

Abstract
Cytochrome c oxidase (COX) is a key mitochondrial enzyme that catalyzes electron transfer at the terminal stage of respiratory chain and is composed of multisubunits. We hypothesize that ethanol withdrawal (EW) impairs the activity of COX and estrogen deprivation exacerbates this problem. Five month-old ovariectomized rats with or without 17β-estradiol (E2) replacement received a control dextrin or a liquid ethanol diet (6.5%, five weeks). They were then sacrificed either during ethanol exposure or at 24 hours of EW (EW group). Mitochondria of the cerebellum and cortex were processed to measure the activities of total COX, COX subunit I, and IV. The effects of EW and E2 on the protein levels of these subunits were also assessed using an immunoblotting method. As compared to the control dextrin and ethanol exposure, EW decreased the activities of total COX, COX I, and COX IV. E2 treatment prevented the effects of EW on the activities of total COX and COX IV but not COX I. Neither EW nor E2 altered the protein levels of the subunits. These findings suggest that a counteracting relationship exists between the effects of EW and E2 on the activity of COX in a subunit specific manner.
Keywords: Cytochrome c oxidase, 17β-estradiol, Ethanol Withdrawal, Mitochondria
Mitochondria produce cellular energy (ATP) through a series of mitochondrial enzyme complexes. Electrons are transferred across the enzyme complexes and create the electrochemical gradient between the mitochondrial membranes. Subsequently, the electrochemical gradient provides force to generate ATP. COX is a terminal enzyme complex among the series of enzymes and plays a key role in the mitochondrial function. Therefore, damage to this enzyme can cause serious clinical consequences. Indeed, the decreased activity of COX has been found in a variety of neurodegenerative illnesses such as Parkinson′s disease and Alzheimer′s disease [1–3].
Accumulated evidence indicates that mitochondria are vulnerable to ethanol/EW toxicity. Reactive oxygen species produced during ethanol metabolism altered mitochondrial function in rodents [4, 5]. The mitochondrial membrane permeability and oxidation were dramatically increased during EW [6, 7], suggesting that ethanol and/or EW preferentially target mitochondria. However, most of the studies did not differentiate between ethanol exposure and EW. Thus, it is not clear whether the observed damage is due to ethanol toxicity or EW. The differentiation is important because the toxic effects of EW are not necessarily identical to those of ethanol per se and can cause more brain damage [8, 9]. In contrast to the deleterious effects of EW on mitochondria, estrogen appears to protect against mitochondrial integrity. Brain mitochondria in male rats produce over 80% more peroxides than those in females [10]. Furthermore, ovariectomy causes an increase in peroxide production by mitochondria in a manner prevented by E2 [10]. Given these findings, estrogen appears to play a role in reducing the oxidative burden in mitochondria [11].
The aim of this study is to elucidate how brain mitochondria respond to EW stress at the level of COX. COX is composed of 13 subunits with subunits I–III encoded by mitochondrial DNA and IV – XIII encoded by nuclear DNA [12]. The large number of subunits is a mystery because of no differences in spectral properties and proton-pumping activities between the 13-subunit enzyme [13, 14]. To gain better understanding of the effects of EW on this enzyme, we measured 1) the activities of total COX, COX I, and COX IV and 2) mRNA expression and protein levels of the COX subunits in the presence and absence of E2. We chose cerebellum and cortex, that are vulnerable to ethanol both in humans and animals [15–17].
Young-adult-female Sprague-Dawley rats (Charles River, Wilmington, MA) were ovariectomized under anesthesia and were subcutaneously implanted with Silastic pellets (30 mm-long, 1.57mm ID, 3.18mm OD) containing either E2 (4 mg/ml) or corn oil [19]. Pellets were replaced every three weeks. All housing and procedures were in accordance with the guidelines of the Institutional Care and Use Committee of the National Institute on Drug Abuse, National Institutes of Health (Institute of Laboratory Animal Resources [18] and were approved by the University of North Texas Health Science Center Animal Care and Use Committee.
Rat groups were 1) the non-ethanol diet (Dextrin/Oil), 2) the ethanol exposure (Ethanol/Oil), 3) the EW without (EW/Oil), or 4) with E2 replacement (EW/E2) groups. The Ethanol/Oil group was sacrificed at the end of the ethanol diet before EW. All other groups were sacrificed at 24 hours of EW. The ethanol dependence was induced by a liquid diet administration such that the amount of dextrin and ethanol was calculated in combination to adjust the concentration of ethanol to 6.5% w/v [20, 21]. A fresh diet was (100 ml) placed in each home cage daily for five weeks. At 24 hours of EW, physical signs of EW such as tremor, rigid tail curve, startle, and vocalization were evaluated by two experimenters who were not aware of a group identity [21]. Immediately thereafter, rats were deeply anesthetized with sodium pentobarbital (200 mg/kg, IP) and then sacrificed by decapitation. To extract proteins, brain tissue was rinsed to remove blood, hand ground in a glass homogenizer, and suspended in isolation buffer. The tissue was then centrifuged at 1330 × g. The pellets were resuspended and centrifuged at 3030 × g. The supernatants were pooled and a standard Biorad protein assay was performed to determine protein concentration.
The activity of total COX was measured spectrophotometrically [22]. Briefly, reduced cytochrome c was prepared by mixing cytochrome c and ascorbic acid in potassium phosphate buffer. The oxidation of cytochrome c was recorded at 550 nm every five minutes for 20 measurements. The activities of COX subunits were measured according to a manufacturer′s instruction (MitoSciences, Eugene, Oregon). A 96 well plate was blocked with dry-milk solution. One hour later, isolated mitochondrial samples were added to each well and incubated for two hours. The subunit specific COX antibody and KH2PO4 were added to each well. Absorbance of each well was measured at 550 nm every five minutes for 20 measurements.
To conduct immunoblotting with monoclonal antibodies (Abcam, Cambridge, MA) for subunits (I and IV), protein samples were separated on SDS-polyacrylamide gel (12%) and transferred to a nitrocellulose membrane. Membranes were blocked by incubation with dry milk in phosphate-buffered saline and Tween 20 (PBST), followed by overnight incubation at 4°C with the primary antibody in blocking buffer. The membrane was then incubated with the horseradish-peroxidase-conjugated secondary antibody, followed by wash in PBST. The membrane was then incubated and developed with an enhanced chemiluminescent kit (Pierce, Rockford, IL).
To extract RNA, the homogenized tissue was centrifuged and lysed in TriZol reagent (Life Technologies, Gaithersburg, MD). The RNA containing aqueous layer was separated by chloroform and incubation on ice for five minutes followed by centrifugation at 12,000 × g. Total RNA was precipitated from the resulting aqueous phase by mixing with an equal volume of isopropyl alcohol, followed by incubation on ice for 15 minutes and centrifugation at 12,000 × g. The precipitated total RNA was resuspended in 70 μl of water and stored at −80°C until assayed.
To make cDNA using reagents from Promega (Madison, WI), total cellular RNA was incubated with random primers at 85°C. RNasin, reverse transcriptase buffer, deoxyribonucleotides, and avian myeloblastosis virus reverse transcriptase were added to the reaction and were incubated at 42°C and then at 94°C. cDNA was stored at −20°C until assayed. The primer sequences for COX I and COX IV were found using Primer 3 program available from MIT as follow; COX I sense primer: 5′GGAGCAGTATTCGCCATCAT3′, COX I antisense primer: 5′CGGCCGTAAGTGAGATGAAT3′, COX IV sense primer: 5′ACTACCCCTTGCCTGATGTG3′, COX IV antisense primer: 5′ACTCATTGGTGCCCTTGTTC3′. Negative controls containing all the reagents except the cDNA were included for each primer pair. Programmable temperature cycling (Mastercycler-Eppendorf-Scientific, Westbury, NY) was performed with the following cycle profile: 1) denaturation at 94°C for two minutes and then at 92°C for two minutes, 2) 40 cycles of annealing at 60°C for 30 seconds and then at 72°C for 90 seconds, and 3) denaturation at 92°C for 45 seconds. Horizontal 1.5% agarose (Omnipure agarose, EMD) gel electrophoresis was performed using PCR reaction product and loading dye per lane with gel run in a Mini-Sub cell electrophoresis (Bio-Rad, Hercules, CA). A 100-base pair DNA ladder (Promega, Madison, WI) was used for molecular size standards [23].
All activity data were expressed as the mean ± S.E.M. A one-way ANOVA followed by Tukey post hoc was conducted to determine the main effects and group differences. All data were from at least two separate determinations using the tissues collected from the same 5 or 6 rats for the COX activity and three rats for the PCR and Western blot.
All ethanol withdrawn rats exhibited EW signs. Dextrin rats and ethanol exposure rats did not show any EW signs [9, 24].
Figure 1A illustrates that EW more than two fold decreased the activity (nM/min/mg protein) of total COX (p < 0.01) in both cerebellum and cortex; whereas, ethanol exposure per se (Ethanol/Oil) did not significantly decrease the activity compared to the dextrin case (Dextrin/Oil). Figure 1B illustrates that the activity of total COX did not significantly differ between the Dextrin/Oil and EW/E2 groups, indicating that E2 prevented the suppressing effects of EW on the total COX activity in both brain areas.
Figure 1.
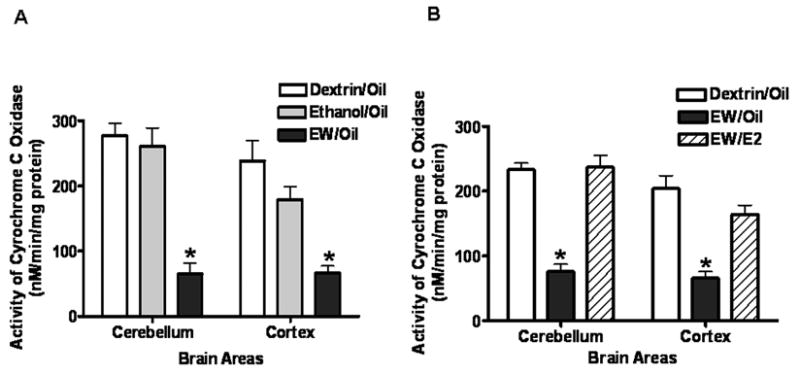
Figure 1A. Effects of EW on the activity of total COX. Ovariectomized rats implanted with oil pellets received an ethanol diet (6.5%, 5 weeks) and then sacrificed either during ethanol exposure or at 24 hours of EW. The COX activity during EW was significantly lower than those during control dextrin and ethanol exposure in both the cerebellum and cortex. N = 5 or 6 animals/group. *(p < 0.01) indicates a difference from the Dextrin/Oil group.
Figure 1B The effects of E2 on the activity of total COX in ethanol withdrawn rats. Ovariectomized rats implanted with oil or E2 pellets received a dextrin or an ethanol diet (6.5%, 5 weeks) and then sacrificed at 24 hours of EW. EW decreased the activity of total COX in a manner protected by E2 treatment. N = 5 or 6 animals/group. *(p < 0.01) indicates a difference from the Dextrin/Oil or the EW/E2 group.
Figure 2 illustrates that EW decreased the activity (nM/min/mg protein) of COX I (p < 0.01), and COX IV (p < 0.01) in both cerebellum and cortex, but E2 protection was observed only for COX IV (p < 0.05).
Figure 2.
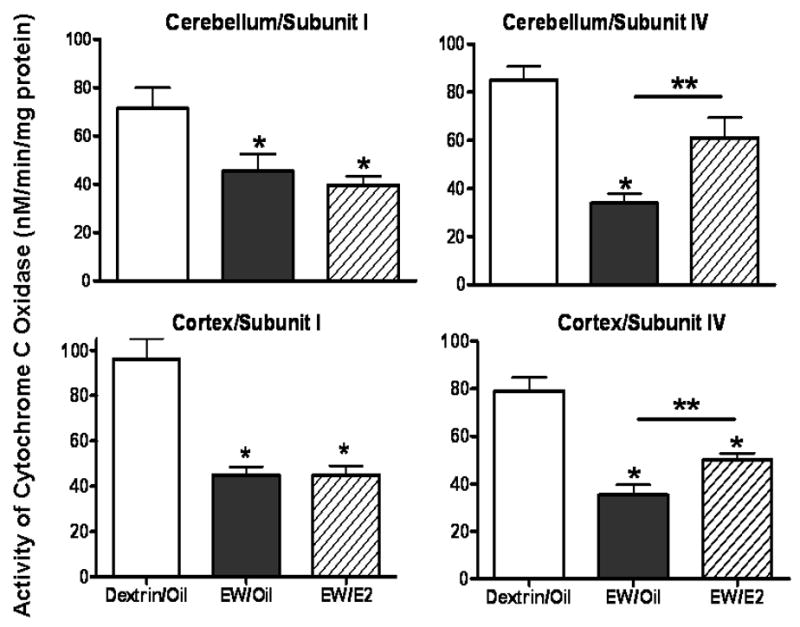
Effects of EW and E2 on the activity of COX I and IV. Ovariectomized rats implanted with oil or E2 pellets received a dextrin or an ethanol diet (6.5%, 5 weeks) and then sacrificed at 24 hours of EW. EW decreased the activity of the subunit I and the subunit IV of COX in both areas but E2 protection occurred only for COX IV. N = 5 or 6 animals/group. *(p < 0.01) indicates a difference from the Dextrin/Oil group. **(p < 0.05) indicates a difference between EW/Oil and EW/E2 groups.
Figure 3 illustrates mRNA (A) and protein (B) signals for cerebella COX IV that were detected at 188 base pairs and 17 KDa, respectively. When measured by Scion Image (NIH) and normalized by the β-actin signals, the protein densities of the COX IV, relative to the Dextrin/Oil group in the cerebellum were 1.07 ± 0.03 in the Ethanol/Oil, 1 ± 0.02 in the EW/Oil, and 1.1 ± 0.01 in the EW/E2 groups (p = 0.08). The corresponding values for the cortex were 1.01 ± 0.02, 1.04 ± 0.01, and 0.98 ± 0.01(p = 0.07). There was no significant difference in the protein levels of COX IV between the treatment groups. Likewise, no difference in the protein levels was observed in the COX I in the cerebellum or the cortex (data not shown).
Figure 3.
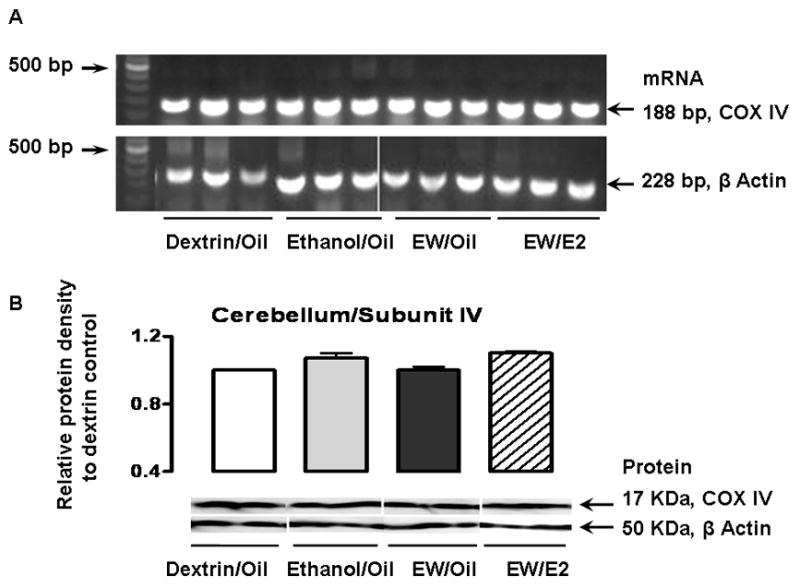
Effects of EW and E2 on the expression of mRNA or proteins of COX IV in the cerebellum and cortex. Ovariectomized rats implanted with oil or E2 pellets received a dextrin or an ethanol diet (6.5%, 5 weeks). Cerebelli and cortex were collected either at the end of ethanol exposure (Ethanol/Oil) or at 24 hours of EW (EW/Oil and EW/E2). In Figure 3A, mRNA of the cerebella COX IV was detected at 188 base pairs. 500 base pairs are indicated as a reference in the left hand side of the panels. In each treatment group, three representative signals from different animals are illustrated. Figure 3B illustrates the density of the COX IV protein that showed no difference between treatment groups either in the cerebellum or in the cortex. In each treatment group, two representative signals from different animals are illustrated.
We demonstrated that abrupt termination of chronic ethanol diet decreased the activity of a key mitochondrial enzyme COX and that a loss of estrogen contributed to this problem. The estrogen protection appeared to be subunit specific because estrogen prevented the effects of EW on the subunit IV but not on the subunit I.
A growing body of evidence suggests that damage to mitochondria is associated with neurodegerative disorders. Mitochondria isolated from Alzheimer′s platelets showed a 15% decrease in COX activity [25] and a cellular model of aging showed COX deficiency associated with oxidant generation [26]. Relevant to the current study, ethanol exposure oxidatively modified COX, resulting in a reduced enzyme activity [22] . In addition, COX activity was decreased by the age/EW combination in male rats but not by age per se, suggesting that COX is particularly vulnerable to EW toxicity [27]. In contrast to our findings, COX activity was not altered in the brain tissues of the ethanol-fed male rats [28]. Similarly, no difference in COX profiles was observed between EW seizure-prone and seizure-resistant mice [29]. However, there are important differences in experimental conditions between these studies and ours. While Marin-Garcia et al. [28] used whole brain tissues of male rats during ethanol exposure conditions; we used discrete brain areas of female rats under the EW condition. Likewise, Buckman and Meshul [29] used hippocampal tissues of male mice, whereas we used the cerebellum and cortex of female rats. These studies suggest that the activity of COX may depend on brain region, gender, species, and/or ethanol regimen. In fact, when measured during ethanol exposure, total COX activity was not different between dextrin-rats and ethanol exposure-rats in either cerebellum or cortex in our study. Our model was designed to mimic alcoholism of which EW causes a more serious damage to brain than ethanol exposure per se. Our results in this model indeed suggest that the abrupt termination of chronic ethanol diet can be more dangerous to the integrity of COX than ethanol exposure per se.
COX is composed of 13 subunits and the exact function of each subunit is still under investigation. We selected subunits I and IV because these two subunits are distinctively different from each other. The subunit I is encoded by mitochondrial DNA [12], whereas the subunit IV is encoded by nuclear DNA. In addition, the two subunits interact with each other such that the subunit IV stabilizes the copper center in the subunit I [30, 31, 32]. A previous study reported that the COX IV formed an adduct with 4-hydroxynonenal (product of lipid peroxidation) after ethanol consumption in rats [33], suggesting a vulnerability of this subunit to ethanol stress. It is interesting that EW decreased the activities of both subunits (I and IV) but E2 protection occurred only for the subunit IV (Figure 2). Despite that E2 failed to protect the subunit I, E2 protected total COX activity. This observation raises 6 a possibility that E2 may act upon only certain COX subunits such as COX IV which may play a key role to regulate total activity. It is also possible that there may be other subunits important for E2 action. We initially thought that E2 protects only nuclear-DNA coded subunits not mitochondrial-DNA encoded subunits. However, this possibility less likely accounts for our results because we have observed that E2 did not significantly alter the activity of the subunit V that is also the nuclear-DNA encoded subunit (data not shown). Had the origin of encoding DNA mediated the different effects of E2, the effects of E2 on the protein levels of the subunits would have been also different. Our results argue against this possibility because we found no change in the protein levels of the subunits. Because the employed PCR assay in our study is only semi-quantitative, the effects of EW or EW/E2 on mRNA levels of the COX subunits remain to be determined. Nonetheless, the lack of changes in protein levels of the subunits suggest that the EW-induced decrease in and estrogen protection of COX activity might have occurred posttranslationally. A previous study reported that E2 increased the mRNA levels of COX II and III [34] but the study used more than a ten-fold higher dose of E2 in pituitary tumor cell lines whereas we used physiological concentrations of E2 in rats. Nevertheless, the report raises a possibility that pharmacological doses of E2 may influence a translational process of these proteins. Alternative explanation for estrogen protection is inferred from lipophilicity of estrogen and the structural configuration of these subunits. The subunit I is located in the center of the enzyme complex, thus, unlikely has direct contact with mitochondrial membranes whereas the subunit IV is a transmembrane helix that crosses mitochondrial inner membrane [12]. The lipophilicity of E2 ensures that at all estrogen concentrations, the vast majority of estrogen is associated with lipid membranes. Therefore, we speculate that E2 intercalates into mitochondrial membranes, assesses to the transmembraneous IV, and prevents the subunit IV from being modified by EW stimulus.
Although our data do not give information about whether EW suppresses selective subunits or globally suppresses the COX activity, it seems clear that EW-induced suppression of the COX activity does not alter protein synthesis. There are a few possible mechanisms by which EW decreases the activity of COX. Upon removal of chronic ethanol, inhibitory neurotransmission is also removed, which consequently activates excitotoxic stimulation such as glutamate and intracellular Ca2+. Subsequently, the excitotoxic stimuli can generate free radicals [35] that can modify lysine or histidine residues of COX.
In conclusion, our data suggest a counteracting relationship between EW and estrogen at the levels of COX, which may not alter protein synthesis. Identifying precise molecular mechanisms underlying subunit specific protection of estrogen can contribute to a therapeutic/research strategy for female alcoholism undergoing estrogen deficiency.
Acknowledgments
This work was supported by NIH/NIAAA AA013864 and AA015982. We wish to thank to Andrew Wilson for his technical assistance and Scott Coleman for his editorial support for this manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Beal MF. Does impairment of energy metabolism result in excitotoxic neuronal death in neurodegenerative illnesses? Ann Neurol. 1992;31:119–30. doi: 10.1002/ana.410310202. [DOI] [PubMed] [Google Scholar]
- 2.Davis RE, Miller S, Herrnstadt C, Ghosh SS, Fahy E, Shinobu LA, Galasko D, Thal LJ, Beal MF, Howell N, Parker WD., Jr Mutations in mitochondrial cytochrome c oxidase genes segregate with late-onset Alzheimer disease. Proc Natl Acad Sci U S A. 1997;94:4526–31. doi: 10.1073/pnas.94.9.4526. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 3.Kish SJ, Shannak K, Rajput A, Deck O, Hornykiewicz JH. Aging produces a specific pattern of striatal dopamine loss: implications for the etiology of idiopathic Parkinson's disease. J Neurochem. 1992;58:642–8. doi: 10.1111/j.1471-4159.1992.tb09766.x. [DOI] [PubMed] [Google Scholar]
- 4.Minana JB, Gomez-Cambronero L, Lloret A, Pallardo FV, Del Olmo J, Escudero A, Rodrigo JM, Pelliin A, Vina JR, Vina J, Sastre J. Mitochondrial oxidative stress and CD95 ligand: a dual mechanism for hepatocyte apoptosis in chronic alcoholism. Hepatology. 2002;35:1205–14. doi: 10.1053/jhep.2002.32969. [DOI] [PubMed] [Google Scholar]
- 5.Mansouri A, Demeilliers C, Amsellem S, Pessayre D, Fromenty B. Acute ethanol administration oxidatively damages and depletes mitochondrial dna in mouse liver, brain, heart, and skeletal muscles: protective effects of antioxidants. J Pharmacol Exp Ther. 2001;298:737–43. [PubMed] [Google Scholar]
- 6.French SW, Todoroff T. Effect of chronic ethanol ingestion and withdrawal on brain mitochondria. Res Commun Chem Pathol Pharmacol. 1971;2:206–15. [PubMed] [Google Scholar]
- 7.Hosein EA, Lee H, Hofmann I. The influence of chronic ethanol feeding to rats on liver mitochondrial membrane structure and function. Can J Biochem. 1980;58:1147–55. doi: 10.1139/o80-154. [DOI] [PubMed] [Google Scholar]
- 8.Phillips SC, Cragg BG. Chronic consumption of alcohol by adult mice: effect on hippocampal cells and synapses. Exp Neurol. 1983;80:218–26. doi: 10.1016/0014-4886(83)90018-3. [DOI] [PubMed] [Google Scholar]
- 9.Jung ME, Rewal M, Perez E, Wen Y, Simpkins JW. Estrogen protects against brain lipid peroxidation in ethanol-withdrawn rats. Pharmacol Biochem Behav. 2004;79:573–86. doi: 10.1016/j.pbb.2004.09.007. [DOI] [PubMed] [Google Scholar]
- 10.Borras C, Sastre J, Garcia-Sala D, Lloret A, Pallardo FV, Vina J. Mitochondria from females exhibit higher antioxidant gene expression and lower oxidative damage than males. Free Radic Biol Med. 2003;34:546–52. doi: 10.1016/s0891-5849(02)01356-4. [DOI] [PubMed] [Google Scholar]
- 11.Chen M, Zsengeller Z, Xiao CY, Szabo C. Mitochondrial-to-nuclear translocation of apoptosis-inducing factor in cardiac myocytes during oxidant stress: potential role of poly(ADP-ribose) polymerase-1. Cardiovasc Res. 2004;63:682–8. doi: 10.1016/j.cardiores.2004.04.018. [DOI] [PubMed] [Google Scholar]
- 12.Lonergan KM, Gray MW. Expression of a continuous open reading frame encoding subunits 1 and 2 of cytochrome c oxidase in the mitochondrial DNA of Acanthamoeba castellanii. J Mol Biol. 1996;257:1019–30. doi: 10.1006/jmbi.1996.0220. [DOI] [PubMed] [Google Scholar]
- 13.Pardhasaradhi K, Ludwig B, Hendler RW. Potentiometric and spectral studies with the two-subunit cytochrome aa3 from Paracoccus denitrificans. Comparison with the 13-subunit beef heart enzyme. Biophys J. 1991;60:408–14. doi: 10.1016/S0006-3495(91)82066-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hendler RW, Pardhasaradhi K, Reynafarje B, Ludwig B. Comparison of energy-transducing capabilities of the two- and three-subunit cytochromes aa3 from Paracoccus denitrificans and the 13-subunit beef heart enzyme. Biophys J. 1991;60:415–23. doi: 10.1016/S0006-3495(91)82067-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Forster MJ, Dubey A, Dawson KM, Stutts WA, Lal H, Sohal RS. Age-related losses of cognitive function and motor skills in mice are associated with oxidative protein damage in the brain. Proc Natl Acad Sci U S A. 1996;93:4765–9. doi: 10.1073/pnas.93.10.4765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Paula-Barbosa MM, Brandao F, Andrade JP, Madeira MD, Zimmer J, Cadete-Leite A. Intracerebral grafting impedes hippocampal cell loss during withdrawal after long-term alcohol consumption in rats. Alcohol Alcohol. 1991;26:177–90. doi: 10.1093/oxfordjournals.alcalc.a045099. [DOI] [PubMed] [Google Scholar]
- 17.Pentney RJ, Alletto JJ, Acara MA, Dlugos CA, Fiel RJ. Small animal magnetic resonance imaging: a means of studying the development of structural pathologies in the rat brain. Alcohol Clin Exp Res. 1993;17:1301–8. doi: 10.1111/j.1530-0277.1993.tb05245.x. [DOI] [PubMed] [Google Scholar]
- 18.I.o.L.A.R. Guide for the Care and Use of Laboratory Animals. Washington, DC: National Academy Press; 1996. [Google Scholar]
- 19.Jung ME, Yang SH, Brun-Zinkernagel AM, Simpkins JW. Estradiol protects against cerebellar damage and motor deficit in ethanol-withdrawn rats. Alcohol. 2002;26:83–93. doi: 10.1016/s0741-8329(01)00199-9. [DOI] [PubMed] [Google Scholar]
- 20.Dodd H, Shorey-Kutschke RL. Ethanol lowers heart carnitine in the methionine and choline deficient rat. Alcohol. 1987;4:395–9. doi: 10.1016/0741-8329(87)90073-5. [DOI] [PubMed] [Google Scholar]
- 21.Lal H, Harris CM, Benjamin D, Springfield AC, Bhadra S, Emmett-Oglesby MW. Characterization of a pentylenetetrazol-like interoceptive stimulus produced by ethanol withdrawal. J Pharmacol Exp Ther. 1988;247:508–18. [PubMed] [Google Scholar]
- 22.Chen J, Robinson NC, Schenker S, Frosto TA, Henderson GI. Formation of 4-hydroxynonenal adducts with cytochrome c oxidase in rats following short-term ethanol intake. Hepatology. 1999;29:1792–8. doi: 10.1002/hep.510290611. [DOI] [PubMed] [Google Scholar]
- 23.Agarwal N, Mehta K. Possible involvement of Bcl-2 pathway in retinoid × receptor alpha-induced apoptosis of HL-60 cells. Biochem Biophys Res Commun. 1997;230:251–3. doi: 10.1006/bbrc.1996.5937. [DOI] [PubMed] [Google Scholar]
- 24.Jung ME, Jacobs S, Rewal M, Wilson A, Simpkins JW. Estradiol protects against alteration of protein kinase C(epsilon) in a binge model of ethanol dependence and withdrawal. Eur J Pharmacol. 2005;515:62–72. doi: 10.1016/j.ejphar.2005.03.038. [DOI] [PubMed] [Google Scholar]
- 25.Cardoso SM, Proenca MT, Santos S, Santana I, Oliveira CR. Cytochrome c oxidase is decreased in Alzheimer's disease platelets. Neurobiol Aging. 2004;25:105–10. doi: 10.1016/s0197-4580(03)00033-2. [DOI] [PubMed] [Google Scholar]
- 26.Xin MG, Zhang J, Block ER, Patel JM. Senescence-enhanced oxidative stress is associated with deficiency of mitochondrial cytochrome c oxidase in vascular endothelial cells. Mech Ageing Dev. 2003;124:911–9. doi: 10.1016/s0047-6374(03)00163-5. [DOI] [PubMed] [Google Scholar]
- 27.Jaatinen P, Riikonen J, Riihioja P, Kajander O, Hervonen A. Interaction of aging and intermittent ethanol exposure on brain cytochrome c oxidase activity levels. Alcohol. 2003;29:91–100. doi: 10.1016/s0741-8329(03)00002-8. [DOI] [PubMed] [Google Scholar]
- 28.Marin-Garcia J, Ananthakrishnan R, Goldenthal MJ. Heart mitochondria response to alcohol is different than brain and liver. Alcohol Clin Exp Res. 1995;19:1463–6. doi: 10.1111/j.1530-0277.1995.tb01008.x. [DOI] [PubMed] [Google Scholar]
- 29.Buckman JF, Meshul CK. Glial differences between naive withdrawal seizure-prone and -resistant mice. Alcohol Clin Exp Res. 1999;23:1905–13. [PubMed] [Google Scholar]
- 30.Iwata S. Structure and function of bacterial cytochrome c oxidase. J Biochem (Tokyo) 1998;123:369–75. doi: 10.1093/oxfordjournals.jbchem.a021946. [DOI] [PubMed] [Google Scholar]
- 31.Babcock GT, Wikstrom M. Oxygen activation and the conservation of energy in cell respiration. Nature. 1992;356:301–9. doi: 10.1038/356301a0. [DOI] [PubMed] [Google Scholar]
- 32.Shapleigh JP, Hosler JP, Tecklenburg MM, Kim Y, Babcock GT, Gennis RB, Ferguson-Miller S. Definition of the catalytic site of cytochrome c oxidase: specific ligands of heme a and the heme a3-CuB center. Proc Natl Acad Sci U S A. 1992;89:4786–90. doi: 10.1073/pnas.89.11.4786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen J, Petersen DR, Schenker S, Henderson GI. Formation of malondialdehyde adducts in livers of rats exposed to ethanol: role in ethanol-mediated inhibition of cytochrome c oxidase. Alcohol Clin Exp Res. 2000;24:544–52. [PubMed] [Google Scholar]
- 34.Van Itallie CM, Dannies PS. Estrogen induces accumulation of the mitochondrial ribonucleic acid for subunit II of cytochrome oxidase in pituitary tumor cells. Mol Endocrinol. 1988;2:332–7. doi: 10.1210/mend-2-4-332. [DOI] [PubMed] [Google Scholar]
- 35.Tsai GE, Ragan P, Chang R, Chen S, Linnoila VM, Coyle JT. Increased glutamatergic neurotransmission and oxidative stress after alcohol withdrawal. Am J Psychiatry. 1998;155:726–32. doi: 10.1176/ajp.155.6.726. [DOI] [PubMed] [Google Scholar]