

Abstract
The rational protein design algorithm DEZYMER was used to introduce the active site of nonheme iron superoxide dismutase (SOD) into the hydrophobic interior of the host protein, Escherichia coli thioredoxin (Trx), a protein that does not naturally contain a transition metal-binding site. Reconstitution of the designed protein, Trx-SOD, showed the incorporation of one high-affinity metal-binding site. The electronic spectra of the holoprotein and its N3− and F− adducts are analogous to those previously reported for native {Fe3+}SOD. Activity assays showed that {Fe3+}Trx-SOD is capable of catalyzing the dismutation of the superoxide anion; comparative studies with the unrelated wild-type E. coli iron SOD indicated that {Fe3+}Trx-SOD catalyzes the dismutation reaction at a rate on the order of 105 M−1s −1. The ability to design catalytically competent metalloenzymes allows for the systematic investigation of fundamental mechanistic questions concerning catalysis at transition metal centers.
Keywords: rational protein design, metalloenzymes, superoxide ion dismutation, nonheme iron
Protein design methodologies (1–3) have recently focused on incorporating transition metal ions into proteins (3–6) to take advantage of their well-documented catalytic or electron transfer functions. However, this long-term objective of rationally engineering metalloproteins to introduce novel catalytic functions has heretofore met with only limited success (5–8). We have used the rational protein design algorithm DEZYMER (9) to test our ability to design catalytically active metalloenzymes by constructing a five-coordinate nonheme iron active site analogous to that found in iron-dependent superoxide dismutase (SOD) (10, 11) in a host protein, thioredoxin (Trx) (Fig. 1). The construction of this site represents a significantly different challenge from the design of coordinatively saturated, enzymatically inert, metal sites (6, 8, 40) by attempting to build a site that catalyzes the inner sphere transfer of electrons both to and from small molecule substrates. For this chemistry to occur, the availability of an open coordination position, or one occupied by a labile ligand such as H2O, is required, but the site must be sufficiently buried to prevent protein dimerization via a metal bridge. Introduction of a cavity for anion binding, as well as other factors relating to the attraction and transport of the anion to the active site, need to be considered as well.
Figure 1.
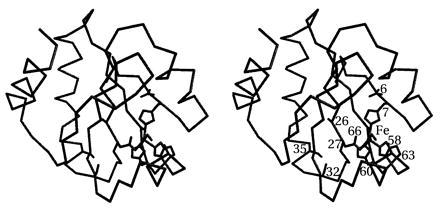
Location of the designed {Fe3+}Trx-SOD metal-binding site is formed by Leu-7-His, Phe-27-Asp, Ile-60-His, and Asn-63-His. Other mutations include His-6-Asn (eliminates potential alternative metal-binding residue), Cys-32-Ser and Cys-35-Ser (prevents disulfide formation), Leu-58-Ala and Thr-66-Ala (rebuilds complementary surface), and Asp-26-Ala (increases global stability of host protein).
The DEZYMER algorithm makes predictions based on strict geometric principles without explicit consideration of binding thermodynamics or protein dynamics. The rational design approach used here is based on the placement of an active site into the framework of a known protein fold. Active sites are described as geometrical arrangements of functionally important amino acids around a ligand, in this case a transition metal center. In the first phase of the search, the DEZYMER algorithm systematically examines a protein structure to identify backbone positions that are arranged in such a way that appropriate rotamers of the residues in the binding-site definition can be placed to satisfy the desired ligand geometry. In the second phase, additional changes may be introduced to ensure steric complementarity of the placed site with the surrounding protein matrix. This approach therefore tests two premises: first, that the proposed mutations retain the original protein fold by maintaining sterically reasonable packing interactions (12); second, that correct geometrical presentation of ligands is a sufficiently powerful approximation to reliably predict the construction of binding sites. In this report, we successfully demonstrate that these approximations have sufficient predictive power to redesign the hydrophobic interior of a protein (Trx) and introduce an iron center that can catalyze the dismutation of superoxide anion.
MATERIALS AND METHODS
Molecular Simulations.
The DEZYMER algorithm (9) was used to perform the calculations for the site design and refinement on a NeXT computer.
Cloning and Protein Expression.
Trx-SOD was constructed by standard PCR mutagenesis techniques using Pfu DNA polymerase (Stratagene), and the modified gene was inserted into the expression vector pKK-T7E downstream of a T7 promoter and sequenced in full. Escherichia coli (HMS174) cells carrying pTrx-SOD were grown in 2xYT media supplemented with 4 ml/liter glycerol to A600 = 2.0 and induced with 0.4 mM isopropyl β-d-thiogalactoside; the cells were harvested 4 hr post induction.
Purification of Apoprotein (apo) Trx-SOD.
The harvested cells from 10 liters of culture were resuspended in 100 ml of 20 mM Tris⋅Cl (pH 8), 100 mM NaCl, 5 mM EDTA, and lysed by sonication. The cleared lysate was then brought to 45% saturation with solid ammonium sulfate. After centrifugation, the pellet was redissolved in a minimum volume of 20 mM Tris⋅Cl (pH 8)/25 mM NaCl, adjusted to 50 mM 2-mercaptoethanol and 1 mM each o-phenanthroline, TIRON (4,5-dihydroxy-m-benzenedisulfonic acid) and EDTA to remove endogenous metals, and dialyzed overnight against 20 mM Tris⋅Cl (pH 8)/25 mM NaCl.
The sample was then loaded onto a 200-ml DEAE–cellulose column, washed with one column volume of 20 mM Tris⋅Cl (pH 8)/25 mM NaCl, and eluted with a 25–500 mM salt gradient in 20 mM Tris⋅Cl, pH 8. Peak fractions were further purified by Superdex 200 column chromatography in 20 mM Hepes⋅Cl (pH 8)/25 mM NaCl. This purification procedure resulted in the isolation of approximately 1.2 g of Trx-SOD of >99% purity from a single 10 liter fermentor growth (Fig. 2).
Figure 2.
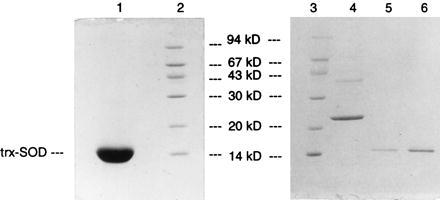
Twenty percent SDS gel of purified apo Trx-SOD. Lanes: 1, 28 μg of purified Trx-SOD; 5, 0.28 μg of purified Trx-SOD; 6, 0.78 μg of purified Trx-SOD; 2 and 3, molecular weight markers as indicated; 4, 1.8 μg of E. coli iron SOD (Sigma).
A final purification step takes advantage of the unusual ability of the Trx host protein to thermally denature and renature reversibly; aliquots of purified apo protein were heated to 90°C for 20 min, spun in a microcentrifuge to remove any precipitated material, and rechromatographed on a Superdex S75 column in 20 mM Hepes⋅Cl (pH 8)/25 mM NaCl, where only the properly folded Trx-SOD is isolated.
Iron Reconstitution of Trx-SOD.
Reconstitution was readily accomplished by the addition of one equivalent of ferrous ammonium sulfate from a concentrated (100 mM) aqueous stock solution prepared immediately before use. Development of a yellow chromophore, indicative of iron oxidation, was complete in minutes. The same reconstitution procedure was followed for both normal and heat-treated enzymes. Iron-to-protein ratios were determined by atomic absorption (Varian SpectrAA-20) and UV/vis (Perkin–Elmer Lambda 6 spectrophotometer; ɛM,280nm = 13,700 based on total amino acid analysis) spectroscopy.
Reconstitution was also performed under anaerobic conditions to assess the level of iron binding. Iron-binding stoichiometry was measured by varying the initial ferrous ammonium sulfate-to-Trx-SOD ratio (0.1–10). After extended incubation (5 hr) in 20 mM Hepes (pH 8.0)/25 mM NaCl at 21°C under strict anaerobic conditions in an inert atmosphere (argon) box, the reconstituted samples were dialyzed overnight against 20 mM Tris·Cl (pH 8.0)/25 mM NaCl (no EDTA). No oxidation of the iron was observed, as judged by the absence of any visible chromophore. After air oxidation (1 hr), iron-to-Trx-SOD ratios were determined by atomic absorption and UV spectrophotometric methods; oxidation, however, was complete in minutes as judged by color development.
Activity Measurements.
The SOD activity of holo Trx-SOD was determined by a standard indirect colorimetric assay that measures the level of inhibition of superoxide-induced reduction of colorless nitroblue tetrazolium (NBT, Sigma) dye to its oxidized blue formazan form (13). One unit of SOD activity is defined as that amount of enzyme that inhibits the rate of NBT reduction, under the specified conditions, by 50%. Several dilutions of one enzyme solution were investigated to accurately extrapolate to the 50% inhibition level. All assay reactions were performed in triplicate in 50 mM 2-(N-cyclohexylamino)ethanesulfonic acid (CHES) (pH 9.0), 0.5 mM xanthine, 0.5 mM EDTA, 40 mg/ml NBT, at 21°C. Generation of the superoxide anion was accomplished enzymatically by the reaction of xanthine and xanthine oxidase (13). Xanthine oxidase was titrated in the standard cytochrome c assay (13) to determine the amount of enzyme required to give a ΔAbs550nm = 0.025/min; this amount of xanthine oxidase was then used in the NBT assays. Parallel studies were also performed with E. coli iron SOD obtained from Sigma.
RESULTS
Rational Design.
The iron SOD site definition, derived from the x-ray crystal structures of iron SOD from E. coli (11) and Pseudomonas ovalis (10), consisted of a trigonal bipyramidal iron center with axial-histidine and exogenous-ligand binding sites (for superoxide or other anions) and equatorial (N-His)2(O-Asp) ligands. A bound superoxide ligand in the axial position was added to the site definition, based upon the idealized geometry of a Fe-O2−· adduct (14). The DEZYMER algorithm generated six solutions for the iron SOD active site definition; we constructed and characterized one solution (Trx-SOD): Leu-7-His, Phe-27-Asp, Ile-60-His, Asn-63-His (Fig. 1). This site was initially selected due to the relatively short axial distance between the iron center and the protein surface (≈9Å), the favorable orientation of the “open” coordination site that is aligned toward the protein surface and not directed toward the protein interior, and the lack of any protein main-chain atoms sterically hindering access to the iron site.
Trx, a relatively small (108 amino acid residues) monomeric protein that contains a single redox active disulfide bond (Cys-32 and Cys-35) (15), was chosen (16) as our initial scaffold protein, since the gene for Trx has been cloned, sequenced, and expressed at high levels (17). Trx is a stable protein (oxidized Trx Tm, 86°C; reduced Trx Tm, 75°C) (15) that accommodates conservative as well as some nonconservative mutations (18). In addition to the availability of a high-resolution (1.68 Å) x-ray structure (19), there are several solution structures (oxidized and reduced) derived from NMR spectroscopic methods (20, 21), indicating the feasibility of obtaining structural information on the designed protein in the absence of crystals.
Several other mutations were introduced into Trx-SOD: His-6-Asn, to prevent competition between the designed His-7 site and the naturally occurring His-6 residue, the only histidine residue in native thioredoxin; Leu-58-Ala, Thr-66-Ala to improve packing around the designed site owing to differences in volume between the naturally occurring residues of Trx and those of the designed iron site; and Asp-26-Ala to improve the global stability of the protein (the Asp-26-Ala mutant of thioredoxin is 3 kcal stabilized as compared with wild type) (22, 23). We also used the well-characterized Cys-32-Ser/Cys-35-Ser mutant of Trx to eliminate the native disulfide bond thereby precluding the possibility of sulfur coordination to the metal center and to facilitate maintenance of monomeric Trx.
Iron Reconstitution of Purified Trx-SOD.
The optical spectra of apo and {Fe3+}Trx-SOD are presented in Fig. 3. The apo protein sample has virtually no absorbance at wavelengths longer than 320 nm. The addition of one equivalent of Fe2+ leads to the rapid incorporation and oxidation of iron to produce the 1:1 holo {Fe3+}Trx-SOD protein. Immediately evident is the broad absorption maximum centered at approximately 350 nm with a long absorption tail extending beyond 500 nm. This feature, which is not observed for either apo or {Fe2+}Trx-SOD, is assigned as a His-to-Fe3+ charge-transfer transition and can only arise from Fe3+ binding to the designed histidine-rich site. The iron is tightly bound (Kd < 1 μM, as determined by titration of Fe2+ assayed by fluorescence, data not shown) and cannot be removed from the protein even by extended incubation in the presence of 50 mM EDTA, suggesting that it is buried in the interior of the protein. Treatment of heat denatured {Fe3+}Trx-SOD with EDTA, however, results in the formation of Fe3+EDTA. The Fe2+ center of reduced {Fe2+}Trx-SOD is readily removed by addition of excess o-phenanthroline, a neutral iron chelator.
Figure 3.
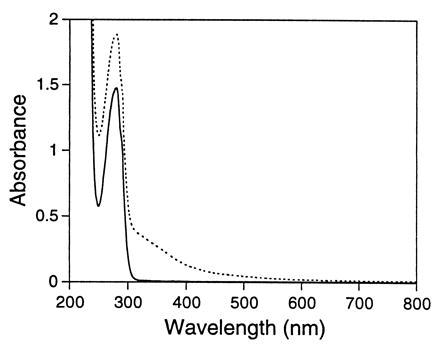
UV/vis spectrum of apo (solid line) and reconstituted (dashed line) {Fe3+}Trx-SOD. The spectra of Trx-SOD (1.1 × 10−4 M) in 20 mM Hepes (pH 8.0)/25 mM NaCl at 21°C, before and after iron reconstitution with ferrous ammonium sulfate in air are shown.
Fe3+ (d5) has relatively slow exchange kinetics and thus it is possible, if oxidation is rapid relative to complete reconstitution, that reconstitution in air does not yield the thermodynamically favored metal-binding site. To address this issue, extended incubation of apo Trx-SOD with as many as 10 equivalents of Fe2+ followed by overnight dialysis against gel filtration buffer (no EDTA present) was carried out under strictly anaerobic conditions. Despite the excess of metal used, only one equivalent of iron is bound after dialysis, indicating that there is only one high-affinity iron-binding site in this protein. Furthermore, the sample develops the characteristic His-to-Fe3+ ligand-to-metal charge transfer (LMCT) bands in the optical spectrum immediately upon exposure to air, whereas stock solutions of ferrous ammonium sulfate oxidize only slowly. The final ɛM350 values of samples reconstituted aerobically and anaerobically are virtually identical (<5% discrepancy). These results indicate that iron oxidation occurs only after incorporation into the histidine-rich site and that the thermodynamically favored iron site (Fe2+/anaerobic conditions) is the same as that found when stoichiometric reconstitution is performed in the presence of dioxygen.
The addition of exogenous anionic ligands such as N3− or F− to {Fe3+}Trx-SOD results in immediate spectral changes (Fig. 4 A and B, respectively) suggesting that these ligands are able to enter the primary coordination sphere of the iron center and form coordination complexes. Azide binding induces a shift in the LMCT envelope to lower energy with the band now observed at 400 nm, whereas fluoride binding results in a reduction of the intensity of the 350-nm absorption and in an apparent blue shift in the LMCT envelope to higher energy with the charge transfer transition now found at 330 nm. Both the magnitude and direction of these shifts are analogous to those reported for E. coli iron SOD (24). These data provide compelling evidence that the electronic structure of the designed iron site resembles E. coli iron SOD, has an open coordination position necessary for anion binding, and is accessible to endogenous anions.
Figure 4.
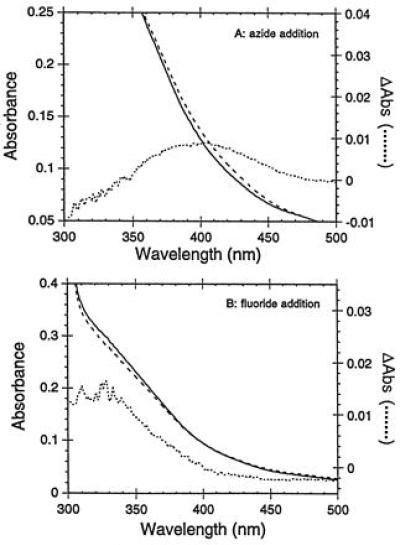
Anion binding. The UV/vis spectrum of {Fe3+}Trx-SOD (1.1 × 10−4 M) in 20 mM Hepes (pH 8.0)/25 mM NaCl at 21°C (solid line) was recorded. The sample was then adjusted to 100 mM sodium azide (A, broken line) or 100 mM sodium fluoride (B, broken line). Dotted lines represent difference spectra in A and B.
Catalytic Dismutase Activity.
SODs are usually assayed indirectly owing to the extreme instability of the superoxide ion in aqueous solution. A common assay method uses xanthine oxidase and xanthine to generate the superoxide ion, which then reduces NBT, a reaction that can be followed spectrophotometrically; inhibition of this reduction is a measure of SOD activity (13). The NBT assay is extremely reproducible provided there is sufficient total protein present in the assay to keep the formazan product of NBT reduction soluble. The results of the SOD assay for heat-treated {Fe3+}Trx-SOD at a single fixed pH (9.0) are given in Fig. 5. {Fe3+}Trx-SOD inhibits the reduction of NBT in proportion to the amount of enzyme added over a large concentration range, indicating that the designed protein does indeed show SOD activity.
Figure 5.
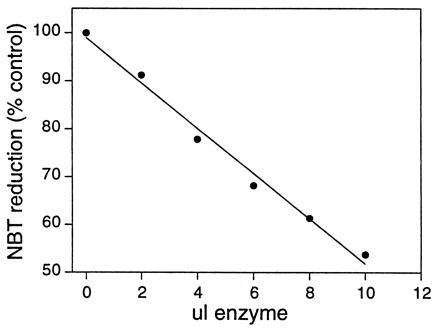
Assay of {Fe3+}Trx-SOD for SOD activity. Each point represents the average of three assays; the correlation coefficient of the least-squares fit is >0.99.
To address the relative rates of catalysis between the native and designed enzymes, samples of E. coli iron SOD were assayed in parallel with {Fe3+}Trx-SOD. This direct comparison indicated that the designed enzyme catalyzes the dismutation reaction at a rate ≈104-fold slower than native iron SOD, demonstrating that {Fe3+}Trx-SOD is a highly active protein able to catalyze the dismutation reaction with k ≈ 105 M−1 −1. Apo Trx-SOD does not show any activity in this assay, nor does {Fe3+}Trx-SOD that has been boiled for 5 min and centrifuged immediately prior to assay. To control against the possibility that free metal ions in solution could contribute to the observed activity, EDTA was included in all assay buffers. Furthermore, Fe3+-EDTA demonstrated no activity when assayed, in agreement with previous reports (25).
As native E. coli iron SOD catalyzes superoxide dismutation at a rate on the order of 109 M−1 s−1 (26), it is essential to demonstrate that the observed enzymatic activity of {Fe3+}Trx-SOD cannot be attributed to trace contamination with E. coli SOD. Several lines of evidence strongly contradict this possibility. First, as discussed above, our preparations of apo Trx-SOD do not show any dismutase activity as isolated. Second, the activity of E. coli iron SOD is reduced at least 1000-fold (lower limit) by the heat-treatment step. Native E. coli iron SOD was heated to 90°C for 10 min, spun to remove the large precipitate, and then assayed for activity. No dismutase activity was observed in the heat-treated native E. coli enzyme, even when assayed at 1000 times the normal enzyme concentration. Reconstitution of apo Trx-SOD by our standard protocol either before or after heat-treatment results in enzymes with little or no difference in dismutase activity, indicating that the observed activity cannot arise from contamination by the heat-labile native E. coli enzyme. Furthermore, treatment of E. coli iron SOD with reductant and chelators (a routine part of our purification procedure) yields an enzyme that still precipitates upon heat treatment (and is thus easily removed), ruling out the possibility that the apo form of the E. coli dismutase enzyme might be carried along in a soluble form that can be reconstituted in parallel with Trx-SOD.
A third argument countering the possibility of trace contamination of our samples takes into account the high degree of purity of our Trx-SOD preparations (see Fig. 2). The activity of Trx-SOD is about 10−4 times that of native E. coli iron SOD, thus, the E. coli enzyme would need to be present at levels of 0.01% to account for the observed activity. However, since it has been demonstrated that heat treatment of E. coli iron SOD reduces its activity by at least 1000-fold (a lower limit), the contaminant would have to comprise 10% of our preparations to account for the observed activity after heat treatment. Examination of Fig. 2 excludes this possibility.
Finally, iron-limiting reconstitution experiments provide positive evidence that it is the iron in the designed site that is catalytically active. Samples of apo heat-treated Trx-SOD (>99% pure; see Fig. 2) were reconstituted to iron:protein ratios of 0, 0.25, 0.50, 0.75, and 0.95 and assayed for activity (Fig. 6). Trx-SOD shows no activity when iron is absent and dismutase activity increases in proportion to the degree of iron incorporation (Fig. 6). Again, these data indicate that the observed activity is associated with the designed protein and is not due to trace amounts of native E. coli iron SOD. Furthermore, the parallel between activity and the intensity of the His-to-Fe3+ LMCT band indicates that it is iron in the designed histidine-rich site that is catalytically active.
Figure 6.
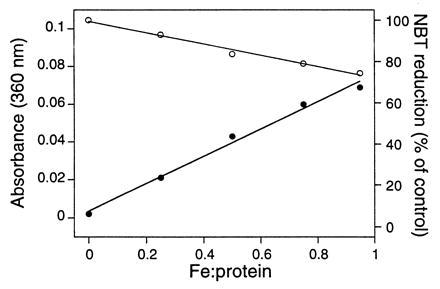
Iron limiting reconstitution and SOD activity. Apo Trx-SOD was reconstituted in air to the following iron:protein ratios: 0, 0.25, 0.50, 0.75, and 0.95. The samples were then assayed for SOD activity (○) and the optical spectra (•) of the samples (20 μl diluted to 1 ml) were recorded. Each point represents the average of three assays; the correlation coefficient of the least-squares fit is >0.99.
DISCUSSION
We have used the rational design algorithm, DEZYMER, to introduce a catalytically active iron SOD site into the hydrophobic core of E. coli Trx, a protein normally devoid of transition metal centers. Inspection of the designed site reveals several favorable properties. Thioredoxin folds to form a hydrophobic core of β-pleated sheet flanked on either side by two α-helices; the structure can be considered as being formed of two domains, βαβαβ from residues 1–59 and ββα from residues 76–108 (19). Two of the designed coordinating residues (His-7 and Asp-27) lie on the β-sheet, the other resides (His-60 and His-63) on an α-helix. The iron coordination sphere is encapsulated by a hydrophobic shell (Phe-12, Val-16, Val-25, Ala-58, Ala-66, Ala-67, Ile-72, and Leu-79), a feature found in native iron SODs and thought to be necessary for activity (10). Finally, the designed axial superoxide coordination position, though buried, is only 7–9 Å from the surface, with no intervening main-chain atoms. It is also possible to modify residues near the iron active site that would allow hydrogen bonding to a bound superoxide ion in further rounds of mutagenesis. Thus, this design contains many of the structural features thought to be necessary for the dismutase reaction.
Three primary chemical features must be realized if the designed iron center is to catalyze the dismutation of superoxide ion (28): (i) the iron center must have a minimum of one coordination site available for binding superoxide in two adjacent oxidation states (Fe2+/Fe3+); (ii) the iron redox couple, governed in part by the primary coordination sphere as well as the surrounding protein matrix, must lie between the redox potentials of superoxide oxidation (0.16 V) and superoxide reduction (0.89 V); and (iii) the iron center must be able to cycle between the Fe2+ and Fe3+ oxidation states more rapidly than the rate of the spontaneous dismutation reaction.
Evidence for iron incorporation into the designed site comes from optical studies of holo Trx-SOD. The electronic absorption spectra of high-spin d5 (Fe3+) centers are typically dominated by the spin-allowed (ɛM > 1000) LMCT transitions originating from relatively easily oxidized ligands such as histidine (28, 29). Electronic transitions arising from within the d-orbital manifold (ligand field) are all spin-forbidden and are therefore much less intense (ɛM < 500). Transitions analogous to those observed in {Fe3+}Trx-SOD (ɛM350 = 2400) are reported for ferric SODs from bacterial and algal sources (ɛM350 = 1675–2860) (29), as well as for ferric soybean lipoxygenase (ɛM350 ≈ 2000) (29). Based on x-ray crystallographic characterization of both Fe3+ SOD (10) and soybean lipoxygenase (30) that show the presence of three and four bound histidine ligands, respectively, the absorption bands observed only in oxidized {Fe3+}Trx-SOD are assigned as endogenous His-to-Fe3+ LMCT transitions, although bound carboxylate may also contribute to this region. Both our qualitative and quantitative data indicate that a single Fe3+ ion binds tightly to a histidine-rich coordination sphere, which is consistent with metal binding occurring in the designed three-histidine site. These spectroscopic techniques, however, do not allow the definitive determination of Asp-27 (carboxylate) coordination.
The addition of anions to solutions of {Fe3+}Trx-SOD produced changes in the optical spectra that also parallel the reported behavior of native iron SOD enzymes (29). A 2.9-Å resolution structure of iron SOD from Pseudomonas ovalis complexed with the inhibitor azide (31) supports the assignment of the origin of the optical LMCT band as arising from the anion active site complex. These data also indicate the ability of small anions to readily bind to the designed active site of {Fe3+}Trx-SOD, a necessary step if an inner-sphere reaction is to occur during the dismutation reaction.
The native E. coli iron SOD falls into the class of “perfectly evolved” enzymes that operate at the diffusion-controlled limit (26). Studies have also shown that copper-, manganese-, and iron-containing complexes can also catalyze the dismutation of superoxide anion at this same diffusion-controlled limit (32–34). Therefore, the chemical basis for the difference in activity between the designed enzyme and either the native SOD enzymes or small molecule model systems is of interest. Previous studies on native bacterial iron SOD have shown that the maximal turnover number is independent of pH over the range 7.5–10.5 (25). However, Km is dramatically influenced by pH; steady-state kinetic studies show the presence of an essential ionizable group close to the iron center in the reduced enzyme with pKa near 9.0, whose acid form appears to promote binding of superoxide to the reduced protein. Kinetic studies of iron SOD in D2O and H2O report a strong dependence of kcat on the solvent isotope present, suggesting that the rate-limiting step of catalysis is proton transfer from solvent H2O to an ionizable active site residue (35). The direct protonation of bound superoxide anion with solvent H2O is unlikely in view of paramagnetic NMR studies, which indicate that water exchange with the active site is too slow to be mechanistically important (36). Recent crystallographic analyzes of azide-inhibited iron SOD are consistent with this interpretation and have implicated conserved tyrosine (Tyr-34) and glutamine (Gln-70) residues as playing possible roles in mediating protonation of superoxide through exchange with solvent (31). Our designed Trx-SOD enzyme model lacks these conserved secondary active-site residues, suggesting that alternative pathways for proton transfer may be operating at slower rates, thereby decreasing the overall rate of superoxide dismutation. The resolution of these issues, which requires detailed studies of the transient features of catalysis as a function of pH and isotopic composition of the solvent, are currently underway.
Unfavorable electrostatic interactions between the Trx-SOD protein surface and the incoming anion may also play a role in diminishing the activity of {Fe3+}Trx-SOD as compared with E. coli FeSOD. Favorable electrostatic attraction of the negatively charged superoxide ion is known to play a role in rapid rate of dismutation the by native SODs (26, 37). A comparison of the surface electrostatic potential of E. coli FeSOD and Trx-SOD reveals that the native enzyme has a strong overall positive charge on the surface near the superoxide coordination position, but that the comparable region in Trx-SOD has a high concentration of negative charge. Thus mutation of surface residues to enhance electrostatic attraction of the negative superoxide ion is expected to boost the overall rate of reaction. Experimental investigations designed to examine the influence of surface electrostatics on the rate of catalysis are in progress. It must be emphasized, however, that the designed enzyme is still highly active, with superoxide dismutation rates on the order of 105 M−1 s−1.
The activity of Trx-SOD is less than that of aqueous Cu2+ ion and several synthetic model systems whose activities are comparable to the SOD enzyme itself (38). However, soluble metal complexes may unpredictably show catalytic dismutase activity ranging from very high to zero, depending on details of the coordination sphere that are not completely defined (32, 34). Although a detailed comparison of Trx-SOD and the functional small molecule SOD mimics will require the completion of both structural characterization and mechanistic/kinetic studies of Trx-SOD, it is reasonable to expect that restricted access to the designed active iron site and the unfavorable electrostatic attraction between Trx-SOD and the negatively charged superoxide anion are responsible for the lower activity of Trx-SOD versus the small molecule models.
In summary, key aspects of our design have been realized: (i) we have successfully created a mononuclear nonheme iron-binding site whose electronic spectrum closely parallels that of the native enzyme, (ii) the iron site is capable of binding anions and exhibiting those spectral features reported for anion adducts of iron SOD, and (iii) the designed site readily catalyzes the dismutation of superoxide anion. In addition, understanding the chemical basis for the differential activity of {Fe3+}Trx-SOD versus wild-type iron SOD will allow insights into the fundamental roles played by the protein matrix in modulating active site catalytic chemistry.
Bioinorganic chemistry has traditionally focused its efforts in two directions, the small molecule, synthetic analogue approach (39) and the study of intact metalloproteins. Small molecules permit the investigation of the effects of varying the primary coordination sphere, but cannot provide the elaborate overall environment or long-range interactions created by insertion into a protein shell. Conversely, study of intact metalloproteins does not frequently provide the opportunity to explore the varied contributions of the protein matrix that are often invoked in mechanistic discussions. This protein design methodology allows us to incorporate both approaches into a single line of research; intact proteins can now be manipulated to vary properties of not only the immediate coordination environment but also the adjacent protein matrix, while maintaining the overall structural or physiological properties of the chosen host protein.
Acknowledgments
A.L.P. gratefully acknowledges Postdoctoral Grant DF95–105 from the Donaghue Foundation. This work was supported in part by National Science Foundation Grant CHE-9419178 (J.P.C.), the Exxon Education Foundation (J.P.C.), the Alfred P. Sloan Foundation (J.P.C.), and National Institutes of Health Grant R29GM49871 (H.W.H.).
ABBREVIATIONS
- SOD
superoxide dismutase
- Trx
thioredoxin
- NBT
nitroblue tetrazolium
- apo
apoprotein
- LMCT
His-to-Fe3+ ligand-to-metal charge transfer
References
- 1.Bryson J W, Betz S F, Lu H S, Suich D J, Zhou H X, O’Neil K T, DeGrado W F. Science. 1995;270:935–941. doi: 10.1126/science.270.5238.935. [DOI] [PubMed] [Google Scholar]
- 2.Desjarlais J R, Handel T M. Curr Opin Biotechnol. 1995;6:460–466. doi: 10.1016/0958-1669(95)80076-x. [DOI] [PubMed] [Google Scholar]
- 3.Barrick D. Curr Opin Biotechnol. 1995;6:411–418. doi: 10.1016/0958-1669(95)80070-0. [DOI] [PubMed] [Google Scholar]
- 4.Higaki J N, Fletterick R J, Craik C S. Trends Biochem Sci. 1992;17:100–104. doi: 10.1016/0968-0004(92)90245-5. [DOI] [PubMed] [Google Scholar]
- 5.Matthews D J. Curr Opin Biotechnol. 1995;6:419–424. doi: 10.1016/0958-1669(95)80071-9. [DOI] [PubMed] [Google Scholar]
- 6.Regan L. Trends Biochem Sci. 1995;20:280–285. doi: 10.1016/s0968-0004(00)89044-1. [DOI] [PubMed] [Google Scholar]
- 7.Tawfik D S, Eshhar Z, Green B S. Mol Biotechnol. 1994;1:87–103. doi: 10.1007/BF02821512. [DOI] [PubMed] [Google Scholar]
- 8.Hellinga H W. In: Design of Metalloproteins. Cleland J L, Craik C S, editors. New York: Wiley–Liss; 1996. pp. 369–398. [Google Scholar]
- 9.Hellinga H W, Richards F M. J Mol Biol. 1991;222:763–785. doi: 10.1016/0022-2836(91)90510-d. [DOI] [PubMed] [Google Scholar]
- 10.Stoddard B L, Howell P L, Ringe D, Petsko G A. Biochemistry. 1990;29:8885–8893. doi: 10.1021/bi00490a002. [DOI] [PubMed] [Google Scholar]
- 11.Stallings W C, Powers T B, Pattridge K A, Fee J A, Ludwig M L. Proc Natl Acad Sci USA. 1983;80:3884–3888. doi: 10.1073/pnas.80.13.3884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Richards F M, Lim W A. Q Rev Biophys. 1993;26:423–498. doi: 10.1017/s0033583500002845. [DOI] [PubMed] [Google Scholar]
- 13.Flohe L, Otting F. Methods Enzymol. 1984;105:93–104. doi: 10.1016/s0076-6879(84)05013-8. [DOI] [PubMed] [Google Scholar]
- 14.Hill A A O, Tew D G. In: Dioxygen, Superoxide and Peroxide as Ligands. Wilkinson G, Gillard R D, McCleverty J A, editors. Vol. 2. Oxford: Pergamon; 1987. pp. 315–333. [Google Scholar]
- 15.Holmgren A. Annu Rev Biochem. 1985;54:237–271. doi: 10.1146/annurev.bi.54.070185.001321. [DOI] [PubMed] [Google Scholar]
- 16.Hellinga H W, Caradonna J P, Richards F M. J Mol Biol. 1991;222:787–803. doi: 10.1016/0022-2836(91)90511-4. [DOI] [PubMed] [Google Scholar]
- 17.Wallace B J, Kushner S R. Gene. 1984;32:399–401. doi: 10.1016/0378-1119(84)90015-5. [DOI] [PubMed] [Google Scholar]
- 18.Wynn R, Richards F M. Protein Sci. 1993;2:395–403. doi: 10.1002/pro.5560020311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Katti S K, LeMaster D M, Eklund H. J Biol Chem. 1990;212:167–184. doi: 10.1016/0022-2836(90)90313-B. [DOI] [PubMed] [Google Scholar]
- 20.Dyson H J, Holmgren A, Wright P E. Biochemistry. 1989;28:7074–7078. doi: 10.1021/bi00443a044. [DOI] [PubMed] [Google Scholar]
- 21.Dyson H J, Gippert G P, Case D A, Holmgren A, Wright P E. Biochemistry. 1990;29:4129–4136. doi: 10.1021/bi00469a016. [DOI] [PubMed] [Google Scholar]
- 22.Langsetmo K, Fuchs J A, Woodward C. Biochemistry. 1991;30:7603–7609. doi: 10.1021/bi00244a032. [DOI] [PubMed] [Google Scholar]
- 23.Langsetmo K, Fuchs J A, Woodward C, Sharp K A. Biochemistry. 1991;30:7609–7614. doi: 10.1021/bi00244a033. [DOI] [PubMed] [Google Scholar]
- 24.Slykhouse T O, Fee J A. J Biol Chem. 1976;251:5472–5477. [PubMed] [Google Scholar]
- 25.Bull C, Fee J A, O’Neill P, Fielden E M. Arch Biochem Biophys. 1982;215:551–555. doi: 10.1016/0003-9861(82)90115-1. [DOI] [PubMed] [Google Scholar]
- 26.Bannister J V, Bannister W H, Rotilio G. CRC Crit Rev Biochem. 1987;22:111–180. doi: 10.3109/10409238709083738. [DOI] [PubMed] [Google Scholar]
- 27.Fee J A. In: Superoxide, Superoxide Dismutases and Oxygen Toxicity. Spiro T A, editor. Vol. 2. New York: Wiley Interscience; 1980. pp. 209–239. [Google Scholar]
- 28.Zhang Y, Gebhard M S, Solomon E I. J Am Chem Soc. 1991;113:5162–5175. [Google Scholar]
- 29.Averill B A, Vincent J B. Methods Enzymol. 1993;226:33–51. doi: 10.1016/0076-6879(93)26004-s. [DOI] [PubMed] [Google Scholar]
- 30.Boyington J C, Gaffney B J, Amzel L M. Science. 1993;260:1482–1486. doi: 10.1126/science.8502991. [DOI] [PubMed] [Google Scholar]
- 31.Stoddard B L, Ringe D, Petsko G A. Protein Eng. 1990;4:113–119. doi: 10.1093/protein/4.2.113. [DOI] [PubMed] [Google Scholar]
- 32.Weiss R H, Flickinger A G, Rivers W J, Hardy M M, Aston K W, Ryan U S, Riley D P. J Biol Chem. 1993;268:23049–23054. [PubMed] [Google Scholar]
- 33.Goldstein S, Czapski G, Meyerstein D. J Am Chem Soc. 1990;112:6489–6492. [Google Scholar]
- 34.Allen A O, Bielski B H J. In: Formation and Disappearance of Superoxide Radicals in Aqueous Solution. Oberly L W, editor. Vol. 1. Boca Raton, FL: CRC; 1982. pp. 125–142. [Google Scholar]
- 35.Bannister J V, Bannister W H, Rotilio G. CRC Crit Rev Biochem. 1987;22:329–332. doi: 10.3109/10409238709083738. [DOI] [PubMed] [Google Scholar]
- 36.Villafranca J J, Yost F, Jr, Fridovich I. J Biol Chem. 1974;249:3532–3536. [PubMed] [Google Scholar]
- 37.Getzoff E D, Cabelli D E, Fisher C L, Parge H E, Viezzoli M S, Banci L, Hallewell R A. Nature (London) 1992;358:347–351. doi: 10.1038/358347a0. [DOI] [PubMed] [Google Scholar]
- 38.Valentine J S. In: Dioxygen Reactions. Bertini I, Gray H B, Lippard S J, Valentine J S, editors. Mill Valley, CA: University Science Books; 1994. pp. 253–314. [Google Scholar]
- 39.Ibers J A, Holm R H. Science. 1980;209:223–235. doi: 10.1126/science.7384796. [DOI] [PubMed] [Google Scholar]
- 40.Coldren, C. D., Hellinga, H. W. & Caradonna, J. P. (1997) Proc. Natl. Acad. Sci. USA, in press. [DOI] [PMC free article] [PubMed]