

Abstract
The cII gene product of bacteriophage λ is unstable and required for the establishment of lysogenization. Its intracellular amount is important for the decision between lytic growth and lysogenization. Two genetic loci of Escherichia coli are crucial for these commitments of infecting λ genome. One of them, hflA encodes the HflKC membrane protein complex, which has been believed to be a protease degrading the cII protein. However, both its absence and overproduction stabilized cII in vivo and the proposed serine protease-like sequence motif in HflC was dispensable for the lysogenization control. Moreover, the HflKC protein was found to reside on the periplasmic side of the plasma membrane. In contrast, the other host gene, ftsH (hflB) encoding an integral membrane ATPase/protease, is positively required for degradation of cII, since loss of its function stabilized cII and its overexpression accelerated the cII degradation. In vitro, purified FtsH catalyzed ATP-dependent proteolysis of cII and HflKC antagonized the FtsH action. These results, together with our previous finding that FtsH and HflKC form a complex, suggest that FtsH is the cII degrading protease and HflKC is a modulator of the FtsH function. We propose that this transmembrane modulation differentiates the FtsH actions to different substrate proteins such as the membrane-bound SecY protein and the cytosolic cII protein. This study necessitates a revision of the prevailing view about the host control over λ lysogenic decision.
Keywords: proteolytic control, membrane protein, AAA ATPase
Upon infection of λ phage to the Escherichia coli cell, the λ genome undergoes either vegetative replication (lytic growth) or integration into the host chromosome as a prophage (lysogenization). The cII gene of λ encodes a transcription regulator (cII) that activates transcription of the genes for lysogeny establishment (1, 2). Intracellular concentration of cII, the critical determinant for lysogenization, is controlled not only at the level of transcription, but at the level of protein stability. The cII protein is unstable in vivo and its stability is believed to be governed by two host genes that can mutate to give a high frequency lysogenization (Hfl) phenotype. One of them is hflA, which consists of two open reading frames, hflK and hflC. These genes, together with promoter-proximally located hflX of unknown function, constitute the hflA operon (3). HflK and HflC, membrane proteins with a single transmembrane segment, form a complex (HflKC) (4–6). Because hflA mutations stabilized cII (7, 8) and a purified HflKC preparation exhibited a protease activity against cII (5), it has generally been believed that HflKC is a cII-degrading protease (9). In accordance with this notion, it was claimed that HflC contains a sequence motif similar to that found in the active site region of the ClpP protease (3).
Another hfl locus is hflB or ftsH (8, 10). The FtsH protein is a plasma membrane protein having two transmembrane segments at its N terminus and a large cytoplasmic domain, which includes a conserved ≈200 amino acid module characteristic of the members of the AAA family of ATPases (11) as well as an HEXXH motif characteristic of the zinc-binding site of metalloproteases (12). We found that a loss of ftsH function altered membrane topology of a SecY–PhoA fusion protein and suggested that FtsH might have a chaperone-like activity (13–15). On the other hand, some unstable proteins have been shown to be stabilized in the ftsH mutants (8, 12, 16–18). In vitro experiments directly showed that FtsH has an ATP-dependent proteolytic activity against the σ32 protein (12) and the SecY protein (19).
Although it was reported that FtsH and HflKC participate in independent pathways of degradation of the cII protein (8, 10), we found that FtsH and HflKC form a complex (20). Moreover, HflKC was shown to be inhibitory against the SecY-degrading function of FtsH in vivo and in vitro (20). The close relationship between HflKC and FtsH, as well as the apparent discrepancy (enhancement vs. inhibition of proteolysis) in the function of the HflKC protein, prompted us to reinvestigate the roles played by these proteins in degradation of cII protein and lysogenization control of bacteriophage λ. We now show several lines of evidence indicating that FtsH is the primary protease that degrades cII, whereas HflKC somehow modulates the proteolysis from the periplasmic side of the plasma membrane.
MATERIALS AND METHODS
Bacterial Strains.
E. coli K-12 strains AD202 (Δlac araD thiA rpsL relA ompT::kan; ref. 21), AD16 (Δpro-lac thi/F′ lacIq ZM15 Y+ pro+; ref. 17), AK990 (AD16, ΔhflK-hflC::kan; ref. 20), AK525 (AD16, zgj-460::Tn5 zgj-525::IS1A; ref. 17), AK863 (hflC9; ref. 20), AK865 (hflK13; ref. 20), and Y1089 (hflA150; ref. 21) were described previously. AK1301 (AD16, ΔhflK-hflC::tet zgj-460::Tn5 zgj-525::IS1A) was a zgj-460::Tn5 zgj-525::IS1A transductant of AK1129 (20). AK1339 (AD16, zjf-803::Tn5 hflA150) was constructed using zjf-803::Tn5 (20) as a selective marker in transduction. AK1127 (AD16, hflC::tet) was constructed by linear DNA (the 7.2-kb AvaI fragment pKH215) transformation of a recD strain (FS1576; ref. 22), followed by P1 transduction of the hflC::tet marker into AD16.
Media.
L medium (23), TB medium (24), and M9 medium (25) were used. Ampicillin (50 μg/ml) and/or chloramphenicol (20 μg/ml) were included for growing plasmid-bearing strains.
Plasmids.
Plasmid ptac-cIIY42, provided by C. Herman (Institut Jacques Monod), was a pBR322 derivative carrying cIIcy42 (λ cII gene with its internal promoter pE inactivated; ref. 26) under the tac promoter control. pKH146 (hflC+) and pKH178 (hflK+-hflC+) were described previously (20). pKH274 (carrying cII under the ara promoter control) was constructed by cloning a 0.5-kb EcoRI–HindIII fragment of ptac-cIIY42 into pBAD18 (27). pKH215 (hflX-hflK-hflC::tet) was constructed by cloning a 1.5-kb blunt-ended XbaI–AvaI tet fragment of pACYC184 into the blunt-ended SalI site of pKH169 (20).
pKH339 carried hflCΔ165–200, in which an internal segment of 108 nucleotides (for amino acid residues 165–200 of HflC) had been deleted from hflC. The 1.3-kb HpaI–EcoRI hflC fragment from pHflA100 (5) was cloned into SmaI–EcoRI-digested M13mp18, and the internal deletion was introduced (28) using a mutagenic primer (5′-GAACTCCGGTTCTGCGGGTACAGAAGATCTGGGTATTGAAGTTGTCGATGTGCG-3′). The mutated hflC gene was transferred to pMW119 (a pSC101-based lac promoter vector obtained from Nippon Gene, Toyama, Japan) after HindIII–EcoRI digestions. The mutation was confirmed by sequencing.
Preparation of Radiolabeled cII Protein.
Cells of AK1301/ptac-cIIY42 were grown at 37°C to an early-logarithmic phase in 20 ml of M9 medium supplemented with 18 amino acids (20 μg/ml each, other than Met and Cys), 2 μg/ml thiamine, 0.4% glycerol, and 50 μg/ml ampicillin. The synthesis of cII was induced with 1 mM isopropyl β-d-thiogalactopyranoside (IPTG) for 10 min and cells were labeled with 3.7 MBq of [35S]methionine (>29.6 TBq/mmol; American Radiolabeled Chemicals, St. Louis) for 1 min. The culture was then chilled and mixed with 200 μl of 2% NaN3 and 800 μl of 2.5 mg/ml chloramphenicol. Cells were collected by centrifugation and suspended in 640 μl of buffer I [50 mM Tris⋅HCl (pH 8.1) containing 2 mM EDTA, 5% glycerol, and 1 mM dithiothreitol (DTT)], supplemented with 1 mM Pefabloc (Boehringer Mannheim), and incubated on ice with lysozyme (200 μg/ml) for 30 min. Cells were disrupted by sonication and aggregates of the cII protein were collected by centrifugation at 5 × 105 × g for 45 min and dissolved in 400 μl of buffer I supplemented with 0.05% deoxycholate, 1 M NaCl, and 0.1 M MgCl2 with incubation at 4°C for 1 hr (5, 29). After centrifugation (as above), supernatant was subjected to buffer exchange using NAP-10 column (Pharmacia) equilibrated with 50 mM Tris⋅HCl (pH 7.2) containing 10% glycerol, 5 mM MgCl2, 30 mM KCl, and 1 mM DTT. This preparation gave a single radioactive band (apparent molecular mass 11 kDa) upon SDS/PAGE. Identity of this protein with cII was shown by its tac promoter-specific and plasmid-specific appearance.
In Vitro cII Degradation Assay.
The radioactive cII preparation (1–1.5 × 104 cpm) was incubated with FtsH-His6-Myc (20) in a standard reaction mixture containing 50 mM Tris⋅HCl (pH 7.2), 10% glycerol, 0.1% Nonidet P-40, 5 mM MgCl2, 5 mM ATP, 25 mM KCl, 1 mM DTT, 25 μM zinc acetate, and 0.5 mg/ml bovine serum albumin. Preincubation of FtsH-His6-Myc with HflKC was done as described (20). The radioactivities of the cII protein, separated by SDS/PAGE, were determined using Fujix bioimaging analyzer BAS2000 (Fuji).
Examination of in Vivo Stability of cII.
Stability of cloned and overproduced cII was examined by growing cells harboring pKH274 (para-cII) to a logarithmic phase in M9 medium supplemented with 18 amino acids (20 μg/ml, other than Met and Cys), 2 μg/ml thiamine, 0.4% glycerol, and 50 μg/ml ampicillin, inducing cII with 0.4% arabinose for 10 min, and pulse-labeling the cells with [35S]methionine for 0.5 min and chasing with unlabeled l-methionine (200 μg/ml) for indicated periods. SDS/PAGE (15% gel; ref. 30) of total cell proteins, directly precipitated with 10% trichloroacetic acid, gave a well-separated band of cII. Stability of cII in λ-infected cells was determined by pulse-chase experiments as described above, using UV-irradiated and λ+-infected cells (multiplicity of infection, about 5) as described (31). The band of cII under the latter conditions was minor but distinct. In both cases, intensities of cII were quantitated by using Fujix BAS2000 bioimaging analyzer.
Hfl Phenotype Tests.
Lysogenization frequency of λ was measured essentially as described (8, 10). Briefly, cells were infected with λ+ at a multiplicity of infection of about 0.1 and infectious centers were determined by plating with himA indicator cells, whereas lysogens were determined by plating with an excess of λcI60 phage. The frequency of lysogenization was the ratio of lysogens to infective centers. For qualitative tests of Hfl phenotype, propagation of λc17 phage was examined as described (8, 10). For these tests, cells had been pre-grown in the following media: TB medium containing 0.4% maltose for cells without plasmid; TB medium containing 1 mM IPTG and 50 μg/ml ampicillin for cells harboring pMW119, pKH146, or pKH339; TB medium containing ampicillin with 0.4% arabinose added 1 hr before λ infection for cells harboring pBAD18 or pKH178. After the establishment of lysis–lysogeny commitment, TB agar was used for plating all the samples.
Determination of the Membrane Orientation of HflKC.
Spheroplasts and inverted plasma membrane vesicles were prepared from AD202 as described (32). They were incubated with 1 mg/ml of proteinase K at 0°C for 2 hr, followed by termination of the digestion with phenylmethylsulfonyl fluoride (final concentration, 1 mM) and precipitation of proteins with trichloroacetic acid. Samples were then subjected to SDS/PAGE and immunoblotting. Antisera against the purified HflKC complex and against the HflK subunit (its purification will be described elsewhere) were raised in rabbits by standard procedures. Anti-HflKC serum was affinity purified. Anti-GroEL serum was described previously (13).
RESULTS
In Vitro Proteolytic Activities of FtsH and HflKC Against the λ cII Protein.
Our previous studies showed that FtsH has a protease activity against the SecY protein, whereas HflKC inhibits FtsH (20). We examined whether these properties of FtsH and HflKC can be extended to the proteolysis of λ cII protein in vitro. Partially purified and radiolabeled cII protein was incubated with the purified preparation of FtsH-His6-Myc (19, 20). It was degraded in the presence of ATP (Fig. 1A, lanes 2 and 3), but not in its absence (Fig. 1A, lanes 8 and 9) or in the presence of a nonhydrolyzable analog (ATPγS; Fig. 1A, lanes 5 and 6). Thus, FtsH proteolyzes cII in an ATP hydrolysis-dependent manner. The reaction occurred slightly faster at 37°C than at 30°C (Fig. 1B). No appreciable degradation of cII occurred without FtsH-His6-Myc (data not shown). The cII-degrading activity of FtsH, like its σ32-degrading activity (12), was inhibited by EDTA but not by phenylmethylsulfonyl fluoride (data not shown). Recently, Y. Shotland, S. Koby, D. Teff, N. Mansur, D. A. Oren, K. Tatematsu, T. Tomoyasu, M. Kessel, B. Bukau, T. Ogura, and A. B. Oppenheim (personal communication) reached the same conclusion that FtsH degrades cII in vitro. Our preparation of purified HflKC protein (20) did not significantly degrade cII under several different buffer conditions (data not shown). When FtsH was preincubated with HflKC, the cII degradation activity of FtsH was abolished (Fig. 1C). During this preincubation, degradation of neither FtsH-His6-Myc nor HflKC occurred (data not shown). These results raise a serious question about the validity of the proposal that HflKC is a protease acting against cII (5, 7).
Figure 1.
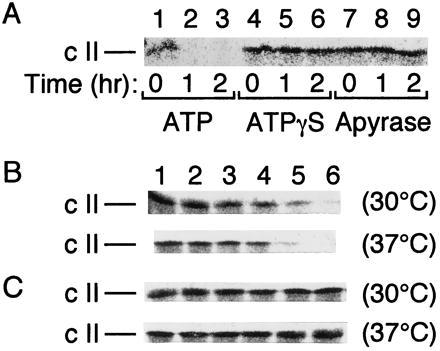
FtsH degrades cII and HflKC antagonizes the degradation in vitro. (A) Purified FtsH-His6-Myc (5 μg) and [35S]methionine-labeled cII (about 15,000 cpm) were incubated at 37°C in the presence of 5 mM ATP (lanes 1–3), 5 mM ATPγS (lanes 4–6), or 50 units/ml apyrase (lanes 7–9) for 0 (lanes 1, 4, and 7), 1 (lanes 2, 5, and 8), and 2 (lanes 3, 6, and 9) hr. (B and C) Purified FtsH-His6-Myc (0.8 μg) alone (B) or FtsH-His6-Myc (0.8 μg) and HflKC (3.2 μg) (C) were preincubated at 0°C for 1 hr. [35S]Methionine-labeled cII (about 10,000 cpm) was then added to each sample and incubated for 0 (lane 1), 5 (lane 2), 10 (lane 3), 20 (lane 4), 40 (lane 5), and 80 (lane 6) min at 30°C or at 37°C, as indicated at right. After SDS/PAGE, cII was visualized by autoradiography.
In Vivo Roles of FtsH and HflKC in Degradation of λ cII Protein.
Hoyt et al. (7) as well as Banuett et al. (8) showed that λ cII protein that was expressed from the cloned cII gene (under the control of the λ pL promoter) was stabilized in the hflA150, the hflA1, and the hflB29 mutants. Similar experiments using a plasmid with cII cloned under the ara promoter control showed that cII was markedly stabilized in the zgj-525::IS1A mutant (17) in which the expression level of FtsH was decreased (Fig. 2A, solid triangles). In contrast, cII remained unstable in the strain deleted for the hflK-hflC segment of the chromosome (open diamonds). The ΔhflK-hflC mutation did not further affect the cII stability when combined with the zgj-525::IS1A mutation (compare open with solid triangles). Thus, the absence of HflKC did not affect degradation of cII. In our experiment, the hflA150 mutation did not stabilize cII either (Fig. 2A, open circles). The hflA150 Mu phage insertion (7) disrupts the hflC open reading frame at the Val-234 codon (unpublished results). A. Oppenheim, Y. Shotland, S. Koby, and M. Gottesman (personal communication) also observed that cII degradation was unaffected by the hflA class of Hfl mutations.
Figure 2.
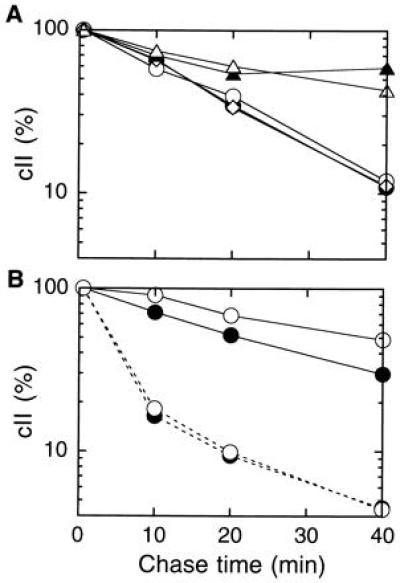
Stability of cII overexpressed from the cloned gene. Cells of AD16 (hflK+ hflC+; •), AK1339 (hflA150; ○), AK990 (ΔhflK-hflC; ⋄), AK525 (zgj-525::IS1A; ▴), and AK1301 (ΔhflK-hflC zgj-525::IS1A; ▵) were transformed with pKH274 (para-cII), induced for the ara transcription, and pulse-labeled with [35S]methionine for 0.5 min followed by chase for 0.5, 10, 20, and 40 min. After SDS/PAGE separation, radioactivities associated with cII were quantitated. Values relative to that at 0.5-min chase point are shown for each curve. (A) Cells were grown at 37°C throughout. (B) Cells were grown first at 30°C, shifted to 43°C for 4 min, pulse-labeled at this temperature, and chased at 30°C (solid lines) or at 43°C (broken lines).
We grew cells at 37°C in all the above experiments, but the previous experiments used the temperature shift conditions (7, 8). We indeed observed a slight stabilization of cII in the hflA150 mutant at 30°C or after a shift of 30°C → 43°C → 30°C (Fig. 2B, compare open and closed circles with the solid line). At 43°C, there was no hflA150 effect (Fig. 2B, broken lines). These results are difficult to interpret in terms of the proteolytic function of HflKC. In contrast, stabilization of cII by the ftsH-lowering zgj-525::IS1A mutation was much clearer and observed at any temperature conditions examined.
To examine the roles of FtsH and HflKC in the stability control of cII under more physiological conditions, UV-irradiated cells were infected with λ and cII was pulse-labeled and chased. Under these conditions, cII in wild-type cells was degraded with a half life of about 1.5 min at 37°C (Fig. 3A, solid circles). It was stabilized to half lives of about 5 min both in the zgj-525::IS1A (solid triangles) and in the ΔhflK-hflC cells (solid squares). At 30°C, where the zgj-525::IS1A mutant had severer cellular defects (17), cII in this mutant was nearly completely stabilized (Fig. 3B, solid triangles). Stabilization of cII in λ-infected hflA mutant cells was also reported previously (7). Curiously, however, when HflKC was overproduced in wild-type cells, cII was stabilized to a similar extent as observed in the ΔhflK-hflC strain (Fig. 3A, open diamonds). When an FtsH overproducer was infected with λ, cII was hardly detected even at the 0.5-min chase point, due presumably to an accelerated degradation. These results are fully consistent with FtsH being the cII degrading protease. However, the HflA effects were more complicated, since both its absence and overproduction gave similar cII-stabilizing effects.
Figure 3.
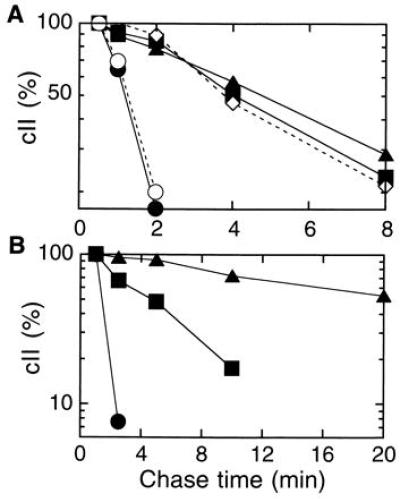
Stability of cII in λ-infected cells. Cells of AD16 (wild-type; •), AK990 (ΔhflK-hflC; ▪), AK525 (zgj-525::IS1A; ▴), AD16/pBAD18 (vector; ○), and AD16/pKH178 (para-hflKC; ◊) were grown at 37°C (A) or at 30°C (B) to a mid-logarithmic phase. To the latter two strains, 0.4% arabinose was added 1 hr before UV irradiation. Cells were UV-irradiated and infected with λ. After 5 min (37°C) or 8 min (30°C), cells were pulse-labeled for 1 min with [35S]methionine and chased as indicated. Total labeled proteins were precipitated by trichloroacetic acid and separated by SDS/PAGE. Radioactivities associated with the cII band were quantitated, and values relative to those without chase are presented.
Paradoxical Effects of hflKC Mutations and HflKC Overexpression on the Lysogenic Decision.
We measured λ lysogenization frequencies using different host strains. As reported previously by Herman et al. (10), the ΔhflK-hflC mutation increased the lysogenization frequency from a few percent in wild type to about 40% (Table 1, experiment 2). The zgj-525::IS1A mutation (pre-grown at 30°C) with a decreased FtsH content did so much more pronouncedly (about 80% lysogenization; Table 1, experiment 5). Interestingly, the HflKC-overproducing plasmid, in contrast to the empty vector, also increased the lysogenization to a similar extent as the hflK-hflC deletion (Table 1, experiment 7). These results agree well with the cII stability data obtained in the λ-infected cells. The hflC9 and the hflK13 mutations, which had been identified as SecY-stabilizing and partially dominant mutations (20), did not significantly increase the λ lysogenization frequency (Table 1, experiments 3 and 4). They did not stabilize the overproduced cII protein (data not shown). All the results of lysogenization were confirmed by λc17 growth tests (Table 1; refs. 8 and 10). We constructed an hflX deletion mutant and found that it did not give any Hfl phenotype (A.K., unpublished results).
Table 1.
Effect of hfl mutations on lysogenization of λ
| Exp. | Strain | Relevant genotype | Plasmid carried | Lysogenization frequency, % | λc17 propagation† |
|---|---|---|---|---|---|
| 1 | AD16 | WT | — | 2.4 | + |
| 2 | AK990 | ΔhflKC | — | 40.6 | − |
| 3 | AK863 | hflC9 | — | 1.7 | + |
| 4 | AK865 | hflK13 | — | 1.5 | + |
| 5 | AK525 | zgj-525::IS1A | — | 81.9 | − |
| 6 | AD16 | WT | pBAD18 (vector) | 3.0 | + |
| 7 | AD16 | WT | pKH178 (hflK+C+) | 42.8 | − |
| 8 | AK1127 | hflC::tet | pMW119 (vector) | 31.3 | − |
| 9 | AK1127 | hflC::tet | pKH146 (hflC+) | 4.5 | + |
| 10 | AK1127 | hflC::tet | pKH339 (hflCΔ165–200) | 6.7 | + |
Cells were pregrown either at 30°C (for experiments 1–5) or at 37°C (for experiments 6–10), but lysogenization frequencies were all measured at 37°C. See Materials and Methods for further details about the media and other growth conditions before infection.
The Proposed Serine Protease Active-Site Region Can Be Deleted from HflC Without Major Dysfunction.
It was noted that E. coli HflC has a ClpP-like sequence motif and that the Ser-197 residue could serve as a serine protease active site (3). Recently, genes for homologs of HflK and HflC were discovered in Haemophilus influenzae (33). Although the HflC homolog in H. influenzae shows high overall sequence similarity to the E. coli counterpart (58% identity and 82% similarity), it lacks a 36 residue segment that overlaps the ClpP-like domain. We deleted this 36 amino acid segment (Glu-165 to Ala-200) from HflC by constructing the hflCΔ165–200 mutation on a plasmid. This plasmid was able to lower the λ lysogenization frequency observed in the hflC::tet strain from 31% (with the empty vector) to 6.7% (Table 1, experiment 10). The hflC+ plasmid gave a lysogenization frequency of 4.5%. Thus, HflC with the Δ165–200 internal deletion retains the ability to maintain λ losogenization frequency to a low level. The ClpP-like domain in HflC is dispensable for its function as a lysogenization controller.
The Major Domains of HflK and HflC Are Periplasmically Oriented.
Both the HflK protein and the HflC protein contain, at their N-terminal regions, a hydrophobic stretch that can traverse the membrane. However, their orientations have not been determined experimentally. The protease hypothesis implicitly assumes that the C-terminally located main hydrophilic domains of these proteins are located in the cytoplasmic side, since cII is a cytosolic protein. We directly examined orientations of the HflK and the HflC proteins by protease digestion experiments. When intact spheroplasts were treated with proteinase K, both HflK (45.5 kDa) and HflC (37.6 kDa) disappeared (Fig. 4A, lane 2). Since GroEL, an internal control for a cytosolic protein, remained undigested unless Triton X-100 was added (Fig. 4A Bottom), it was suggested that HflK and HflC are located externally. When inverted membrane vesicles, prepared by passing the spheroplasts through the French pressure cell, were treated with proteinase K, a membrane-protected 40-kDa band (HflK* in Fig. 4B) was produced that reacted with anti-HflK (Fig. 4B Lower). This is consistent with a digestion of a part of the N-terminal hydrophilic segment (79 residues) from the cytoplasmic side. The HflC band was almost completely protected from digestion in the inverted membrane vesicles, consistent with the presence of only three amino acids that are located N terminally to the transmembrane segment. Thus, the HflC protein, the suspected protease subunit, contains virtually no cytosolic residues. These results, that the large hydrophilic domain of each of these proteins resides in the periplasmic space, indicate that HflKC exerts transmembrane modulation over the proteolytic activity of FtsH.
Figure 4.
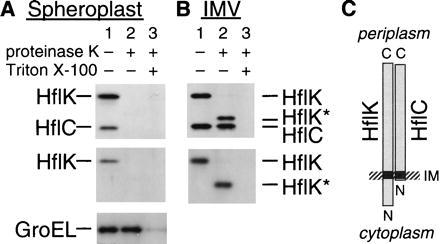
HflK and HflC reside on the periplasmic side of the membrane. Spheroplasts (A) and inverted membrane vesicles (B) prepared from AD202 were incubated at 0°C for 2 hr in the absence (lane 1) or presence (lanes 2 and 3) of 1 mg/ml proteinase K. Lanes 3 received 1% Triton X-100 before digestion. Proteins were analyzed by SDS/PAGE and immunoblotting using antisera directed against the HflKC complex (Upper), the HflK subunit (A Middle) or (B Lower), and the GroEL protein (A Bottom). (C) Schematic representation of the topographical arrangements of HflK and HflC.
DISCUSSION
It is clear that FtsH (HflB) plays an essential role in degradation of λ cII protein, the crucial determinant for the lysogenic pathway. In vitro, it catalyzes ATP-dependent proteolysis of cII. In vivo, a lowered cellular content of FtsH markedly stabilizes the cII protein, which was synthesized either from a cloned cII gene in the absence of other λ gene products or from the λ genome under the conditions of infection. Overproduction of FtsH accelerates the cII degradation. These observations, together with the results of Oppenheim and colleagues (Shotland et al., personal communication), establish that cII is degraded by the FtsH protease.
In contrast, the roles played by the HflKC protein in the lysogenic decision and cII degradation control are much more complicated. We were unable to reproduce the results that HflKC had a serine–protease-like activity to degrade cII (5). The exact reason for this discrepancy is unknown, but it is generally difficult to exclude a possibility of contamination. We detected a serine–protease-like activity that degrades cII from detergent extracts of membranes from the ΔhflK-hflC strain. Such a protease might have contributed to the proteolysis observed previously (5). Although it was proposed that HflC contains a serine–protease-like sequence motif (13), the H. influenzae HflC homolog (33) lacks it. Although the role of the HflC homolog in H. influenzae is totally unknown, we demonstrated that the removal of the proposed protease active site from the E. coli HflC does not abolish the function of HflC as a λ lysogenization controller. The compelling evidence against the notion that HflK and HflC proteins act as a protease for cytosolic proteins came form our direct determination of their topography. Contrary to the expectation, both the HflK and HflC proteins have their large hydrophilic domains exposed to the periplasmic space. Thus, they could not catalyze proteolysis of cII.
We showed that HflKC is actually inhibitory against the FtsH-dependent proteolysis of cII in vitro. Similar results were obtained previously for the proteolytic degradation of the SecY protein (20). Both HflK and HflC, when expressed as a single subunit, are unstable, but this degradation is not FtsH dependent, as expected from their localization (A.K., unpublished results). HflKC is not an in vitro substrate of FtsH either. Thus, the possibility that HflKC is simply competing with the substrate can be excluded. In vivo mutational and overproduction effects of HflKC were complicated. Its deletion increased lysogenization and stabilized cII under the conditions of λ phage infection. Puzzlingly, overproduction of HflKC also increased the λ lysogenization (Table 1) and stabilized cII in the λ-infected cells (Fig. 3) as well as in cells with the cloned cII (data not shown).
There is a clear distinction in the actions of HflKC with respect to stability control of SecY and cII. Whereas the deletion of hflK-hflC accelerated the degradation of the SecY24 mutant protein (20), it stabilized cII at least under the conditions of λ phage infection. The hflK13 and the hflC9 mutations stabilized SecY (20) but did not give an Hfl phenotype. These results, taken together, suggest that HflKC modulates the substrate specificity of FtsH. This action should be exerted from the periplasmic side of the membrane and transmitted to the cytosolic protease domain of FtsH. However, HflKC could interact directly with a periplasmic domain or a transmembrane segment of a membrane protein as well. This could explain the differential effects of HflKC exerted to SecY and cII. In this regard, the in vitro inhibitory action of HflKC against the proteolytic action of FtsH needs some reservation, since HflKC and a cytosolic protein can meet directly in vitro but not in vivo.
Why do both the absence and the excess presence of HflKC stabilize cII? In view of the in vitro FtsH-inhibiting ability of HflKC, its high concentration may lead to general inhibition of FtsH. In contrast, at a physiological concentration, HflKC may differentially inhibit the protease activities against different substrates. There might be two classes of FtsH substrates. The SecY class of proteins will be destabilized, whereas the cII class of proteins will be stabilized in the absence of HflKC. Cheng and Echols (34) reported the results of two-dimensional gel electrophoresis showing that some 13 proteins were stabilized in the hflA mutant. These could be cII-like substrates of FtsH. Although these authors did not discuss the following, their results also indicate some proteins, especially those at the basic region of the gel, decreased or disappeared in the mutant cells. The latter proteins could be the SecY class of the FtsH substrates. We propose that HflKC, at the normal concentration, preferentially suppresses the proteolytic activity of FtsH against the SecY class of proteins. If hflK-hflC is deleted, now FtsH is directed toward the SecY-like substrates and, as a consequence, cII-like substrates have decreased opportunities of degradation.
The cII protein synthesized from the cloned gene in the absence of other λ gene products (or the λ genome) was not clearly affected by the hflK-hflC deletion, and the effects were somewhat variable at different temperatures. The variable results could be explained by the different expression levels as well as by the presence or absence of other λ gene products and the λ DNA to which cII may bind. In particular, the λ cIII gene product is another inhibitor of FtsH (7, 12, 18) and should affect the cII stability. It should also be noted that FtsH is subject to heat shock regulation (18), and protein degradation in vivo can be affected by concentrations of various chaperones as well. Potentially, FtsH itself might exert a chaperone-like activities against some proteins (13–15). In spite of these complexities, it became clearer that FtsH is a regulated protease. Our findings that the protease activity of FtsH is modulated by the periplasmically located HflKC protein complex suggest that intracellular proteolysis for biological regulation can be controlled by events that occur on the surfaces of the cell.
Acknowledgments
We thank T. Ogura and C. Herman for bacterial strains and plasmids, A. B. Oppenheim for communicating unpublished results, T. Yoshihisa for instruction in the preparation of anti-HflKC serum, and K. Mochizuki for laboratory supplies. This work was supported by grants from the Ministry of Education, Science and Culture (Japan) and from the Human Frontier Science Program Organization. A.K. was supported by a Japan Society for the Promotion of Science Fellowship for Japanese Junior Scientists.
References
- 1.Shimatake H, Rosenberg M. Nature (London) 1981;292:128–132. doi: 10.1038/292128a0. [DOI] [PubMed] [Google Scholar]
- 2.Ho Y S, Wulff D L, Rosenberg M. Nature (London) 1983;304:703–708. doi: 10.1038/304703a0. [DOI] [PubMed] [Google Scholar]
- 3.Noble J A, Innis M A, Koonin E V, Rudd K E, Banuett F, Herskowitz I. Proc Natl Acad Sci USA. 1993;90:10866–10870. doi: 10.1073/pnas.90.22.10866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Banuett F, Herskowitz I. J Bacteriol. 1987;169:4076–4085. doi: 10.1128/jb.169.9.4076-4085.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cheng H C, Muhlrad P J, Hoyt M A, Echols H. Proc Natl Acad Sci USA. 1988;85:7882–7886. doi: 10.1073/pnas.85.21.7882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zorick T S, Echols H. J Bacteriol. 1991;173:6307–6310. doi: 10.1128/jb.173.19.6307-6310.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hoyt M A, Knight D M, Das A, Miller H I, Echols H. Cell. 1982;31:565–573. doi: 10.1016/0092-8674(82)90312-9. [DOI] [PubMed] [Google Scholar]
- 8.Banuett F, Hoyt M A, McFarlane L, Echols H, Herskowitz I. J Mol Biol. 1986;187:213–224. doi: 10.1016/0022-2836(86)90229-9. [DOI] [PubMed] [Google Scholar]
- 9.Gottesman S, Maurizi M R. Microbiol Rev. 1992;56:592–621. doi: 10.1128/mr.56.4.592-621.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Herman C, Ogura T, Tomoyasu T, Hiraga S, Akiyama Y, Ito K, Thomas R, D’Ari R, Bouloc P. Proc Natl Acad Sci USA. 1993;90:10861–10865. doi: 10.1073/pnas.90.22.10861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Confalonieri F, Duguet M. BioEssays. 1995;17:639–650. doi: 10.1002/bies.950170710. [DOI] [PubMed] [Google Scholar]
- 12.Tomoyasu T, Gamer J, Bukau B, Kanemori M, Mori H, Rutman A J, Oppenheim A B, Yura T, Yamanaka K, Niki H, Hiraga S, Ogura T. EMBO J. 1995;14:2551–2560. doi: 10.1002/j.1460-2075.1995.tb07253.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Akiyama Y, Ogura T, Ito K. J Biol Chem. 1994;269:5218–5224. [PubMed] [Google Scholar]
- 14.Akiyama Y, Shirai Y, Ito K. J Biol Chem. 1994;269:5225–5229. [PubMed] [Google Scholar]
- 15.Shirai Y, Akiyama Y, Ito K. J Bacteriol. 1996;178:1141–1145. doi: 10.1128/jb.178.4.1141-1145.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gama M-J, Toussaint A, Pato M L. Mol Microbiol. 1990;4:1891–1897. doi: 10.1111/j.1365-2958.1990.tb02038.x. [DOI] [PubMed] [Google Scholar]
- 17.Kihara A, Akiyama Y, Ito K. Proc Natl Acad Sci USA. 1995;92:4532–4536. doi: 10.1073/pnas.92.10.4532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Herman C, Thévenet D, D’Ari R, Bouloc P. Proc Natl Acad Sci USA. 1995;92:3516–3520. doi: 10.1073/pnas.92.8.3516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Akiyama Y, Kihara A, Tokuda H, Ito K. J Biol Chem. 1996;271:31196–31201. doi: 10.1074/jbc.271.49.31196. [DOI] [PubMed] [Google Scholar]
- 20.Kihara A, Akiyama Y, Ito K. EMBO J. 1996;15:6122–6131. [PMC free article] [PubMed] [Google Scholar]
- 21.Akiyama Y, Ito K. Biochem Biophys Res Commun. 1990;167:711–715. doi: 10.1016/0006-291x(90)92083-c. [DOI] [PubMed] [Google Scholar]
- 22.Stahl F W, Kobayashi I, Thaler D, Stahl M M. Genetics. 1986;113:215–227. doi: 10.1093/genetics/113.2.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Davis R W, Bostein D, Roth J R. Advanced Bacterial Genetics. Plainview, NY: Cold Spring Harbor Lab. Press; 1980. [Google Scholar]
- 24.Arber W, Enquist L, Hohn B, Murray N E, Murry K. In: Lambda II. Hendrix R W, Roberts J W, Stahl F W, Weisberg R A, editors. Plainview, NY: Cold Spring Harbor Lab. Press; 1983. pp. 433–466. [Google Scholar]
- 25.Silhavy T J, Berman M L, Enquist L W. Experiments with Gene Fusions. Plainview, NY: Cold Spring Harbor Lab. Press; 1984. [Google Scholar]
- 26.Schwarz E, Scherer G, Hobom G, Kössel H. Nature (London) 1978;272:410–414. doi: 10.1038/272410a0. [DOI] [PubMed] [Google Scholar]
- 27.Guzman L-M, Belin D, Carson M J, Beckwith J. J Bacteriol. 1995;177:4121–4130. doi: 10.1128/jb.177.14.4121-4130.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kunkel T A, Roberts J D, Zakour R A. Methods Enzymol. 1987;154:367–382. doi: 10.1016/0076-6879(87)54085-x. [DOI] [PubMed] [Google Scholar]
- 29.Ho Y S, Lewis M, Rosenberg M. J Biol Chem. 1982;257:9128–9134. [PubMed] [Google Scholar]
- 30.Laemmli U K. Nature (London) 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 31.Gottesman S, Gottesman M, Shaw J E, Pearson M L. Cell. 1981;24:225–233. doi: 10.1016/0092-8674(81)90518-3. [DOI] [PubMed] [Google Scholar]
- 32.Yoshihisa T, Ito K. J Biol Chem. 1996;271:9429–9436. doi: 10.1074/jbc.271.16.9429. [DOI] [PubMed] [Google Scholar]
- 33.Fleischmann R D, Adams M D, White O, Clayton R A, Kirkness E F, Kerlavage A R, Bult C J, Tomb J-F, Dougherty B A, Merrick J M, McKenney K, Sutton G, FitzHugh W, Fields C A, Gocayne J D, Scott J D, Shirley R, Liu L-I, Glodek A, Kelley J M, Weidman J F, Phillips C A, Spriggs T, Hedblom E, Cotton M D, Utterback T R, Hanna M C, Nguyen D T, Saudek D M, Brandon R C, Fine L D, Fritchman J L, Fuhrmann J L, Geoghagen N S M, Gnehm C L, McDonald L A, Small K V, Fraser C M, Smith H O, Venter J C. Science. 1995;269:496–512. doi: 10.1126/science.7542800. [DOI] [PubMed] [Google Scholar]
- 34.Cheng H H, Echols H. J Mol Biol. 1987;196:737–740. doi: 10.1016/0022-2836(87)90046-5. [DOI] [PubMed] [Google Scholar]