rientsma, L.C. Oomen, and D. Roos. 1999. Vascular-endothelial-cadherin modulates endothelial monolayer permeability. J. Cell Sci. 112:1915–1923. [DOI] [PubMed] [Google Scholar]
- Kohler, K., D. Louvard, and A. Zahraoui. 2004. Rab13 regulates PKA signaling during tight junction assembly. J. Cell Biol. 165:175–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komatsu, Y., H. Shibuya, N. Takeda, J. Ninomiya-Tsuji, T. Yasui, K. Miyado, T. Sekimoto, N. Ueno, K. Matsumoto, and G. Yamada. 2002. Targeted disruption of the Tab1 gene causes embryonic lethality and defects in cardiovascular and lung morphogenesis. Mech. Dev. 119:239–249. [DOI] [PubMed] [Google Scholar]
- Kreis, T.E., and W. Birchmeier. 1980. Stress fiber sarcomeres of fibroblasts are contractile. Cell. 22:555–561. [DOI] [PubMed] [Google Scholar]
- Kreis, T.E., K.H. Winterhalter, and W. Birchmeier. 1979. In vivo distribution and turnover of fluorescently labeled actin microinjected into human fibroblasts. Proc. Natl. Acad. Sci. USA. 76:3814–3818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwiatkowski, A.V., D.A. Rubinson, E.W. Dent, J.E. van Veen, J.D. Leslie, J. Zhang, L.M. Mebane, U. Philippar, E.M. Pinheiro, A.A. Burds, et al. 2007. Ena/VASP is required for neuritogenesis in the developing cortex. Neuron. In press. [DOI] [PubMed]
- Lambrechts, A., A.V. Kwiatkowski, L.M. Lanier, J.E. Bear, J. Vandekerckhove, C. Ampe, and F.B. Gertler. 2000. cAMP-dependent protein kinase phosphorylation of EVL, a Mena/VASP relative, regulates its interaction with actin and SH3 domains. J. Biol. Chem. 275:36143–36151. [DOI] [PubMed] [Google Scholar]
- Lanier, L.M., M.A. Gates, W. Witke, A.S. Menzies, A.M. Wehman, J.D. Macklis, D. Kwiatkowski, P. Soriano, and F.B. Gertler. 1999. Mena is required for neurulation and commissure formation. Neuron. 22:313–325. [DOI] [PubMed] [Google Scholar]
- Latinkic, B.V., B. Cooper, S. Smith, S. Kotecha, N. Towers, D. Sparrow, and T.J. Mohun. 2004. Transcriptional regulation of the cardiac-specific MLC2 gene during Xenopus embryonic development. Development. 131:669–679. [DOI] [PubMed] [Google Scholar]
- Lawrence, D.W., K.M. Comerford, and S.P. Colgan. 2002. Role of VASP in reestablishment of epithelial tight junction assembly after Ca2+ switch. Am. J. Physiol. Cell Physiol. 282:C1235–C1245. [DOI] [PubMed] [Google Scholar]
- Lazarides, E., and K. Burridge. 1975. Alpha-actinin: immunofluorescent localization of a muscle structural protein in nonmuscle cells. Cell. 6:289–298. [DOI] [PubMed] [Google Scholar]
- Lebrand, C., E.W. Dent, G.A. Strasser, L.M. Lanier, M. Krause, T.M. Svitkina, G.G. Borisy, and F.B. Gertler. 2004. Critical role of Ena/VASP proteins for filopodia formation in neurons and in function downstream of netrin-1. Neuron. 42:37–49. [DOI] [PubMed] [Google Scholar]
- Li, B., J.R. Dedman, and M.A. Kaetzel. 2006. Nuclear Ca2+/calmodulin-dependent protein kinase II in the murine heart. Biochim. Biophys. Acta. 1763:1275–1281. [DOI] [PubMed] [Google Scholar]
- Li, S., B.P. Chen, N. Azuma, Y.L. Hu, S.Z. Wu, B.E. Sumpio, J.Y. Shyy, and S. Chien. 1999. Distinct roles for the small GTPases Cdc42 and Rho in endothelial responses to shear stress. J. Clin. Invest. 103:1141–1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin-Padura, I., S. Lostaglio, M. Schneemann, L. Williams, M. Romano, P. Fruscella, C. Panzeri, A. Stoppacciaro, L. Ruco, A. Villa, et al. 1998. Junctional adhesion molecule, a novel member of the immunoglobulin superfamily that distributes at intercellular junctions and modulates monocyte transmigration. J. Cell Biol. 142:117–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitic, L.L., and J.M. Anderson. 1998. Molecular architecture of tight junctions. Annu. Rev. Physiol. 60:121–142. [DOI] [PubMed] [Google Scholar]
- Miyake, Y., N. Inoue, K. Nishimura, N. Kinoshita, H. Hosoya, and S. Yonemura. 2006. Actomyosin tension is required for correct recruitment of adherens junction components and zonula occludens formation. Exp. Cell Res. 312:1637–1650. [DOI] [PubMed] [Google Scholar]
- Nusrat, A., M. Giry, J.R. Turner, S.P. Colgan, C.A. Parkos, D. Carnes, E. Lemichez, P. Boquet, and J.L. Madara. 1995. Rho protein regulates tight junctions and perijunctional actin organization in polarized epithelia. Proc. Natl. Acad. Sci. USA. 92:10629–10633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orr, A.W., M.H. Ginsberg, S.J. Shattil, H. Deckmyn, and M.A. Schwartz. 2006. Matrix-specific suppression of integrin activation in shear stress signaling. Mol. Biol. Cell. 17:4686–4697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papadopoulos, N., and M.T. Crow. 1993. Transcriptional control of the chicken cardiac myosin light-chain gene is mediated by two AT-rich cis-acting DNA elements and binding of serum response factor. Mol. Cell. Biol. 13:6907–6918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pokutta, S., and W.I. Weis. 2002. The cytoplasmic face of cell contact sites. Curr. Opin. Struct. Biol. 12:255–262. [DOI] [PubMed] [Google Scholar]
- Price, C.J., and N.P. Brindle. 2000. Vasodilator-stimulated phosphoprotein is involved in stress-fiber and membrane ruffle formation in endothelial cells. Arterioscler. Thromb. Vasc. Biol. 20:2051–2056. [DOI] [PubMed] [Google Scholar]
- Reinhard, M., M. Halbrugge, U. Scheer, C. Wiegand, B.M. Jockusch, and U. Walter. 1992. The 46/50 kDa phosphoprotein VASP purified from human platelets is a novel protein associated with actin filaments and focal contacts. EMBO J. 11:2063–2070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridley, A.J., and A. Hall. 1992. The small GTP-binding protein rho regulates the assembly of focal adhesions and actin stress fibers in response to growth factors. Cell. 70:389–399. [DOI] [PubMed] [Google Scholar]
- Rosenberger, P., J. Khoury, T. Kong, T. Weissmuller, A.M. Robinson, and S.P. Colgan. 2007. Identification of vasodilator-stimulated phosphoprotein (VASP) as an HIF-regulated tissue permeability factor during hypoxia. FASEB J. 21:2613–2621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rotrosen, D., and J.I. Gallin. 1986. Histamine type I receptor occupancy increases endothelial cytosolic calcium, reduces F-actin, and promotes albumin diffusion across cultured endothelial monolayers. J. Cell Biol. 103:2379–2387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rottner, K., B. Behrendt, J.V. Small, and J. Wehland. 1999. VASP dynamics during lamellipodia protrusion. Nat. Cell Biol. 1:321–322. [DOI] [PubMed] [Google Scholar]
- Scott, J.A., A.M. Shewan, N.R. den Elzen, J.J. Loureiro, F.B. Gertler, and A.S. Yap. 2006. Ena/VASP proteins can regulate distinct modes of actin organization at cadherin-adhesive contacts. Mol. Biol. Cell. 17:1085–1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seebach, J., P. Dieterich, F. Luo, H. Schillers, D. Vestweber, H. Oberleithner, H.J. Galla, and H.J. Schnittler. 2000. Endothelial barrier function under laminar fluid shear stress. Lab. Invest. 80:1819–1831. [DOI] [PubMed] [Google Scholar]
- Shen, T.L., A.Y. Park, A. Alcaraz, X. Peng, I. Jang, P. Koni, R.A. Flavell, H. Gu, and J.L. Guan. 2005. Conditional knockout of focal adhesion kinase in endothelial cells reveals its role in angiogenesis and vascular development in late embryogenesis. J. Cell Biol. 169:941–952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shikata, Y., A. Rios, K. Kawkitinarong, N. DePaola, J.G. Garcia, and K.G. Birukov. 2005. Differential effects of shear stress and cyclic stretch on focal adhesion remodeling, site-specific FAK phosphorylation, and small GTPases in human lung endothelial cells. Exp. Cell Res. 304:40–49. [DOI] [PubMed] [Google Scholar]
- Sieminski, A.L., R.P. Hebbel, and K.J. Gooch. 2004. The relative magnitudes of endothelial force generation and matrix stiffness modulate capillary morphogenesis in vitro. Exp. Cell Res. 297:574–584. [DOI] [PubMed] [Google Scholar]
- Sobieszek, A. 1977. Ca-linked phosphorylation of a light chain of vertebrate smooth-muscle myosin. Eur. J. Biochem. 73:477–483. [DOI] [PubMed] [Google Scholar]
- Sotiropoulos, A., D. Gineitis, J. Copeland, and R. Treisman. 1999. Signal-regulated activation of serum response factor is mediated by changes in actin dynamics. Cell. 98:159–169. [DOI] [PubMed] [Google Scholar]
- Sporbert, A., K. Mertsch, A. Smolenski, R.F. Haseloff, G. Schonfelder, M. Paul, P. Ruth, U. Walter, and I.E. Blasig. 1999. Phosphorylation of vasodilator-stimulated phosphoprotein: a consequence of nitric oxide- and cGMP-mediated signal transduction in brain capillary endothelial cells and astrocytes. Brain Res. Mol. Brain Res. 67:258–266. [DOI] [PubMed] [Google Scholar]
- Stockton, R.A., E. Schaefer, and M.A. Schwartz. 2004. p21-activated kinase regulates endothelial permeability through modulation of contractility. J. Biol. Chem. 279:46621–46630. [DOI] [PubMed] [Google Scholar]
- Svitkina, T.M., I.G. Surguchova, A.B. Verkhovsky, V.I. Gelfand, M. Moeremans, and J. De Mey. 1989. Direct visualization of bipolar myosin filaments in stress fibers of cultured fibroblasts. Cell Motil. Cytoskeleton. 12:150–156. [DOI] [PubMed] [Google Scholar]
- Symons, M.H., and T.J. Mitchison. 1991. Control of actin polymerization in live and permeabilized fibroblasts. J. Cell Biol. 114:503–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanoue, T., and M. Takeichi. 2004. Mammalian Fat1 cadherin regulates actin dynamics and cell–cell contact. J. Cell Biol. 165:517–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyofuku, T., H. Zhang, A. Kumanogoh, N. Takegahara, M. Yabuki, K. Harada, M. Hori, and H. Kikutani. 2004. Guidance of myocardial patterning in cardiac development by Sema6D reverse signalling. Nat. Cell Biol. 6:1204–1211. [DOI] [PubMed] [Google Scholar]
- van Nieuw Amerongen, G.P., and V.W. van Hinsbergh. 2001. Cytoskeletal effects of rho-like small guanine nucleotide-binding proteins in the vascular system. Arterioscler. Thromb. Vasc. Biol. 21:300–311. [DOI] [PubMed] [Google Scholar]
- Vasioukhin, V., C. Bauer, M. Yin, and E. Fuchs. 2000. Directed actin polymerization is the driving force for epithelial cell-cell adhesion. Cell. 100:209–219. [DOI] [PubMed] [Google Scholar]
- Waldmann, R., M. Nieberding, and U. Walter. 1987. Vasodilator-stimulated protein phosphorylation in platelets is mediated by cAMP- and cGMP- dependent protein kinases. Eur. J. Biochem. 167:441–448. [DOI] [PubMed] [Google Scholar]
- Waschke, J., S. Burger, F.R. Curry, D. Drenckhahn, and R.H. Adamson. 2006. Activation of Rac-1 and Cdc42 stabilizes the microvascular endothelial barrier. Histochem. Cell Biol. 125:397–406. [DOI] [PubMed] [Google Scholar]
- Wei, L., S. Muller, J. Ouyang, J.F. Stoltz, and X. Wang. 2003. Changes of vasodilator-stimulated phosphoprotein (VASP) and its phosphorylation in endothelial cells exposed to laminar flow. Clin. Hemorheol. Microcirc. 28:113–120. [PubMed] [Google Scholar]
- Wu, M.H. 2005. Endothelial focal adhesions and barrier function. J. Physiol. 569:359–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wysolmerski, R., and D. Lagunoff. 1985. The effect of ethchlorvynol on cultured endothelial cells. A model for the study of the mechanism of increased vascular permeability. Am. J. Pathol. 119:505–512. [PMC free article] [PubMed] [Google Scholar]
- Wysolmerski, R., D. Lagunoff, and T. Dahms. 1984. Ethchlorvynol-induced pulmonary edema in rats. An ultrastructural study. Am. J. Pathol. 115:447–457. [PMC free article] [PubMed] [Google Scholar]
- Yonemura, S., M. Itoh, A. Nagafuchi, and S. Tsukita. 1995. Cell-to-cell adherens junction formation and actin filament organization: similarities and differences between non-polarized fibroblasts and polarized epithelial cells. J. Cell Sci. 108:127–142. [DOI] [PubMed] [Google Scholar]
- Yoshigi, M., L.M. Hoffman, C.C. Jensen, H.J. Yost, and M.C. Beckerle. 2005. Mechanical force mobilizes zyxin from focal adhesions to actin filaments and regulates cytoskeletal reinforcement. J. Cell Biol. 171:209–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}