

SUMMARY
While the core splicing machinery is highly conserved between budding yeast and mammals, the absence of alternative splicing in Saccharomyces cerevisiae raises the fundamental question of why introns have been retained in ~5% of the 6000 genes. Because ribosomal protein-encoding genes (RPGs) are highly overrepresented in the set of intron-containing genes, we tested the hypothesis that splicing of these transcripts would be regulated under conditions in which translation is impaired. Using a microarray-based strategy, we find that, within minutes after the induction of amino acid starvation, the splicing of the majority of RPGs is specifically inhibited. In response to an unrelated stress, exposure to toxic levels of ethanol, splicing of a different group of transcripts is inhibited, while the splicing of a third set is actually improved. We propose that regulation of splicing, like transcription, can afford rapid and specific changes in gene expression in response to the environment.
INTRODUCTION
Messenger RNA splicing is an essential part of eukaryotic gene expression as the coding regions of most eukaryotic genes are interrupted by noncoding introns. In higher eukaryotes, the process of splicing is utilized to regulate both qualitative and quantitative aspects of gene expression (reviewed in Black, 2000; Blencowe, 2006). Alterations in the pattern of splice site usage in multi-intronic transcripts can produce a spectrum of protein isoforms from a single genomic locus, greatly expanding the genetic repertoire of an organism. Furthermore, when coupled to nonsense-mediated decay, alternative splicing can function as an “on/off” switch by introducing premature termination codons, thereby directing mRNA degradation (Lewis et al., 2003; Mitrovich and Anderson, 2000).
By contrast, only ~5% of genes in the budding yeast Saccharomyces cerevisiae are intron containing, and with few exceptions these genes have only a single intron (Spingola et al., 1999). Splice site sequences in yeast introns generally conform to a strict consensus, and documented instances of alternative splicing are rare (Davis et al., 2000). Nonetheless, a number of examples of regulated splicing have been demonstrated in yeast, including a set of transcripts that are constitutively transcribed but are efficiently spliced only during meiosis (Davis et al., 2000; Juneau et al., 2007; Nandabalan et al., 1993). These introns, as well as the autoregulated introns in Rpl30 and Yra1, all contain nonconsensus splice sites, which are required for regulation (Eng and Warner, 1991; Preker et al., 2002). However, because most yeast introns contain consensus splice site sequences, it has remained unknown whether splicing could function as a more general regulator of gene expression.
Interestingly, the set of intron-containing genes in yeast includes many metabolic regulators and is highly enriched for ribosomal protein genes (RPGs). Of the 139 RPGs encoded in the yeast genome, 102 are interrupted by at least one intron, making this by far the largest functional category. It has been previously established that starvation for amino acids leads to transcriptional repression of RPG synthesis, repression of rRNA synthesis, a general repression of translation, and an upregulation of enzymes involved in amino acid biosynthesis through a process controlled by the nonessential kinase Gcn2 (Chen and Powers, 2006; Cherkasova and Hinnebusch, 2003; Dever et al., 1992; Hinnebusch, 2005). Because of the overrepresentation of translational components among the intron-containing genes, we hypothesized that the splicing of these transcripts might also be regulated in response to amino acid starvation.
Here we have taken a microarray-based strategy to examine the transcript-specific splicing changes resulting from exposure to two unrelated but environmentally relevant stresses: amino acid starvation and ethanol toxicity. We find that the splicing of the majority of RPGs is inhibited within minutes of inducing amino acid starvation. By comparison, exposure to toxic levels of ethanol, which is not known to induce a global repression of translation, has little effect on the splicing of the RPG transcripts. Rather, in response to the latter stress, the splicing of a different set of transcripts is downregulated, while the splicing efficiency of a third group of transcripts is improved. The specificity of these responses and the speed of their onset argue that splicing provides an important opportunity for regulation of gene expression in response to environmental stress. Furthermore, the capacity for transcription-independent regulation may explain the evolutionary retention of introns in these genes.
RESULTS
A Genome-wide Approach to Examining Pre-mRNA Splicing
We have employed a microarray-based strategy to monitor quantitative changes in the splicing of ~250 intron-containing yeast transcripts in response to alterations in the environment. The microarrays contain three different oligonucleotide probes targeting each intron-containing gene, as described previously (Clark et al., 2002; Pleiss et al., 2007). These probes allow for the independent determination of differences in the levels of the precursor mRNA species (probe P), mature mRNA species (probe M), and total transcript (probe T) between two samples (Figure 1). Whereas analysis of a traditional expression array hinges upon a single measurement of the relative abundance of a transcript in an experimental and reference sample, analysis of changes in the splicing behavior of a transcript requires the simultaneous consideration of the relative behaviors of all three types of probes. By examining the behavior of both the precursor species and the mature species against the background of changes in the total level of transcript, splicing efficiency can be distinguished from overall changes in transcript levels (Pleiss et al., 2007). Thus, a splicing profile can be generated and used to identify transcripts whose splicing is affected by an experimental treatment.
Figure 1. Splicing-Specific Microarrays.
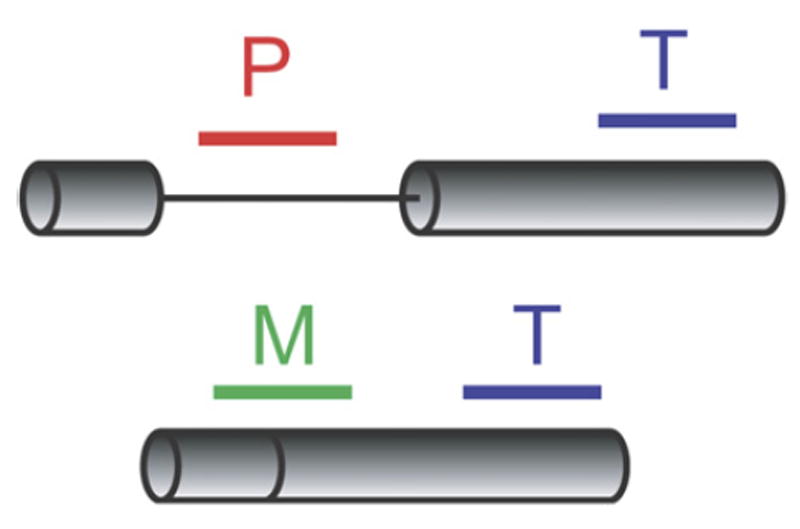
Probes targeting intron-containing genes are designed to detect the precursor species (P), the mature species (M), or the total transcript (T).
Changes in Pre-mRNA Splicing in Response to Amino Acid Starvation
We began by examining the cellular response to amino acid starvation. Amino acid starvation was induced by addition of the histidine-mimic 3-aminotriazole (3AT) (Hinnebusch, 2005). Cells were collected at several short times after either a mock treatment or exposure to 50 mM 3AT. Figure 2A shows the time-resolved splicing profiles for each intron-containing gene obtained from microarray experiments comparing RNA isolated from cells exposed to these two treatments. Genes have been divided into two groups: RPGs and non-RPGs. Within these groups, the genes are ordered based on the similarity of the behavior of their three feature types across the amino acid starvation time course. Notably, the splicing of the majority of RPG transcripts is rapidly reduced, as evidenced by the increased abundance of precursor mRNA in starved relative to unstarved cells. Not surprisingly, because RPG transcripts exhibit a strong partitioning toward their spliced form (Pleiss et al., 2007), little decrease is seen in the level of mature mRNA for these species during the short time course of 3AT exposure. By comparison, little change is seen in the level of pre-mRNA for any of the non-RPG transcripts. Interestingly, several non-RPG transcripts do show a rapid decrease in the level of total mRNA and mature mRNA present, suggesting that the mature forms of these transcripts may be rapidly destabilized in response to amino acid starvation.
Figure 2. Regulation of Pre-mRNA Splicing in Response to Amino Acid Starvation.
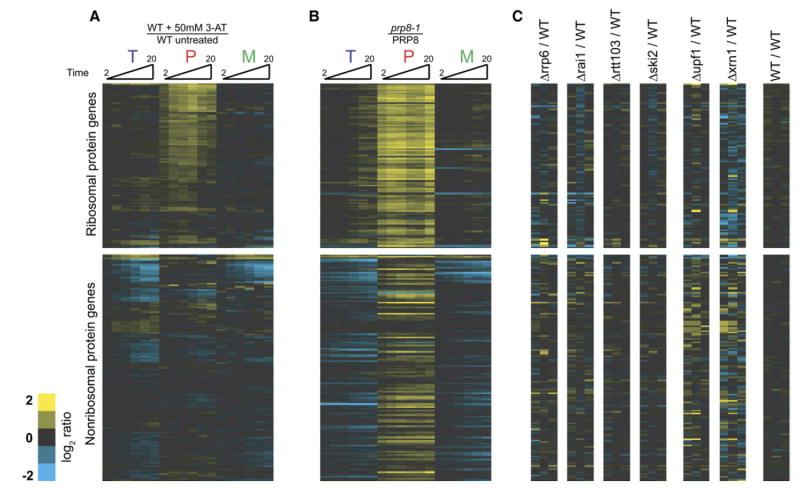
Time-resolved splicing profiles resulting from comparisons of (A) wild-type cells either treated with 50 mM 3AT or mock treated for 2, 4, 6, 10, 15, and 20 min; (B) prp8-1 and wild-type cells shifted from 25°C to 37°C for 5, 10, 15, 20, 25, and 30 min; and (C) wild-type cells compared to cells deleted for either RRP6, RAI1, RTT103, SKI2, UPF1, or XRN1. Also included is a comparison of two wild-type strains. The transcripts are ordered identically in all images.
Quantitative RT-PCR Validation of Splicing Inhibition
To validate the observed response to amino acid starvation, quantitative RT-PCR experiments using both intron- and exon-specific primers were employed. As seen in Figure 3, results from the quantitative PCR assay are in good agreement with the microarray data. Both methods show accumulation of the RPG pre-mRNAs corresponding to Rpl21a and Rpl17b at very short time points after induction of amino acid starvation. As seen in Figure S1 in the Supplemental Data available with this article online, nearly equal levels of accumulation are detected when using primers that include the intron and the entire second exon, demonstrating that the accumulating product is in fact an unspliced species. By contrast, no defect is detected in the splicing of either the U3 pre-snRNA or the Tef4 pre-mRNA.
Figure 3. Quantitative RT-PCR Validates the Rapid, Transcript-Specific Downregulation of Splicing in Response to Amino Acid Starvation.
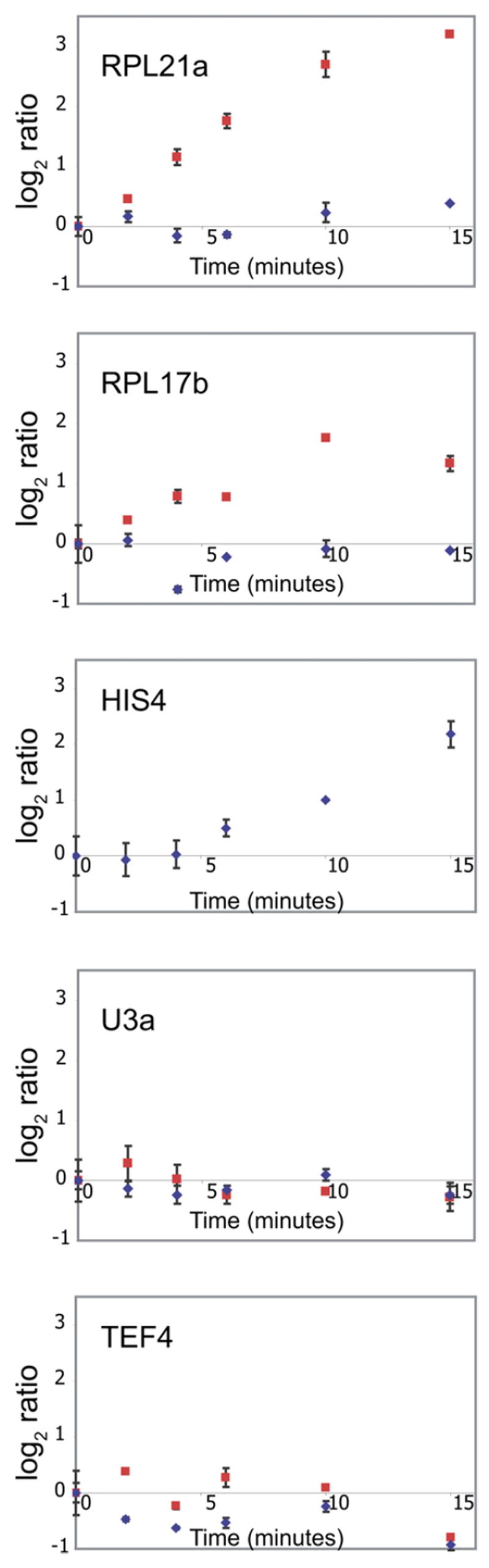
The behaviors of RPL21a, RPL17b, HIS4, U3 snRNA, and TEF4 were examined using primers specific to intron regions (red squares) or exon regions (blue diamonds). Error bars are the result of triplicate measurements from a single biological sample.
Comparison with a General Inhibition of Splicing
Because ribosomal proteins are highly transcribed (Holstege et al., 1998), it was important to demonstrate that the observed RPG-specific phenotype truly results from an RPG-specific change in splicing and does not simply suggest that these pre-mRNAs are more likely to show accumulation given their high level of transcription. Thus, we compared the splicing profile derived from amino acid starvation to that seen after inactivation of a core spliceosomal factor. Figure 2B shows the splicing profile resulting from shifting a strain containing the temperature-sensitive prp8-1 mutant to the nonpermissive temperature. PRP8 encodes a stable component of the U5 small nuclear RNP, and mutations in Prp8 are expected to result in defects in the splicing of most actively transcribed pre-mRNAs. In response to inactivation of prp8-1, the fold increases in pre-mRNA levels are nearly equal for RPG and non-RPG transcripts alike. This strongly suggests that amino acid starvation does not impart a global defect in splicing but instead that the splicing of RPG transcripts is specifically downregulated. Importantly, these experiments also demonstrate the relative stability of the mature forms of the RPG mRNAs in response to defects in splicing; whereas the decreases in mature levels for many non-RPG transcripts are apparent within minutes of inactivation of Prp8, very little decrease in the level of mature RPG transcripts can be observed during the prp8-1 time course.
Reduced RNA Turnover Is Unlikely to Account for the Increased Pre-mRNA Levels
Considered a priori, two different mechanisms could account for the observed accumulation of RPG transcript pre-mRNA during amino acid starvation. First, a reduction in the splicing efficiency of these transcripts could lead to an increase in their pre-mRNA levels. Alternatively, some fraction of these pre-mRNAs may normally be targeted to a decay pathway prior to their processing by the spliceosome. In this case, the specific inhibition of their decay could result in an increase in pre-mRNA levels. To discriminate between these possibilities, we asked whether RPG pre-mRNAs as a class are specifically stabilized in the absence of any one of a number of different components involved in RNA turnover. Figure 2C shows the splicing profiles resulting from deletion of the following: RRP6, a component of the nuclear exosome (Houseley et al., 2006); RAI1 and RTT103, two components of the Rat1 nuclear decay pathway (Kim et al., 2004); SKI2, a regulator of 3′ to 5′ decay (Frischmeyer et al., 2002; van Hoof et al., 2002); UPF1, a component of the nonsense-mediated degradation pathway (Leeds et al., 1991); or XRN1, a cytoplasmic 5′ to 3′ exonuclease (Fillman and Lykke-Andersen, 2005). Notably, while the genome-wide changes in mRNA levels resulting from deletion of many of these factors have previously been reported (He et al., 2003; Lelivelt and Culbertson, 1999; Wyers et al., 2005), our experiments specifically address the behavior of the pre-mRNA species.
Importantly, there is no evidence from these experiments that RPG pre-mRNAs as a class are constitutively degraded at significant levels under normal growth conditions. By contrast, our data validate several pre-mRNAs that were previously shown to be stabilized in response to mutations in these decay pathways. For example, the RPL28 pre-mRNA is strongly stabilized in the absence of UPF1 (He et al., 1993). Also, as we had previously shown (Preker and Guthrie, 2006), the YRA1 pre-mRNA is strongly stabilized in the absence of XRN1. Several additional pre-mRNAs are also stabilized in response to each of these mutations. While we cannot formally rule out the possibility that a different, completely nonoverlapping degradation pathway may contribute to the phenotype observed in response to amino acid starvation, our results strongly suggest that the increased levels of pre-mRNA result from a failure of these transcripts to be efficiently spliced.
Gcn2 Is Not Required for the Spliceosomal Response to Amino Acid Starvation
Given the well-defined role of Gcn2 in regulating the response to amino acid starvation, it was important to determine whether the observed splicing changes were also dependent upon the activity of this nonessential kinase. The accumulation of uncharged tRNAs resulting from amino acid starvation is known to activate Gcn2, which subsequently phosphorylates the initiation factor eIF-2α, causing a general decrease in translation initiation (Dong et al., 2000). By a mechanism that is now well understood, this results in the paradoxical increase in translation of the transcription factor Gcn4 (Hinnebusch, 1997), which leads to the transcriptional upregulation of numerous genes involved in amino acid biosynthesis (Natarajan et al., 2001). Interestingly, as seen in Figure 3, the onset of the splicing response appears to precede the transcriptional upregulation of one Gcn4 target, HIS4, suggesting that the changes in splicing are independent of the Gcn4-mediated transcriptional response.
Using a strain deleted for GCN2, we repeated our experiment by comparing an untreated sample with one exposed to 3AT. As expected, the Δgcn2 strain is largely unable to effect the upregulation of the biosynthetic genes in response to amino acid starvation (Figures 4A and 4B). Nonetheless, the capacity of this strain to downregulate RPG splicing is nearly unchanged. Indeed, a direct experimental comparison of the amino acid starvation responses of a wild-type and a Δgcn2 strain both treated with 3AT demonstrates their highly divergent transcriptional responses but nearly identical splicing responses (Figure 4C). Interestingly, slight differences are apparent between the two strains at the longer time points, with more pre-mRNA accumulation apparent in the Δgcn2 strain. We presume this difference reflects the Gcn2-dependent global downregulation of RPG transcription known to accompany amino acid starvation. The onset of this transcriptional program presumably limits pre-mRNA accumulation in the wild-type cells.
Figure 4. The Splicing Response to Amino Acid Starvation Does Not Require the Activity of Gcn2.
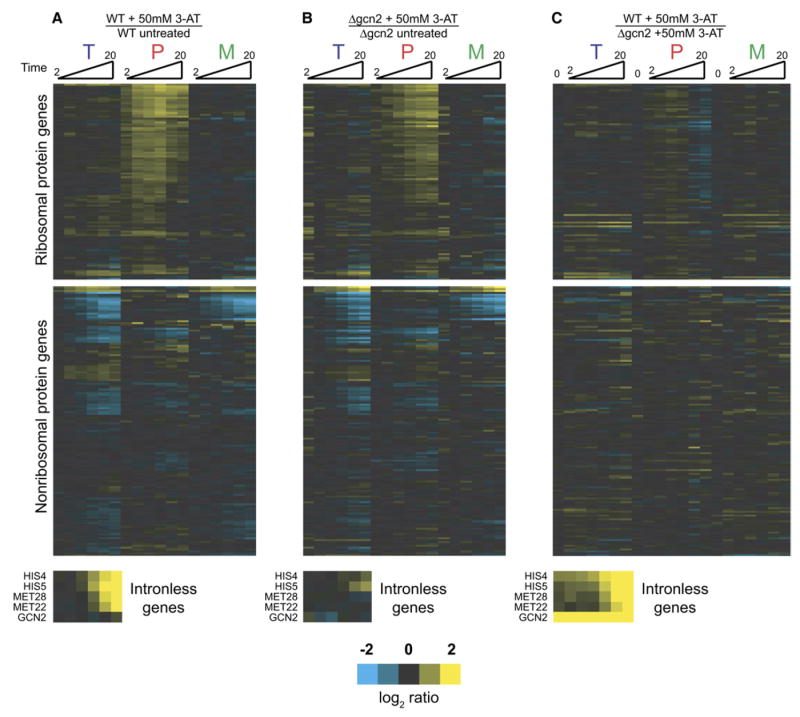
Time-resolved splicing profiles resulting from (A) wild-type cells treated with 50 mM 3AT compared to mock-treated, (B) cells deleted for GCN2 treated with 50 mM 3AT compared to mock-treated, or (C) wild-type cells treated with 50 mM 3AT compared to cells deleted for GCN2 treated with 50 mM 3AT. Included in the final set is a comparison of the wild-type strain with the GCN2-deleted strain in the absence of 3AT.
Distinct Splicing Regulation Is Observed in Response to Ethanol Toxicity
To determine whether splicing changes affecting the RPG transcripts are specific to the cellular response to amino acid starvation or reflect a more general response to unfavorable growth conditions, we examined the effects of exposure to toxic levels of ethanol. Figure 5A shows the kinetic response of wild-type cells to ethanol toxicity. Interestingly, in response to ethanol exposure, a small number of transcripts decrease in splicing efficiency (marked with a red bar in Figure 5A and highlighted in the top of Figure 5B). As seen in Figure 5C, these transcripts show no splicing defect in response to amino acid starvation. Notably, the splicing efficiency of a second small subset of transcripts actually increases in response to ethanol (marked with a green bar in Figure 5A and highlighted at the bottom of Figure 5B). These transcripts likewise showed no change in splicing efficiency in response to amino acid starvation. A full comparison of the responses to amino acid starvation and toxic ethanol shown in Figure 6 reveals their nonoverlapping nature, demonstrating that the splicing changes are specifically tailored to each of these particular environmental stresses.
Figure 5. Regulation of Pre-mRNA Splicing in Response to Ethanol Toxicity.
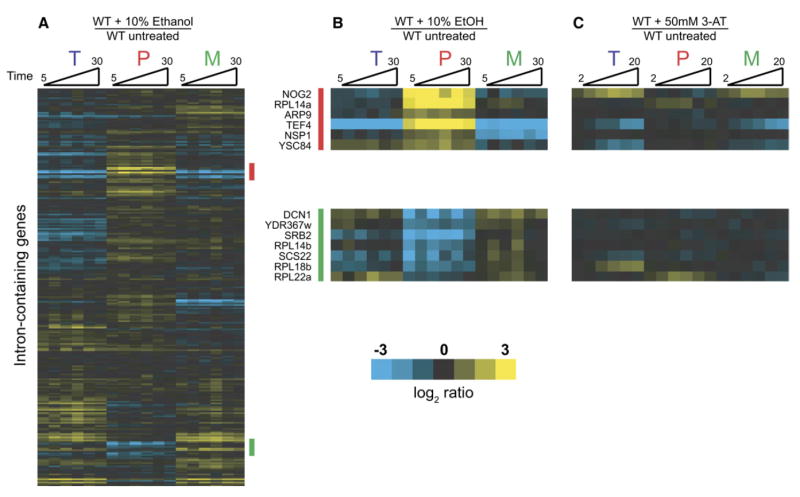
(A) Time-resolved splicing profiles resulting from comparisons of wild-type cells either treated with 10% ethanol or mock treated for 5, 10, 15, 20, 25, and 30 min.
(B) Genes whose splicing is downregulated (top panel, also highlighted with a red bar in [A]) or upregulated (bottom panel, also highlighted with a green bar in [A]).
(C) Behavior of genes highlighted in (B) in response to amino acid starvation.
Figure 6. Comparison of Splicing Responses to Amino Acid Starvation and Ethanol Toxicity.
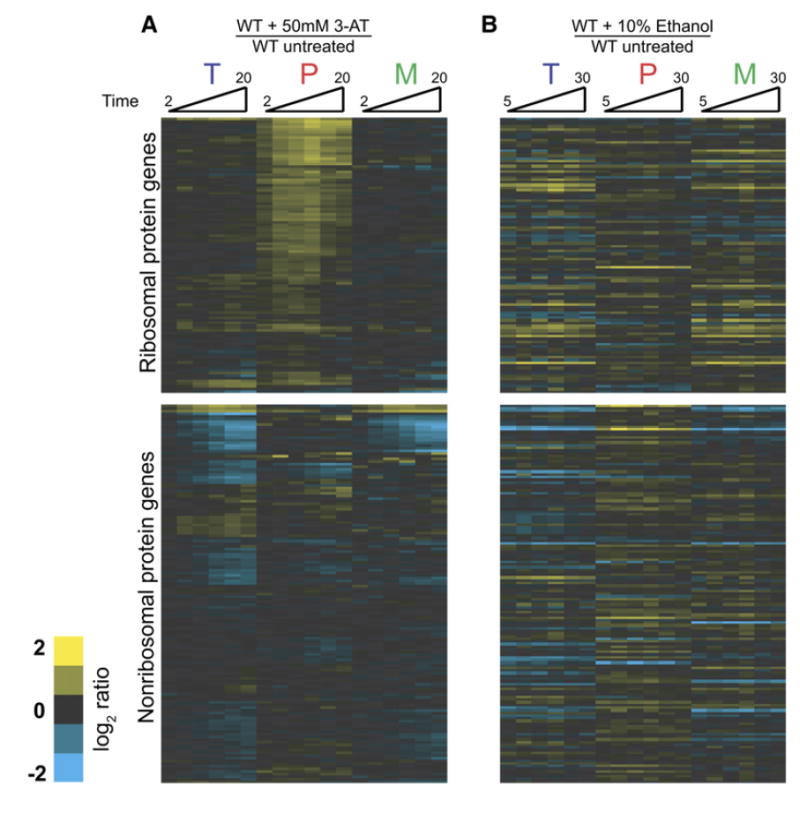
The order of genes is identical to that shown in Figure 2.
DISCUSSION
Here we have taken a microarray-based approach to examine the capacity of pre-mRNA splicing in the budding yeast, S. cerevisiae, to be regulated in response to changes in environment. In response to amino acid starvation, the splicing efficiency of nearly all RPG transcripts is rapidly downregulated. Interestingly, these changes in splicing efficiency, unlike previously described responses to amino acid starvation, are not dependent upon the activity of the Gcn2 kinase. Furthermore, this response appears specific to amino acid starvation, as a different environmental stress, exposure to toxic levels of ethanol, does not globally repress RPG splicing. Instead, ethanol toxicity induces a unique spliceosomal response wherein the splicing efficiency of a different set of transcripts is downregulated while the splicing efficiency of another group of transcripts is improved.
The splicing architecture present in yeast is simple relative to higher eukaryotes. Very few genes in yeast contain multiple introns, and yeast introns tend to conform to tight consensus sequences at their splice sites, precluding the process of alternative splicing so rampant in metazoans. As such, it has been unclear whether yeast has retained the capacity to extensively regulate the activity of this step in gene expression. However, our experiments demonstrate that the splicing efficiency of distinct sets of transcripts can be rapidly and specifically modulated in response to changing environmental conditions. We have recently reported that mutations in core spliceosomal components can lead to transcript-specific defects in splicing (Pleiss et al., 2007), demonstrating that the yeast splicing architecture exhibits the physical capacity to differentiate among transcripts. Importantly, the results of our current experiments suggest that there must exist in yeast mechanisms to differentiate splicing activity even among the many transcripts that contain strong consensus splice sites.
The Biology of Splicing Regulation
The splicing changes observed in response to amino acid starvation can be readily rationalized: under conditions in which translational resources are limiting, the spliceosome can rapidly downregulate the synthesis of new ribosomal components. Under normal growth conditions, wild-type yeast devote a significant fraction of their metabolic resources to the synthesis of new ribosomes. Many steps in this pathway are known to be tightly regulated, presumably reflecting the importance of optimizing the process of ribosome production (Warner, 1999). Our results suggest an explanation for the prevalence of introns in RPGs in that they may offer an additional level at which translational capacity can be coordinately regulated.
Moreover, the provocative result that the splicing of certain transcripts can be rapidly upregulated in response to ethanol toxicity suggests that not all yeast transcripts are processed by the spliceosome at maximal efficiency at all times. Indeed, upregulation of the splicing of these genes potentially allows for a rapid increase in protein expression without the need for de novo transcription. An analogy can be drawn with the unfolded protein response, in which induction of splicing of the Hac1 transcription factor, in this case by a nonspliceosomal mechanism, allows for a rapid response to the stress in the absence of new transcription (Sidrauski and Walter, 1997). Remarkably, one of the transcripts whose splicing is improved after ethanol exposure encodes Srb2, a nonessential component of the Mediator complex of transcription factors. Our results suggest that expression of this conserved coactivator of transcription can also be regulated at the level of splicing and that splicing can in turn act to regulate transcription.
Mechanisms of Splicing Regulation
While it is true that yeast splice sites generally adhere to a strict consensus, all previously documented cases of regulated splicing in yeast rely on the presence of suboptimal splice site sequences. For example, a nonconsensus 5′ splice site in the Rpl30 transcript is necessary to maintain a splicing-based autoregulatory feedback loop (Eng and Warner, 1991). Likewise, nonconsensus 5′ splice sites in the transcripts of MER2 and MER3 are necessary for their positive regulation during meiosis (Nandabalan et al., 1993). However, splice site sequences alone are insufficient to explain the transcript-specific changes we have observed during amino acid starvation, as no unique sequences have been identified within this group of transcripts (Figure S2). Another potential explanation is based on the observation that, as a class, RPG introns tend to be significantly larger in size than introns in other yeast genes: the median RPG intron is ~400 nucleotides in length, whereas the median non-RPG intron is only ~130 nucleotides. Nevertheless, size alone is also insufficient to explain the specificity, as the splicing of non-RPGs such as Act1, whose intron contains 308 nucleotides, is unaffected during amino acid starvation. Thus, other currently unknown cis-acting features must contribute to the observed specificity.
In higher eukaryotes, members of the large SR and hnRNP families of regulatory proteins are known to play major roles in regulating alternative splicing decisions. By comparison, the spectrum of yeast proteins belonging to these families is reduced, and it remains unknown whether these proteins play any role in regulating splicing activity in yeast. Instead, recent evidence suggests that “specificity” factors will include core components of the spliceosome itself. RNAi-mediated knockdowns in flies of several essential splicing factors have been shown to have differential effects on the splicing of alternative exons (Park et al., 2004). Likewise, our recent microarray analyses of point mutations in conserved, core splicing components (Pleiss et al., 2007) also revealed remarkably transcript-specific effects that were not readily accounted for by splice site sequences per se. In particular, we previously demonstrated that the spliceosome interacts with RPG transcripts in a fundamentally different way than it does with non-RPG transcripts. The similar behavior of RPG pre-mRNAs in response to perturbations in essential, core spliceosomal components may well underlie the behavior of these pre-mRNAs in response to amino acid starvation.
We previously proposed that the rate of cotranscriptional loading of spliceosomal components would be higher for RPG transcripts than for non-RPG transcripts (Pleiss et al., 2007). Consistent with this proposal, recent genome-wide chromatin immunoprecipitation experiments demonstrated a higher density of the U1 snRNP on RPGs than nonribosomal protein genes (Tardiff et al., 2006). Conceivably, modification of a common spliceosomal factor that affects the cotranscriptional engagement of the spliceosome could be responsible for the RPG-specific reduction of splicing efficiency seen in response to amino acid starvation. By comparison, a separate genome-wide experiment (Moore et al., 2006) showed that the transcript encoding Srb2 is one of a small number of intron-containing transcripts that fails to engage with the spliceosome cotranscriptionally. The improved splicing seen for this transcript in response to toxic ethanol may therefore also depend upon its ability to efficiently recruit active spliceosomes. For example, these data suggest that under normal growth conditions the Srb2 transcript is inefficiently engaged by the spliceosome but that under conditions of ethanol toxicity spliceosomal recruitment is increased, allowing for more efficient splicing. Further characterization of the behavior of this transcript will likely provide important insights into the general mechanism by which transcripts engage spliceosomes.
Connecting the Spliceosome to Cellular Environment
An important question for the future regards the mechanisms by which environmental signals are transduced to the splicing machinery. The unique molecular profiles observed in response to two unrelated physiological stresses suggest that at least two independent pathways connect the spliceosome with the cellular environment. The rapid onset of the molecular defects in response to amino acid starvation suggests that posttranslational modifications of spliceosomal components might likely be involved in this regulation. Interestingly, recent evidence suggests a potential role for ubiquitin in regulating the splicing cycle (Bellare et al., 2006). Alternatively, phosphorylation and methylation of regulatory proteins play an important role in regulating splicing in higher eukaryotes and would be obvious candidates in yeast as well (Shin and Manley, 2004). Nevertheless, the candidate most likely to control such a signaling pathway in response to amino acid starvation, the Gcn2 kinase, is not essential for the spliceosomal response. Rather, our results imply the existence of a previously uncharacterized signaling pathway linking amino acid starvation with transcript-specific changes in pre-mRNA splicing.
Implications
The findings presented here argue strongly that pre-mRNA splicing can play an important role in regulating gene expression even in a system that lacks the infrastructure necessary for alternative splicing. Further, these results argue against the notion that most transcripts interact equally with the spliceosome, suggesting instead that transcript identity is an important factor in determining splicing activity. Given the high level of conservation of the basic splicing machinery, we predict that the mechanisms utilized by yeast to regulate pre-mRNA splicing will also be important in higher eukaryotes, where they likely underlie or augment the regulation of alternative splice site choices.
EXPERIMENTAL PROCEDURES
Sample Collection and Preparation
Amino acid starvation experiments utilized either the wild-type strain (yMB1) or the Δgcn2 strain (yMB2). The strain yMB1 was constructed by repairing the HIS3 locus in the BY4742 strain (Brachmann et al., 1998) using standard methods. The strain yMB2 was constructed by disrupting the GCN2 locus of yMB1 with LEU2. A single master culture of these strains was grown at 30°C in 250 ml of minimal medium lacking histidine (Guthrie and Fink, 2002) until its optical density was between A600 = 0.5 and A600 = 0.7. An initial 15 ml sample was collected by filtration using Millipore HAWP0025 filters (http://www.millipore.com) prior to initiation of the time course. The filters were immediately frozen in N2(l). Experiments were initiated by splitting the master culture into two 100 ml aliquots and adding either 5 ml of water or 1 M 3AT. At the appropriate times, 15 ml of cells were collected.
For experiments examining ethanol toxicity, a single master culture of the strain BY4742 was grown at 30°C in 300 ml of YEPD (Guthrie and Fink, 2002) until its optical density was between A600 = 0.5 and A600 = 0.7. Samples were then collected in a 96-well format as previously described (Pleiss et al., 2007). Briefly, 180 μl of water or 100% ethanol was added to each well of a 96-well plate. A reverse time course was initiated by addition of 1.6 ml aliquots of culture to the appropriate wells at the appropriate times. At the completion of the time course, cells were collected by centrifugation at 5000 × g for 5 min.
For experiments examining inactivation of Prp8, master cultures of both the wild-type (yEJS1) and prp8-1 (yEJS17) strain were grown at 25°C in 100 ml of complete minimal medium until their optical densities were between A600 = 0.5 and A600 = 0.7. Cells were shifted from 25°C to 37°C, and 15 ml samples were collected by filtration (as above) at the indicated times.
For experiments examining the nonessential RNA turnover mutants, samples of each mutant strain (all derivatives of BY4741 [Brachmann et al., 1998]) and the wild-type strain (BY4742) were grown at 30°C in 50 ml of complete minimal medium until their optical densities were between A600 = 0.5 and A600 = 0.7. Cells were then collected by centrifugation at 3000 × g for 5 min.
Samples for microarray analysis were isolated and prepared as previously described (Pleiss et al., 2007). Briefly, 25 μg of total cellular RNA was collected from each sample and converted into cDNA for each individual microarray. Data presented as figures are derived from independent biological replicates, which were utilized to perform dye-flipped microarray replicates. Additional experiments examining distinct biological replicates have been performed and yield similar results. Microarray images were collected and analyzed as previously described (Pleiss et al., 2007).
Quantitative PCR
Total cellular RNA for quantitative RT-PCR experiments was treated with DNase I (Fermentas, http://www.fermentas.com) according to the manufacturer’s protocol prior to use. The cDNA was then produced as previously described (Pleiss et al., 2007). Quantitative PCR was then performed using an Opticon from MJ Research (http://www.mjresearch.com). Primer sets used in this study are shown in Table S1. Error bars in Figure 3 are the result of triplicate measurements from a single biological sample. Standard deviations were only slightly higher when comparing biological replicates. A series of 10-fold dilutions of genomic DNA covering a total range of 106 molecules was used to generate the standard curves. Genomic DNA was purified using a ZR Fungal DNA kit (Zymo Research) according to the manufacturer’s protocol.
Supplementary Material
Supplemental Data include two figures and two tables and can be found with this article online at http://www.molecule.org/cgi/content/full/27/6/928/DC1/.
Acknowledgments
We thank J. DeRisi, A. Carroll, and M. Ares for technical assistance with the microarrays; and J. Abelson, J. Steitz, A. Johnson, H. Madhani, M. McMahon, M. Inada, and members of the C.G. lab for helpful discussions and critical comments on the manuscript. This work was supported by funds from the Damon Runyon Cancer Research Foundation (J.A.P.), the National Institutes of Health (NIH) and the National Science Foundation (G.B.W.), the Howard Hughes Medical Institute (M.B.), and NIH grant GM21119 (C.G.). C.G. is an American Cancer Society Research Professor of Molecular Genetics.
Footnotes
Accession Numbers Microarray data are available under the series accession number GSE8817 at the Gene Expression Omnibus (GEO) repository at the National Center for Biotechnology Information (http://www.ncbi.nlm.nih.gov/geo).
References
- Bellare P, Kutach AK, Rines AK, Guthrie C, Sontheimer EJ. Ubiquitin binding by a variant Jab1/MPN domain in the essential pre-mRNA splicing factor Prp8p. RNA. 2006;12:292–302. doi: 10.1261/rna.2152306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Black DL. Protein diversity from alternative splicing: a challenge for bioinformatics and post-genome biology. Cell. 2000;103:367–370. doi: 10.1016/s0092-8674(00)00128-8. [DOI] [PubMed] [Google Scholar]
- Blencowe BJ. Alternative splicing: new insights from global analyses. Cell. 2006;126:37–47. doi: 10.1016/j.cell.2006.06.023. [DOI] [PubMed] [Google Scholar]
- Brachmann CB, Davies A, Cost GJ, Caputo E, Li J, Hieter P, Boeke JD. Designer deletion strains derived from Saccharomyces cerevisiae S288C: a useful set of strains and plasmids for PCR-mediated gene disruption and other applications. Yeast. 1998;14:115–132. doi: 10.1002/(SICI)1097-0061(19980130)14:2<115::AID-YEA204>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- Chen JC, Powers T. Coordinate regulation of multiple and distinct biosynthetic pathways by TOR and PKA kinases in S. cerevisiae. Curr Genet. 2006;49:281–293. doi: 10.1007/s00294-005-0055-9. [DOI] [PubMed] [Google Scholar]
- Cherkasova VA, Hinnebusch AG. Translational control by TOR and TAP42 through dephosphorylation of eIF2alpha kinase GCN2. Genes Dev. 2003;17:859–872. doi: 10.1101/gad.1069003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark TA, Sugnet CW, Ares M., Jr Genomewide analysis of mRNA processing in yeast using splicing-specific microarrays. Science. 2002;296:907–910. doi: 10.1126/science.1069415. [DOI] [PubMed] [Google Scholar]
- Davis CA, Grate L, Spingola M, Ares M., Jr Test of intron predictions reveals novel splice sites, alternatively spliced mRNAs and new introns in meiotically regulated genes of yeast. Nucleic Acids Res. 2000;28:1700–1706. doi: 10.1093/nar/28.8.1700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dever TE, Feng L, Wek RC, Cigan AM, Donahue TF, Hinnebusch AG. Phosphorylation of initiation factor 2 alpha by protein kinase GCN2 mediates gene-specific translational control of GCN4 in yeast. Cell. 1992;68:585–596. doi: 10.1016/0092-8674(92)90193-g. [DOI] [PubMed] [Google Scholar]
- Dong J, Qiu H, Garcia-Barrio M, Anderson J, Hinnebusch AG. Uncharged tRNA activates GCN2 by displacing the protein kinase moiety from a bipartite tRNA-binding domain. Mol Cell. 2000;6:269–279. doi: 10.1016/s1097-2765(00)00028-9. [DOI] [PubMed] [Google Scholar]
- Eng FJ, Warner JR. Structural basis for the regulation of splicing of a yeast messenger RNA. Cell. 1991;65:797–804. doi: 10.1016/0092-8674(91)90387-e. [DOI] [PubMed] [Google Scholar]
- Fillman C, Lykke-Andersen J. RNA decapping inside and outside of processing bodies. Curr Opin Cell Biol. 2005;17:326–331. doi: 10.1016/j.ceb.2005.04.002. [DOI] [PubMed] [Google Scholar]
- Frischmeyer PA, van Hoof A, O’Donnell K, Guerrerio AL, Parker R, Dietz HC. An mRNA surveillance mechanism that eliminates transcripts lacking termination codons. Science. 2002;295:2258–2261. doi: 10.1126/science.1067338. [DOI] [PubMed] [Google Scholar]
- Guthrie C, Fink GR. Guide to Yeast Genetics and Molecular and Cell Biology. Amsterdam: Academic Press; 2002. [Google Scholar]
- He F, Peltz SW, Donahue JL, Rosbash M, Jacobson A. Stabilization and ribosome association of unspliced pre-mRNAs in a yeast upf1 – mutant. Proc Natl Acad Sci USA. 1993;90:7034–7038. doi: 10.1073/pnas.90.15.7034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He F, Li X, Spatrick P, Casillo R, Dong S, Jacobson A. Genome-wide analysis of mRNAs regulated by the nonsense-mediated and 5′ to 3′ mRNA decay pathways in yeast. Mol Cell. 2003;12:1439–1452. doi: 10.1016/s1097-2765(03)00446-5. [DOI] [PubMed] [Google Scholar]
- Hinnebusch AG. Translational regulation of yeast GCN4. A window on factors that control initiator-trna binding to the ribosome. J Biol Chem. 1997;272:21661–21664. doi: 10.1074/jbc.272.35.21661. [DOI] [PubMed] [Google Scholar]
- Hinnebusch AG. Translational regulation of GCN4 and the general amino acid control of yeast. Annu Rev Microbiol. 2005;59:407–450. doi: 10.1146/annurev.micro.59.031805.133833. [DOI] [PubMed] [Google Scholar]
- Holstege FC, Jennings EG, Wyrick JJ, Lee TI, Hengartner CJ, Green MR, Golub TR, Lander ES, Young RA. Dissecting the regulatory circuitry of a eukaryotic genome. Cell. 1998;95:717–728. doi: 10.1016/s0092-8674(00)81641-4. [DOI] [PubMed] [Google Scholar]
- Houseley J, LaCava J, Tollervey D. RNA-quality control by the exosome. Nat Rev Mol Cell Biol. 2006;7:529–539. doi: 10.1038/nrm1964. [DOI] [PubMed] [Google Scholar]
- Juneau K, Palm C, Miranda M, Davis RW. High-density yeast-tiling array reveals previously undiscovered introns and extensive regulation of meiotic splicing. Proc Natl Acad Sci USA. 2007;104:1522–1527. doi: 10.1073/pnas.0610354104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim M, Krogan NJ, Vasiljeva L, Rando OJ, Nedea E, Greenblatt JF, Buratowski S. The yeast Rat1 exonuclease promotes transcription termination by RNA polymerase II. Nature. 2004;432:517–522. doi: 10.1038/nature03041. [DOI] [PubMed] [Google Scholar]
- Leeds P, Peltz SW, Jacobson A, Culbertson MR. The product of the yeast UPF1 gene is required for rapid turnover of mRNAs containing a premature translational termination codon. Genes Dev. 1991;5:2303–2314. doi: 10.1101/gad.5.12a.2303. [DOI] [PubMed] [Google Scholar]
- Lelivelt MJ, Culbertson MR. Yeast Upf proteins required for RNA surveillance affect global expression of the yeast transcriptome. Mol Cell Biol. 1999;19:6710–6719. doi: 10.1128/mcb.19.10.6710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis BP, Green RE, Brenner SE. Evidence for the widespread coupling of alternative splicing and nonsense-mediated mRNA decay in humans. Proc Natl Acad Sci USA. 2003;100:189–192. doi: 10.1073/pnas.0136770100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitrovich QM, Anderson P. Unproductively spliced ribosomal protein mRNAs are natural targets of mRNA surveillance in C. elegans. Genes Dev. 2000;14:2173–2184. doi: 10.1101/gad.819900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore MJ, Schwartzfarb EM, Silver PA, Yu MC. Differential recruitment of the splicing machinery during transcription predicts genome-wide patterns of mRNA splicing. Mol Cell. 2006;24:903–915. doi: 10.1016/j.molcel.2006.12.006. [DOI] [PubMed] [Google Scholar]
- Nandabalan K, Price L, Roeder GS. Mutations in U1 snRNA bypass the requirement for a cell type-specific RNA splicing factor. Cell. 1993;73:407–415. doi: 10.1016/0092-8674(93)90239-m. [DOI] [PubMed] [Google Scholar]
- Natarajan K, Meyer MR, Jackson BM, Slade D, Roberts C, Hinnebusch AG, Marton MJ. Transcriptional profiling shows that Gcn4p is a master regulator of gene expression during amino acid starvation in yeast. Mol Cell Biol. 2001;21:4347–4368. doi: 10.1128/MCB.21.13.4347-4368.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park JW, Parisky K, Celotto AM, Reenan RA, Graveley BR. Identification of alternative splicing regulators by RNA interference in Drosophila. Proc Natl Acad Sci USA. 2004;101:15974–15979. doi: 10.1073/pnas.0407004101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pleiss JA, Whitworth GB, Bergkessel M, Guthrie C. Transcript specificity in yeast pre-mRNA splicing revealed by mutations in core spliceosomal components. PLoS Biol. 2007;5:e90. doi: 10.1371/journal.pbio.0050090. 10.1371/journal.pbio.0050090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preker PJ, Guthrie C. Autoregulation of the mRNA export factor Yra1p requires inefficient splicing of its pre-mRNA. RNA. 2006;12:994–1006. doi: 10.1261/rna.6706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preker PJ, Kim KS, Guthrie C. Expression of the essential mRNA export factor Yra1p is autoregulated by a splicing-dependent mechanism. RNA. 2002;8:969–980. doi: 10.1017/s1355838202020046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin C, Manley JL. Cell signalling and the control of pre-mRNA splicing. Nat Rev Mol Cell Biol. 2004;5:727–738. doi: 10.1038/nrm1467. [DOI] [PubMed] [Google Scholar]
- Sidrauski C, Walter P. The transmembrane kinase Ire1p is a site-specific endonuclease that initiates mRNA splicing in the unfolded protein response. Cell. 1997;90:1031–1039. doi: 10.1016/s0092-8674(00)80369-4. [DOI] [PubMed] [Google Scholar]
- Spingola M, Grate L, Haussler D, Ares M., Jr Genome-wide bioinformatic and molecular analysis of introns in Saccharomyces cerevisiae. RNA. 1999;5:221–234. doi: 10.1017/s1355838299981682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tardiff DF, Lacadie SA, Rosbash M. A genome-wide analysis indicates that yeast pre-mRNA splicing is predominantly post-transcriptional. Mol Cell. 2006;24:917–929. doi: 10.1016/j.molcel.2006.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Hoof A, Frischmeyer PA, Dietz HC, Parker R. Exosome-mediated recognition and degradation of mRNAs lacking a termination codon. Science. 2002;295:2262–2264. doi: 10.1126/science.1067272. [DOI] [PubMed] [Google Scholar]
- Warner JR. The economics of ribosome biosynthesis in yeast. Trends Biochem Sci. 1999;24:437–440. doi: 10.1016/s0968-0004(99)01460-7. [DOI] [PubMed] [Google Scholar]
- Wyers F, Rougemaille M, Badis G, Rousselle JC, Dufour ME, Boulay J, Regnault B, Devaux F, Namane A, Seraphin B, et al. Cryptic pol II transcripts are degraded by a nuclear quality control pathway involving a new poly(A) polymerase. Cell. 2005;121:725–737. doi: 10.1016/j.cell.2005.04.030. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Data include two figures and two tables and can be found with this article online at http://www.molecule.org/cgi/content/full/27/6/928/DC1/.