

Abstract
The expression of a number of genes encoding products involved in copper ion uptake in yeast is specifically inhibited by copper ions. We show here that copper metalloregulation occurs through Cu-dependent repression of the transactivation activity of Mac1p. A segment of the yeast transcription factor Mac1p was identified that activated transcription in vivo in a heterologous system using fusion polypeptides with the yeast Gal4 DNA-binding domain. The Gal4/Mac1p hybrid exhibits transactivation activity that is repressed in cells cultured in the presence of copper salts and derepressed in cells with reduced copper uptake. The repressive effect is specific for copper ions. The concentration dependency of the Cu-inactivation of Gal4/Mac1p is similar to that of Cu-inhibition of CTR1 expression, a known Cu-regulated gene in vivo. Copper inhibition of gene expression is not observed with a Gal4/Mac1p chimera containing the MAC1up1 substitution within the transactivation domain. Cells harboring the MAC1up1 allele fail to attenuate FRE1 and CTR1 expression in a Cu-dependent manner. Additional MAC1up alleles exist within the first of two cysteine-rich sequence motifs adjacent to the His → Gln MAC1up1 encoded substitution. Thus, Cu-regulation of Mac1p function arises from a novel Cu-specific repression of the transactivation domain function. Models for the mechanism of Cu-repression of Mac1p function will be discussed.
Keywords: copper, MAC1, transcriptional regulation
Copper is an essential nutrient in cells. In Saccharomyces cerevisiae, copper ions are required for at least three key enzymes: the respiratory cytochrome oxidase, the oxidative defense superoxide dismutase, and the Fet3p ferro-oxidase, which is important for iron uptake (1–3). Excess cell accumulation of copper ions results in toxicity possibly from cell damage caused by Cu-catalyzed formation of the highly reactive hydroxyl radical (4).
Homeostatic mechanisms exist to maintain a positive copper balance in cells and prevent Cu-induced toxicity. Copper homeostasis in yeast is maintained, in part, by Cu-regulation of the biosynthesis of several proteins. Both copper-mediated induction and copper-mediated inhibition of transcription are observed in yeast.
Three genes (CUP1, CRS5, and SOD1) are specifically induced in their expression in response to elevated copper salt levels in the growth medium (5–7). The three genes encode molecules (metallothioneins and superoxide dismutase) that have protective functions in cells. Cu-dependent activation of gene expression is mediated by Ace1p (activator of CUP1 expression) in S. cerevisiae (8–10). Copper induction is achieved through the formation of a tetracopper-thiolate cluster in the copper-regulatory domain of the Ace1 transcription factor (11–13). Polycopper cluster formation converts Ace1p from an inactive molecule into a functional transcriptional activator (9, 11).
The expression of a different subset of genes is inhibited in cells cultured in copper-replete medium by a previously unresolved mechanism. Three genes, FRE1, CTR1, and CTR3, which are involved in copper uptake across the plasma membrane, are down-regulated by copper ions (14–16). FRE1 encodes a NADPH-dependent metalloreductase, whereas CTR1 and CTR3 encode high-affinity plasma membrane copper transporters (16–19). Inhibition of CTR1 expression is specific to copper ions, whereas FRE1 expression is modulated by both copper and iron ions (14, 20, 21). CTR1 and CTR3 are fully expressed only in conditions of low environmental Cu(II) (14–16, 19). Thus, expression of this copper uptake system appears to be a cellular response to inadequate intracellular copper levels. Cu-dependent inhibition of CTR1, CTR3, and FRE1 expression occurs at the level of transcription but is independent of Ace1p (14–16, 22). Cu-attenuation of Ctr1p function also occurs by an independent, posttranslational process (19).
Basal transcription of CTR1 and FRE1 is mediated by the transcription factor Mac1p (metal binding activator) (15, 20). Basal expression of FRE1 is also influenced by the Aft1p transcriptional activator (21, 23). The role of Mac1p in the expression of CTR1 and FRE1 has been demonstrated in studies with mutant MAC1 cells. Metalloreductase activity and copper uptake rates are attenuated in cells containing a frame-shift mutation in MAC1 (mac1–1) (15, 20). In contrast, metalloreductase activity is markedly elevated in cells containing a semi-dominant MAC1 mutation, designated MAC1up1 (15, 20). The MAC1up1 mutant cells exhibit a copper-hypersensitive phenotype, presumably from enhanced copper uptake (15, 20). The lack of apparent copper repression in MAC1up1 cells is consistent with Mac1p also being responsible for Cu-dependent inhibition of CTR1 and FRE1 expression.
Fungal transcriptional activators of genes transcribed by RNA polymerase II typically exhibit two distinct functions, one for DNA binding and the other for assembly of the preinitiation transcription complex (24–26). The second function arises from a segment designated as the transactivation domain (AD). Interactions of the AD with components of TFIID or other basal factors such as TFIIA or TFIIB allow for RNA polymerase binding, creating the preinitiation complex (24–26). Yeast AD are typified by an unusual abundance of acidic residues, prolines, or glutamines (27, 28). The acidic nature of the AD is more a descriptive feature of this class of ADs rather than a functional feature, as no strict correlation exists between the net charge and activation potential (28, 29).
The mutation in MAC1up1 is a single T–A transversion resulting in a His → Gln substitution (20). This substitution lies within a candidate AD. The segment of Mac1 consisting of residues 201–340 exhibits a pI of 3.9. The pI of the intact Mac1p molecule is 6.9. Within this candidate AD lies two cysteine-rich motifs. The Cys-rich motif consisting of a CXCXXXXCXCXXC sequence repeat resembles Cu(I)-binding cysteinyl sequence motifs found in Ace1p and metallothioneins (30). The His → Gln substitution encoded by the MAC1up1 allele is adjacent to the first of the two Cys-rich motifs.
The DNA-binding domain of Mac1 has not been mapped. However, the N-terminal 40 residues of Mac1p are homologous to the N-terminal 40 residues of Ace1p and Amt1p, two Cu-activated DNA-binding proteins (31). The N-terminal 40 residues in Ace1p and Amt1p consist of a conserved Zn(II) domain that functions in base-specific minor groove DNA contacts (32–34).
The proximity of the Mac1p Cys-rich motifs to the candidate AD led us to postulate that the activity of Mac1p ADs may be copper-regulated. To test the prediction that copper regulation of Mac1p is achieved through regulation of AD activity, we created a Gal4/Mac1p fusion protein in which the DNA-binding domain of Gal4p (residues 1–147) was fused in-frame to residues 42–417 of Mac1p. Studies presented here demonstrate that Mac1p contains a novel Cu-regulated transactivation domain.
MATERIALS AND METHODS
Yeast Strains.
The strains used were YPH499 (MATa, ura3–52, lys2–801, ade2–101, trp1-Δ63, his3-Δ200, leu2-Δ1) and YPH499(ΔCTR1::LEU2) (35). YPH499(ΔCTR1) was engineered by transfecting strain YPH499 with a LEU2-based CTR1 disruption vector (ΔMTR) obtained from A. Dancis (University of Pennsylvania; ref. 14). The plasmid was linearized at the unique HindIII site prior to transformation. Transformants selected for leucine prototropy were shown by Southern blot analyses to contain the CTR1 disruption. As expected, the ctr1Δ cells failed to grow on medium with ethanol as the sole carbon source (18). YPH499 and YPH499(ΔCTR1) cells containing an episomal pGAL1/lacZ fusion gene (pRY131) were used in subsequent studies (36).
Growth Conditions.
Yeast were grown on selective media. When moderate copper starvation was required, a modified selective growth medium was made from components of yeast nitrogen base without copper sulfate (Bio 101). To achieve more severe copper starvation, ascorbic acid (1 mM) and the Cu(I) chelator bathocuproinedisulfonic acid (BCS; 33 μM) were present in the medium.
Construction of Plasmids.
The DNA-binding domain of Gal4 is encoded on the plasmid pAS1 (37). The plasmids pYaCu1 and pYaCu31, which contained genomic fragments of MAC1 and MAC1up1, respectively, were gifts from D. Hamer (National Institutes of Health). The plasmid pAS1-MAC1 was constructed by inserting a NcoI/Sau3AI fragment from pYaCu1 into the NcoI/BamHI sites of pAS1. The plasmid pAS1-MAC1up1 was constructed analogously using pYaCu31. The gene products of these two constructs are hybrid proteins containing the N-terminal DNA-binding domain of Gal4p fused in-frame to residues 42–417 of Mac1p or Mac1up1p. Plasmid YEpGAL4 (38) carries the DNA-binding and transactivation domain of GAL4. Plasmid pRY131 (36) carries a pGAL1/lacZ fusion. Plasmid CTR1/lacZ (obtained from A. Dancis, University of Pennsylvania) contains the promoter of CTR1 fused to the lacZ ORF (14).
Mutations.
A NcoI/SalI fragment of pAS1-MAC1 containing MAC1 codons 42–417 was cloned into pAlter (Promega) and subsequently used in site-directed mutagenesis (39). Mutant MAC1 (codons 42–417) sequences were removed as NcoI/SalI fragments and cloned into pAS1 for studies. The predicted mutations were confirmed by DNA sequencing carried out at the DNA Sequencing Core Facility at the University of Utah (Salt Lake City).
Truncations of MAC1 codons 176–417 and MAC1up1 codons 176–417 were constructed by subcloning PCR fragments of MAC1- and MAC1up1-encoding residues 176–417 into pAS1. Truncation of MAC1 codons 42–255 was constructed by subcloning a PCR fragment of MAC1-encoding residues 42–255 followed by an engineered stop codon into pAS1. The 42–255 truncation contains L253P and T254G substitutions as a result of designing a SmaI internal site. The MAC1 truncation (Δ253–340) was constructed by subcloning a PCR fragment of MAC1-encoding residues 341–417 into the 42–255 truncate at the 3′ end using SmaI and BamHI. The (Δ253–340) truncate encodes a Pro–Gly dipeptide between the two protein segments as a result of the design.
β-Galactosidase Activity Assay.
Transformants of strain YPH499 or YPH499(ΔCTR1) were grown to saturation under moderate copper starvation conditions, diluted 100-fold into fresh media containing appropriate additives, and incubated 24 hr before harvesting cells for β-galactosidase activity. Activity was assayed using sodium phosphate buffers and o-nitrophenyl-β-d-galactoside as described (40).
RESULTS
An acidic segment of Mac1p consisting of residues 201–340 contains a candidate copper-binding motif (Fig. 1). We postulated that Cu-regulation of Mac1p function occurred through regulation of AD activity. To test this hypothesis, a GAL4/MAC1 fusion gene was constructed by encoding the DNA-binding domain of Gal4p (residues 1–147) to residues 42–417 of Mac1p. The first 40 codons were deleted because they likely contribute to the DNA-binding function of Mac1p. The fusion gene was placed under the constitutive ADH1 promoter and termination sequences. The resulting plasmid was transfected into YPH499 cells containing a GAL1/lacZ reporter. Expression of the Gal4/Mac1p fusion protein in these cells resulted in expression of β-galactosidase, implying that residues 42–417 of Mac1p contain transcriptional activation activity (Fig. 2). As expected, transfection with a plasmid containing only GAL4 sequences (codons 1–147) failed to induce any expression of β-galactosidase, because this segment of Gal4p contains no transactivation function (Fig. 2).
Figure 1.

Sequence of Mac1p. The boxed N-terminal 40 residues are a candidate DNA-binding motif based on the homology with the Ace1 Zn(II) module (31). The two Cys-rich motifs within the candidate activation domain are indicated by C1 and C2 regions. The codon of the MAC1up1 mutation is marked by a +.
Figure 2.
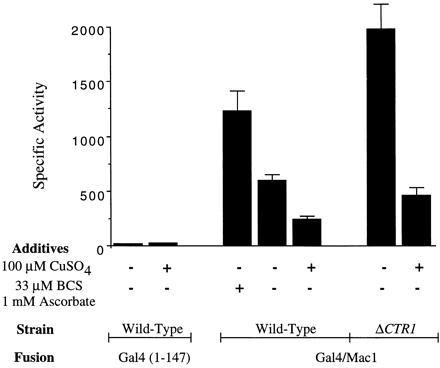
β-Galactosidase-specific activities in cells harboring the GAL4 (codons 1–147) control vector and the GAL4/MAC1 fusion vector. Studies were carried out in wild-type cells and ctr1Δ cells. In experiments indicated, the growth medium contained either BCS/ascorbate or CuSO4. The results are the mean (± SEM) of three independent measurements.
Expression of β-galactosidase was enhanced 2-fold in GAL4/MAC1 cells cultured overnight in Cu-deficient medium containing 0.03 mM bathocuproine disulfonic acid (BCS) and 1 mM ascorbate (Fig. 2). BCS was added to the growth medium to reduce the available copper ion concentration (Fig. 2). Because BCS exhibits a preference for Cu(I) ions, ascorbate was added to reduce Cu(II) in the medium to Cu(I). CTR1 expression is known to be maximal in cells cultured in Cu-deficient growth medium containing BCS (14, 22). In contrast, GAL1/lacZ expression was reduced more than 2-fold in cells cultured in medium containing >10 μM CuSO4 (Fig. 2). Thus, the range of β-galactosidase activity varied 5-fold when comparing Cu-deficient and Cu-supplemented conditions.
The enhancement in lacZ expression in the presence of BCS/ascorbate and attenuation in Cu-supplemented medium was consistent with Cu regulation of Gal4/Mac1p. To more specifically limit copper ion uptake into cells, we disrupted CTR1 encoding the high-affinity transporter in YPH499 cells. Cells (ctr1Δ) transformed with the GAL4/MAC1 and lacZ reporter fusions showed dramatically high β-galactosidase levels. The disruption of CTR1 resulted in high lacZ expression in cells cultured in Cu-deficient medium in the absence of added BCS and ascorbate. β-Galactosidase levels in ctr1Δ cells were 4-fold higher than enzyme levels in CTR1 cells cultured in synthetic medium. Thus, disruption of CTR1 was more effective than addition of BCS/ascorbate to the growth medium in limiting copper ion uptake.
The addition of 100 μM CuSO4 markedly attenuated GAL1/lacZ expression in ctr1Δ cells (Fig. 2). Cu uptake in ctr1Δ cells presumably occurs through low-affinity transporters. β-Galactosidase levels were reduced 4- to 5-fold when comparing Cu-deficient and Cu-replete conditions. The addition of BCS/ascorbate to the growth medium of ctr1Δ cells did not give any further increase in GAL1/lacZ expression (data not shown). The inhibition of lacZ expression by the Gal4/Mac1p fusion polypeptide is specific for copper salts. The addition of 10 μM Co(II), Ni(II), or Zn(II) to the growth medium was without effect on lacZ expression, whereas 10 μM Cu(II) gave marked repression (Fig. 3). The addition of Fe(III) as ferric ammonium citrate gave a modest inhibition in expression, although the extent of inhibition was not reproducible.
Figure 3.

β-Galactosidase-specific activities in cells harboring the GAL4/MAC1 fusion vector cultured in medium devoid of added metal ions or containing 10 μM of the metal ion shown. β-Galactosidase values represent the mean (± SEM) of three independent determinations.
Diminution in GAL1/lacZ expression may arise from cytotoxicity of the added copper salt, inactivation of the DNA-binding domain of Gal4p in the Gal4/Mac1p fusion, or inactivation of the transactivation domain in Gal4/Mac1p. Toxicity is unlikely to contribute to the observed inhibition of expression because the YPH499 cells show no growth impairment in medium containing 10 μM CuSO4. The DNA-binding domain of Gal4p contains a binuclear Zn(II) site (41), so Cu-dependent displacement of Zn(II) ions may inactivate Gal4p. Transfection of YPH499 cells containing the intact GAL4 that encodes the entire 881-residue Gal4p polypeptide did not result in any Cu attenuation in GAL1/lacZ expression (data not shown). The intact Gal4p molecule contains its own transactivation domain. Thus, the observed Cu repression is mediated by Mac1p sequences in the Gal4/Mac1p fusion.
If Mac1p mediates Cu repression of CTR1, the concentration dependency of the Cu-mediated inhibition of GAL1/lacZ expression by Gal4/Mac1p is expected to be similar to the concentration dependency of attenuation of CTR1 expression in vivo. Expression of GAL1/lacZ exhibited a Cu concentration dependency in ctr1Δ cells with half-maximal inhibition occurring near 0.3 μM CuSO4 (Fig. 4). A similar concentration dependency was observed in the down-regulation of CTR1 expression. Cells (ctr1Δ) were transformed with a vector containing a CTR1/lacZ fusion in which the 5′ segment of CTR1 (893 bp) was fused to the lacZ ORF. The resulting construct exhibits Cu-dependent inhibition of lacZ expression as reported previously (22). The copper concentration, which resulted in 50% inhibition in lacZ expression, was near 0.2 μM CuSO4. Cu-dependent repression of CTR1 in these cells occurs at a Cu ion concentration 10- to 20-fold lower than the Cu ion concentration to half-maximally activate Ace1p for Cu-induced expression of CUP1 (Fig. 4).
Figure 4.

Comparison of the copper ion concentration dependency of the expression of GAL1/lacZ to Cu modulation of the expression of CTR1/lacZ and CUP1/lacZ. Expression of GAL1/lacZ is regulated by the GAL4/MAC1 fusion gene, whereas regulation of CTR1/lacZ is modulated by chromosomal MAC1. The Cu activation of CUP1/lacZ expression is regulated by ACE1. The results are the mean (± SEM) of three independent measurements.
One prediction of the model of Cu regulation of the Mac1p transactivation domain is that Cu repression would be impaired in a GAL4/MAC1 fusion containing the MAC1up1 allele. The MAC1up1 mutation that resulted in a His279Gln substitution was engineered in GAL4/MAC1, creating GAL4/MAC1up1. GAL1/lacZ cells (CTR1+) harboring this mutant chimera exhibited high β-galactosidase activity, and enzyme levels were unaffected by the addition of copper salts to the growth medium (Fig. 5). Expression of GAL1/lacZ was elevated even in cells cultured in medium containing 0.1 mM CuSO4 (data not shown). Thus, the loss of Cu regulation in MAC1up1 cells correlates with the loss of Cu regulation in the Gal4/Mac1up1p fusion.
Figure 5.

β-Galactosidase-specific activities in cells harboring the GAL4/MAC1 fusion gene (top construct) to mutant fusion genes. ∗, The presence of the MAC1up1 mutation resulting in the His → Gln substitution (20). The numbers over the Mac1 portion of the fusion indicate the Mac1 residues encoded by the fusion genes. One internal deletion (Δ252–341) was constructed in which residues 252–341 were deleted. The results are the mean (± SEM) of three independent measurements.
To map more precisely the Cu-regulated AD in Mac1p, different Gal4/Mac1p hybrids were engineered with Mac1p truncations. The Gal4/Mac1p fusion used in experiments described above consisted of Mac1p residues 42–417. Three new Gal4/Mac1p hybrids were generated that consisted of Mac1p residues 42–255, 176–417, and an internal Mac1p deletion (Δ252–341) (Fig. 5). YPH499 cells (CTR1+) transfected with plasmids encoding the Gal4/Mac1p truncates were tested for their ability to activate GAL1/lacZ expression. The Gal4/Mac1p (Mac1p residues 42–255) hybrid and the Mac1p internal deletion (Δ252–341) failed to confer any transactivation (Fig. 5). In contrast, the Gal4/Mac1p (Mac1 residues 176–417) fusion gave exceptionally high transactivation that was unaffected by copper salts (Fig. 5). The corresponding Mac1up1 mutation in the GAL4/MAC1 (codons 176–417) fusion did not enhance transactivation as it did in the original fusion [Fig. 5, ∗ (marked construct)].
The presence of two Cys-rich sequence motifs in the Mac1p AD is consistent with Cu(I) binding in this region being related to the mechanism of Cu repression. Mutations were engineered in various cysteinyl codons in the GAL4/MAC1 fusion gene to determine whether any Cys → Ser (C → S) substitutions in the Mac1p segment affected Cu regulation. Various combinations of C → S substitutions were engineered at all Cys sequence positions in the AD. Analyses of the mutant Gal4/Mac1p molecules revealed that C → S substitutions in the first Cys-rich motif (residues 271–276; C1 region in Fig. 1) abolished Cu repression analogous to the MAC1up phenotype (Fig. 6). In contrast, C → S substitutions in the second Cys-rich motif (residues 315–334) did not affect Cu regulation. Whereas the H279Q substitution is the known “up” mutation, the corresponding H337Q substitution adjacent to the second Cys-rich repeat was without significant effect on transactivation and Cu regulation. Substitutions in the CC motifs (codons 69, 70 and 172, 173) upstream as double C → S substitutions of adjacent Cys residues resulted in partial attenuation of Cu repression of Mac1 function. Cu inhibition of CTR1 expression is incomplete. We were unsuccessful in using immunoblotting to verify that variation in transcriptional activities of Gal4/Mac1p fusions did not arise from changes in the cellular concentration of each hybrid.
Figure 6.
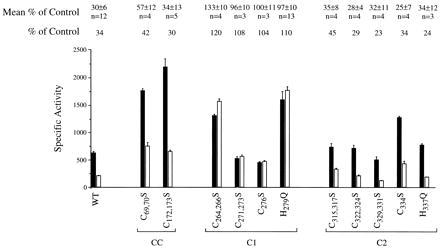
Mapping of residues involved in copper regulation of Mac1 function. Site-specific mutations were engineered in the GAL4/MAC1 fusion gene resulting in a series of single and double substitutions within the two Cys-rich motifs within the Mac1 activation domain (regions C1 and C2) as well as the two Cys–Cys sequences (labeled CC). The codons altered and the resulting substitutions are specified. Also shown is a representative experiment of the effect of mutant Gal4/Mac1p molecules on expression of the GAL1/lacZ reporter gene in cells cultured in the absence (solid bars) and presence (open bars) of 0.1 mM CuSO4. The β-galactosidase-specific activities are the mean and variance of three independent measurements. The ratio of activities of cells cultured in the presence versus the absence of CuSO4 is listed above the figure for the experiment shown as is the mean ratio (and SD) for multiple, related experiments. The number of individual experiments is specified.
DISCUSSION
Much attention has been focused on how promoter-specific eukaryotic transcription factors are regulated. Regulation of activity of transcription factors occurs often through modulation of DNA-binding activity (42–46). Regulation of activation function occurs through intramolecular or intermolecular repressive interactions (45, 47–49). Coordinate regulation of both DNA-binding and activation functions is also known (48, 50).
Results presented here reveal a novel mechanism of Cu-dependent regulation of transactivation activity. The transcriptional activity of Mac1p was tested after being fused to the Gal4p (1–147) DNA-binding domain. The evidence for a Cu-regulated transactivation domain in Mac1p is 3-fold. First, the Gal4/Mac1p chimera exhibits transactivation activity that is repressed in cells cultured in the presence of copper salts. Second, the repressive effect is specific for copper ions and occurs at a similar extracellular Cu(II) concentration that confers repression of the CTR1 promoter. Third, copper repression is abolished in the Gal4/Mac1up1 chimera analogous to the loss of copper repression observed in MAC1up1 cells (20).
The boundaries of the Mac1p activation domain remain ill-defined, but likely exist between residues 255 and 417. Activation activity is abolished with a deletion of residues 252–341. Cu repression requires a larger segment of Mac1p. Deletion of Mac1p residues 42–175 from Gal4/Mac1p results in potent transactivation activity that is unchecked by copper salts. The enhanced activity of Gal4/Mac1p (176–417) may arise from the unmasking of latent activation activity, the removal of AD inhibitory activity, or altered protein levels.
A variety of mechanisms may account for the Cu repression of Mac1p function. First, the regulation may be intramolecular, in which Cu(I) binding to Mac1 may directly inactivate Mac1p. According to this mechanism, Mac1p itself is the Cu(I) sensor. Cu inhibition of Mac1 AD may arise from a Cu-induced structural rearrangement that precludes a productive intermolecular interaction with perhaps a TFIID component. Alternatively, Cu(I) binding may attenuate the concentration of active Mac1p within the nucleus through Cu-dependent export of Mac1p to the cytoplasm or Cu-enhanced proteolytic degradation of Mac1p. Mac1p was shown to be nuclear (20), but its localization may change in Cu-treated cells.
A second candidate mechanism is that the Cu effect is mediated through another molecule analogous to the inhibition of the Gal4 AD function by Gal80 (45, 49). Cu(I) binding in the AD of Mac1 may stabilize a conformation that is recognized by a repressor molecule or a modifying enzyme. The actual inhibition of Mac1 function may be imposed by a secondary molecule, but Cu(I) binding to Mac1 initiates the process. The point mutations that result in constitutive activation may yield substitutions that preclude interaction with a repressor molecule. The clustering of gain-of-function mutations to the first Cys-rich sequence (C1 in Fig. 1) is consistent with this region defining an interface for interaction with a putative repressor. A different region may enfold the interface for a TAF interaction to initiate transcription.
MAC1up alleles map to the first Cys-rich motif within the AD. The clustering of “up” mutations within this motif and lack of gain-of-function mutations in the second Cys-rich motif imply that the first motif is dominant in Cu regulation. This region may serve as an interface for an interaction that is critical for Cu repression of Mac1 function.
Although metal ion repression of activation function has not been reported previously, ligand-dependent regulation of transactivation domain function is known to exist in the yeast Hap1p and Gal4p (51). Hap1p is activated by heme binding through two processes, one of which involves a modest heme-mediated enhancement in the activation activity of Hap1p (51). The transcriptional activity of Gal4p is regulated by the inhibitory protein Gal80p and Gal1p in a galactose-dependent manner (45, 49).
Because yeast is a significant model organism for eukaryotes, it will be of interest to determine how applicable yeast Cu regulation mechanisms are to animal cells and whether similar metal sensors exist in higher eukaryotes.
Acknowledgments
We thank Dr. Andrew Sewell, Dr. David Stillman, Dr. David Eide, and Laran Jensen for helpful discussions. This work was funded by a grant from the National Cancer Institute, National Institutes of Health (CA 61286).
ABBREVIATIONS
- BCS
bathocuproinedisulfonic acid
- AD
transactivation domain
References
- 1.Tsukihara T, Aoyama H, Yamashita E, Tomizaki T, Yamaguchi H, Shinzawa-Itoh K, Hakashima R, Yaono R, Yoshikawa S. Science. 1995;269:1069–1074. doi: 10.1126/science.7652554. [DOI] [PubMed] [Google Scholar]
- 2.Fridovich I. J Biol Chem. 1969;264:7761–7764. [PubMed] [Google Scholar]
- 3.Askwith C, Eide D, Van Ho A, Bernard P S, Li L, Davis-Kaplan S, Sipe D M, Kaplan J. Cell. 1994;76:403–410. doi: 10.1016/0092-8674(94)90346-8. [DOI] [PubMed] [Google Scholar]
- 4.Imlay J A, Linn S. Science. 1988;240:1302–1309. doi: 10.1126/science.3287616. [DOI] [PubMed] [Google Scholar]
- 5.Karin M, Najarian R, Haslinger A, Valenzuela P, Welch J, Fogel S. Proc Natl Acad Sci USA. 1984;81:337–341. doi: 10.1073/pnas.81.2.337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gralla E B, Thiele D J, Silar P, Valentine J S. Proc Natl Acad Sci USA. 1991;88:8558–8562. doi: 10.1073/pnas.88.19.8558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Culotta V C, Howard W R, Liu X F. J Biol Chem. 1994;269:25295–25302. [PubMed] [Google Scholar]
- 8.Thiele D J. Mol Cell Biol. 1988;8:2745–2752. doi: 10.1128/mcb.8.7.2745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Furst P, Hu S, Hackett R, Hamer D. Cell. 1988;55:705–717. doi: 10.1016/0092-8674(88)90229-2. [DOI] [PubMed] [Google Scholar]
- 10.Welch J, Fogel S, Buchman C, Karin M. EMBO J. 1989;8:255–260. doi: 10.1002/j.1460-2075.1989.tb03371.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dameron C T, Winge D R, George G N, Sansone M, Hu S, Hamer D. Proc Natl Acad Sci USA. 1991;88:6127–6131. doi: 10.1073/pnas.88.14.6127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nakagawa K H, Inouye C, Hedman B, Karin M, Tullius T D, Hodgson K O. J Am Chem Soc. 1991;113:3621–3623. [Google Scholar]
- 13.Graden J A, Posewitz M C, Simon J R, George G N, Pickering I J, Winge D R. Biochemistry. 1996;35:14583–14589. doi: 10.1021/bi961642v. [DOI] [PubMed] [Google Scholar]
- 14.Dancis A, Haile D, Yuan D S, Klausner R D. J Biol Chem. 1994;269:25660–25667. [PubMed] [Google Scholar]
- 15.Hassett R, Kosman D J. J Biol Chem. 1995;270:128–134. doi: 10.1074/jbc.270.1.128. [DOI] [PubMed] [Google Scholar]
- 16.Knight S A B, Labbe S, Kwon L F, Kwon L F, Kosman D J, Thiele D J. Genes Dev. 1996;10:1917–1929. doi: 10.1101/gad.10.15.1917. [DOI] [PubMed] [Google Scholar]
- 17.Dancis A, Roman D G, Anderson G J, Hinnebush A G, Klausner R D. Proc Natl Acad Sci USA. 1990;89:3869–3873. doi: 10.1073/pnas.89.9.3869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dancis A, Yuan D S, Halle D, Askwith C, Eide D, Moehle C, Kaplan J, Klausner R D. Cell. 1994;76:393–402. doi: 10.1016/0092-8674(94)90345-x. [DOI] [PubMed] [Google Scholar]
- 19.Ooi C E, Rabinovich E, Dancis A, Bonifacino J S, Klausner R D. EMBO J. 1996;15:3515–3523. [PMC free article] [PubMed] [Google Scholar]
- 20.Jungmann J, Reins H A, Lee J, Romeo A, Hassett R, Kosman D, Jentsch S. EMBO J. 1993;12:5051–5056. doi: 10.1002/j.1460-2075.1993.tb06198.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yamaguchi-Iwai Y, Dancis A, Klausner R D. EMBO J. 1995;14:1231–1239. doi: 10.1002/j.1460-2075.1995.tb07106.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yuan D S, Stearman R, Dancis A, Dunn T, Beeler T, Klausner R D. Proc Natl Acad Sci USA. 1995;92:2632–2636. doi: 10.1073/pnas.92.7.2632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yamaguchi-Iwai Y, Stearman R, Dancis A, Klausner R D. EMBO J. 1996;15:3377–3384. [PMC free article] [PubMed] [Google Scholar]
- 24.Mitchell P J, Tjian R. Science. 1989;245:371–378. doi: 10.1126/science.2667136. [DOI] [PubMed] [Google Scholar]
- 25.Zawel L, Reinberg D. Annu Rev Biochem. 1995;64:533–561. doi: 10.1146/annurev.bi.64.070195.002533. [DOI] [PubMed] [Google Scholar]
- 26.Stargell L A, Struhl K. Science. 1995;269:75–78. doi: 10.1126/science.7604282. [DOI] [PubMed] [Google Scholar]
- 27.Hope I A, Mahadevan S, Struhl K. Nature (London) 1988;333:635–640. doi: 10.1038/333635a0. [DOI] [PubMed] [Google Scholar]
- 28.Hahn S. Cell. 1993;72:481–483. doi: 10.1016/0092-8674(93)90064-w. [DOI] [PubMed] [Google Scholar]
- 29.Leuther K K, Salmeron J M, Johnston S A. Cell. 1993;75:575–585. doi: 10.1016/0092-8674(93)90076-3. [DOI] [PubMed] [Google Scholar]
- 30.Winge D R, Dameron C T, George G N. Adv Inorg Biochem. 1994;10:1–48. [PubMed] [Google Scholar]
- 31.Farrell R A, Thorvaldsen J L, Winge D R. Biochem. 1996;35:1571–1580. doi: 10.1021/bi9517087. [DOI] [PubMed] [Google Scholar]
- 32.Buchman C, Skroch P, Dixon W, Tullius T D, Karin M. Mol Cell Biol. 1990;10:4778–4787. doi: 10.1128/mcb.10.9.4778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dobi A, Dameron C T, Hu S, Hamer D, Winge D R. J Biol Chem. 1995;270:10171–10178. doi: 10.1074/jbc.270.17.10171. [DOI] [PubMed] [Google Scholar]
- 34.Koch K A, Thiele D J. Mol Cell Biol. 1996;16:724–734. doi: 10.1128/mcb.16.2.724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sikorski R S, Hieter P. Genetics. 1989;122:19–27. doi: 10.1093/genetics/122.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yocum R R, Hanley S, West R, Ptashne M. Mol Cell Biol. 1984;4:1985–1998. doi: 10.1128/mcb.4.10.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Durfee T, Becherer K, Chen P-L, Yeh S-H, Yang Y, Kilburn A E, Lee W-H, Elledge S J. Genes Dev. 1993;7:555–569. doi: 10.1101/gad.7.4.555. [DOI] [PubMed] [Google Scholar]
- 38.Nawaz Z, Baniahmad C, Burris T P, Stillman D J, O’Malley B W, Tsai M-J. Mol Gen Genet. 1994;245:724–733. doi: 10.1007/BF00297279. [DOI] [PubMed] [Google Scholar]
- 39.Sewell A K, Jensen L T, Erickson J C, Palmiter R D, Winge D R. Biochemistry. 1995;34:4740–4747. doi: 10.1021/bi00014a031. [DOI] [PubMed] [Google Scholar]
- 40.Ausubel F M, Brent R, Kingston R E, Moore D D, Seidman J G, Smith J A, Struhl K, editors. Current Protocols in Molecular Biology. New York: Wiley; 1995. [Google Scholar]
- 41.Marmorstein R, Harrison S C. Genes Dev. 1994;8:2504–2512. doi: 10.1101/gad.8.20.2504. [DOI] [PubMed] [Google Scholar]
- 42.Vandromme M, Gauthier-Rouviere C, Lamb N, Fernandez A. Trends Biochem Sci. 1996;21:59–64. [PubMed] [Google Scholar]
- 43.Whiteside S T, Goodbourn S. J Cell Sci. 1993;104:949–955. doi: 10.1242/jcs.104.4.949. [DOI] [PubMed] [Google Scholar]
- 44.Zhang L, Guarente L. J Biol Chem. 1994;269:14643–14647. [PubMed] [Google Scholar]
- 45.Suzuki-Fujimoto T, Fukuma M, Yano K-I, Sakurai H, Vonika A, Johnston S A, Fukasawa T. Mol Cell Biol. 1996;16:2504–2508. doi: 10.1128/mcb.16.5.2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Momand J, Zambetti G P, Olson D C, George D, Levine A J. Cell. 1992;69:1237–1245. doi: 10.1016/0092-8674(92)90644-r. [DOI] [PubMed] [Google Scholar]
- 47.Jakobsen B K, Pelham H R B. EMBO J. 1991;10:369–375. doi: 10.1002/j.1460-2075.1991.tb07958.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Li X-Y, Green M R. Genes Dev. 1996;10:517–527. doi: 10.1101/gad.10.5.517. [DOI] [PubMed] [Google Scholar]
- 49.Zenke F T, Engels R, Vollenbroich V, Meyer J, Hollenberg C P, Breunig K D. Science. 1996;272:1662–1665. doi: 10.1126/science.272.5268.1662. [DOI] [PubMed] [Google Scholar]
- 50.Lefstin J A, Thomas J R, Yamamoto K R. Genes Dev. 1994;8:2842–2855. doi: 10.1101/gad.8.23.2842. [DOI] [PubMed] [Google Scholar]
- 51.Zhang L, Guarente L. EMBO J. 1995;14:313–320. doi: 10.1002/j.1460-2075.1995.tb07005.x. [DOI] [PMC free article] [PubMed] [Google Scholar]