

Abstract
SecA, a 102-kDa hydrophilic protein, couples the energy of ATP binding to the translocation of preprotein across the bacterial inner membrane. SecA function and topology were studied with metabolically labeled [35S]SecA and with inner membrane vesicles from cells that overexpressed SecYEGDFyajC, the integral domain of preprotein translocase. During translocation in the presence of ATP and preprotein, a 65-kDa N-terminal domain of SecA is protected from proteolytic digestion through insertion into the membrane, as previously reported for a 30-kDa C-terminal domain [Economou, A. & Wickner, W. (1994) Cell 78, 835–843]. Insertion of both domains occurs at saturable SecYEGDFyajC sites and is rapidly followed by deinsertion. SecA also associates nonsaturably and unproductively with lipid. In the presence of ATP, yet without involvement of preprotein or SecYEG, lipid-bound SecA forms domains that are protease-resistant and that remain so even upon subsequent membrane disruption. Unlike the [35S]SecA that inserts into the membrane at SecYEGDFyajC as it promotes preprotein translocation, lipid-associated [35S]SecA does not chase from its protease-resistant state upon the addition of excess SecA. The finding that two domains of SecA (which together represent most regions of the polypeptide chain) cycle into the membrane during preprotein translocation, as well as the distinction between the membrane association of SecA at translocation sites of SecYEGDFyajC and at nonproductive lipid sites, are fundamental to the study of the role of SecA in preprotein movement.
Keywords: translocase, Escherichia coli, preprotein
Pathways of protein targeting and translocation into noncytoplasmic compartments have been examined in various eukaryotic organelles, including the mitochondria, nucleus, and endoplasmic reticulum, as well as in bacteria (1). With their unparalleled ease of combining biochemistry and genetics, bacteria offer unique advantages for addressing questions of protein translocation (2–5). In Escherichia coli, most protein translocation across the inner membrane occurs posttranslationally, catalyzed by a multicomponent secretory apparatus consisting of the cytosolic, export-specific SecB chaperone, the ATPase protein SecA, and the membrane-embedded heterotrimeric SecYEG complex (6–9). Recent studies have shown that SecDFyajC can be isolated with SecYEG as part of a larger membrane-embedded complex, termed SecYEGDFyajC (10). SecA plays a pivotal role in bacterial protein export by interacting with most, if not all, of the components involved in translocation: SecA recognizes both the leader sequence and as yet unidentified motifs in the mature domain of the preprotein (9, 11, 12). SecA interacts with SecB, thereby assisting with protein targeting (8). SecA also promotes functional interactions between preprotein and translocation sites in the inner membrane through its affinity for SecYEG and the acidic phospholipids of the membrane (8, 9). Translocation of 20–30 residues of the preprotein across the membrane occurs when SecA, in contact with a preprotein, SecYEG, and acidic phospholipids, binds ATP. Hydrolysis of the bound ATP then releases the preprotein from SecA, allowing the electrochemical proton gradient (proton motive force) to drive further translocation. Translocating proteins undergo many such cycles of SecA binding, ATP binding to SecA, limited translocation, ATP hydrolysis, and preprotein release from SecA, followed by proton motive force-driven translocation (13, 14).
Many aspects of how SecA couples the energy of ATP binding to the translocation of preprotein across the bacterial inner membrane remain unclear. In the presence of SecYEG, preprotein, and bound ATP, a 30-kDa domain of SecA inserts into the plane of the membrane where it is protected from proteases added to the cytoplasmic membrane surface (15) as well as to photoactivatable probes dissolved in the lipid phase (16). Cycles of SecA membrane insertion/deinsertion may be responsible for the forward movement of a translocating polypeptide chain across the membrane. Indeed, cross-linking techniques have shown that the preprotein crosses in association with SecA throughout its membrane passage (17). We now report that an N-terminal 65-kDa domain of the 102-kDa SecA protein is protected from proteolytic digestion under the same translocation conditions required for protease protection of a C-terminal 30-kDa domain. Furthermore, both SecA domains participate in membrane insertion/deinsertion cycles at SecYEG sites. Since the high-affinity nucleotide-binding domain (18, 19) as well as a proposed site of preprotein binding (12) are contained within this N-terminal domain of SecA, conformational changes of this domain are likely to be of central importance to preprotein movement.
MATERIALS AND METHODS
Materials.
SecA (20), SecB (6), and proOmpA (21) were prepared as described. Adenylyl-imidophosphate, ampicillin, ATP, 2-mercaptoethanol, chymotrypsin, DTT, soybean trypsin inhibitor, and trypsin were from Sigma; creatine kinase, creatine phosphate, isopropylthio-β-d-galactopyranoside, proteinase K, and Staphylococcus aureus V8 were from Boehringher Mannheim; P11 cellulose phosphate was from Whatman; and [35S]Express protein-labeling mixture and [125I]Na were from Amersham. [125I]SecA (16) was prepared as described.
Cell Growth, [35S]Metabolic Labeling, and Purification of [35S]SecA.
The E. coli SecA overproducing strain BL21 (λDE3)/pT7-secA (22) was grown with aeration at 37°C in 100 ml of M9 medium (23), 1 μg/ml thiamine, 0.2% glucose, 0.5% amino acids (lacking methionine and cysteine), and 50 μg/ml ampicillin (20). At OD600 = 0.5 the cells were induced with 0.5 mM isopropylthio-β-d-galactopyranoside for 30 min, after which 2 mCi (1 Ci = 37 GBq) of [35S]Express protein-labeling mixture were added. Growth was continued for an additional 60 min. The cells were then harvested (3,000 × g at 4°C for 5 min), resuspended in 1 ml of 10% sucrose/50 mM Tris⋅HCl (pH 7.5), and frozen in liquid N2.
Purification of [35S]SecA was based on the procedure of Cunningham et al. (20). Thawed cells (500 μl) were treated with lysozyme (5 μl, 8 mg/ml) for 30 min on ice followed by 3 min at 37°C without shear. The suspension was centrifuged (30,000 rpm for 20 min at 4°C in a TL 120.2 rotor; Beckman Optima TLX ultracentrifuge), and the supernatant was applied to a P11 cellulose phosphate column (0.7 × 2 cm), equilibrated in 10 mM sodium phosphate, pH 6.5. After washing with 20 ml of this buffer, [35S]SecA was eluted with a 20-ml linear gradient of 0.1–0.5 M sodium phosphate, pH 6.5. Fractions (0.5 ml) were collected and the position of the eluted [35S]SecA peak was determined by scintillation counting and confirmed by SDS/PAGE and fluorography. The specific activity of the purified [35S]SecA was 1.95 × 105 cpm/μg.
Inner Membrane Vesicles (IMVs).
Inner membrane vesicles were prepared as previously described (24) from E. coli BL21 that was genetically altered to overexpress SecYEGDFyajC (10), from BL21/pHAsecEYG, an overproducer of SecYEG (24), from wild-type BL21 and from E. coli KM9 (unc-::tn10, relA1, spoT1, metB1; ref. 25). The membranes were treated with 6 M urea (30 min at 0°C) to remove and inactivate endogenous SecA (20).
Formation of Membrane-Inserted SecA.
For formation of the protease-protected SecA domains, 10 μl of 10× buffer B (500 mM KCl/10 mM DTT/500 mM Tris⋅HCl, pH 7.9), 3 μg SecB, water, 1 μl [125I]SecA (100,000 cpm/μl) or 20 μl [35S]SecA (5,000 cpm/μl), an ATP regenerating system (1 μg creatine kinase and 2 μl of 0.5 M phosphocreatine), 10 μg IMVs (or liposomes), 1 μl proOmpA [1.5 mg/ml in urea buffer (6 M urea/1 mM DTT/50 mM Tris⋅HCl, pH 7.9)], and 5 μl of 0.1 M ATP were added in sequence to a final reaction volume of 0.1 ml and incubated at 37°C for 30 min. After transfer to ice, the samples were digested with 10 μl of trypsin (10 mg/ml) for 15 min. The samples were then incubated with 15% trichloroacetic acid (TCA) for 30 min on ice, centrifuged at 4°C in a microfuge, suspended in 1 ml of ice-cold acetone, collected by centrifugation, dissolved in 30 μl of sample buffer containing 2-mercaptoethanol (26), and examined by SDS/PAGE and autoradiography or fluorography.
Limited Proteolytic Digestion of SecA in Solution.
SecA was cleaved into a characteristic set of proteolytic fragments (27) as follows: 15 μg of SecA, in some experiments with either [125I]SecA or [35S]SecA, was incubated with 0–25 μg/ml of trypsin in a final volume of 0.1 ml for 15 min on ice. Proteolysis was terminated by the addition of 0.5 mg/ml soybean trypsin inhibitor. The protein fragments were then TCA precipitated and acetone “washed” as above, heated in sample buffer (3 min, 95°C), and examined by SDS/PAGE. The gels were then stained with Coomassie brilliant blue and dried, and those samples containing radioactive SecA were exposed to film (X-Omat, Kodak).
One-Dimensional Peptide Mapping.
One-dimensional peptide maps were prepared essentially according to Cleveland et al. (28). [35S]SecA was subjected to limited trypsinolysis in solution or used in translocation reactions, both as described above, and examined by SDS/PAGE. After Coomassie staining and destaining, gel regions containing the 65-kDa fragment were excised and incubated with buffer P (1 mM EDTA/0.1% SDS/125 mM Tris⋅HCl, pH 6.8) (10 ml, 30 min). Individual gel slices were then added to the bottoms of the wells of a second gel and overlayed with 10 μl of buffer P containing 20% glycerol. Finally, 10 μl of a solution of buffer P containing 10% glycerol and 0–0.1 μg of S. aureus V8 were added to the wells, and SDS/PAGE was performed as normal except that the current was shut off for 30 min when the bromphenol blue dye front approached the bottom of the stacking gel. The resulting peptide maps were examined by fluorography.
Other Methods.
Protein concentrations were determined using Bradford reagent (Bio-Rad) with BSA as a standard. SDS/PAGE was performed using 15% gels. Densitometry was performed using a Silverscan III scanner (LaCie, Beaverton, OR) and iplabh software.
RESULTS
Radiolabel in [35S]SecA Is Evenly Distributed Throughout the Protein.
Limited tryptic digestion of SecA (102 kDa) leads to the generation of a limited number of major proteolytic fragments of apparent molecular masses of 65, 48, 38, and 30 kDa (27) (Fig. 1 Top). Amino acid sequencing of the 65-kDa and 30-kDa fragments has shown them to begin at residues 5 and 611 of the SecA polypeptide, respectively (27, 29). When [125I]SecA was subjected to identical limited proteolysis, radioactivity was almost exclusively found in the 38-kDa fragment at lower trypsin concentrations, and in the 30-kDa fragment at higher protease levels (Fig. 1 Middle). This is surprising, since the amino acid sequence of SecA reveals an even distribution of tyrosyl residues, the targets of radioiodination (29). This suggests that only tyrosyl residues in the C-terminal third of the protein are accessible for radioiodination. [125I]iodination of SecA in the presence of denaturing agents such as 6 M urea or 8 M guanidine hydrochloride prevented proper SecA refolding (not shown). Therefore, to provide an even distribution of radiolabel throughout properly folded SecA, cells genetically programmed to overproduce SecA were grown in the presence of 35S-labeled methionine and cysteine. SecA contains an even distribution of these sulfur-containing residues throughout its sequence (29). Purification and limited trypsin digestion of the metabolically labeled [35S]SecA confirmed that the radiolabel had been incorporated throughout the protein (Fig. 1 Bottom). As expected, [35S]SecA was fully functional in preprotein translocation reactions (not shown).
Figure 1.
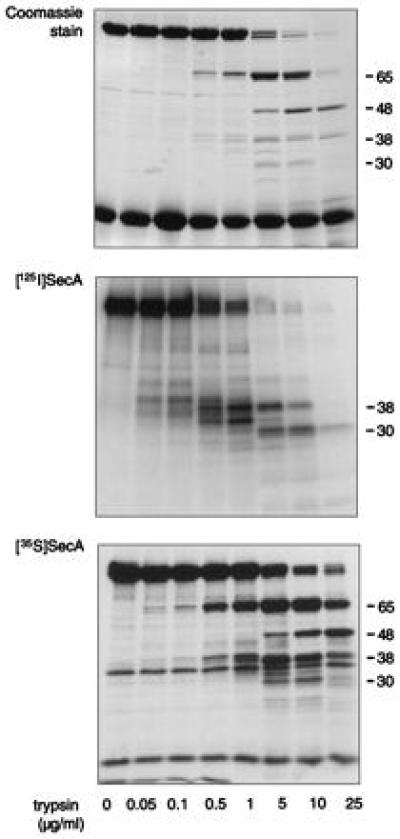
Radiolabel distribution in SecA. A limited trypsin digest of SecA (15 μg) in solution was performed using 0–25 μg/ml trypsin. The proteolytic fragments were analyzed by SDS/PAGE and stained with Coomassie brilliant blue (Top). In some cases, either [125I]SecA or [35S]SecA was included in the trypsin digest reaction and the resulting proteolytic pattern was detected by autoradiography or fluorography (Middle and Bottom, respectively). The molecular masses of the major digestion products are shown on the right.
A 65-kDa Domain of SecA Becomes Protected from Protease During Preprotein Translocation.
We have reported that SecA, upon incubation with inverted inner membrane vesicles, preprotein, and nucleotide, inserts a C-terminal 30-kDa domain into the membrane, where it is protected from proteolytic digestion (15, 27). These studies used [125I]SecA and, therefore, because of the biased distribution of the [125I] label, detected no other protease-protected domains of SecA. To determine whether any other portions of SecA are protected from proteolytic digestion, SecA insertion reactions were performed using [35S]SecA (Fig. 2A). In the absence of membranes, preprotein, or nucleotide, [35S]SecA, a 102-kDa protein (lane 1), is digested to small peptides by trypsin (1 mg/ml, 15 min on ice; lane 2). However, in translocation reactions conducted at physiological temperature with IMVs, the precursor of outer membrane protein A (proOmpA), and ATP, both a 65-kDa domain of [35S]SecA (solid arrow) and the expected 30-kDa domain (open arrow) were protected from externally added trypsin (lane 3). As for the 30-kDa domain, protection of the 65-kDa SecA domain from proteolysis was not observed in the absence of ATP or proOmpA (lanes 4 and 5, respectively) or when the reaction was conducted on ice (not shown). The requirement for SecYEG in the protection of the 65-kDa domain from proteolysis was confirmed by using liposomes in place of IMVs (lanes 6–8). Strikingly, when ATP as well as IMVs or liposomes were present, additional SecA fragments survived the proteolytic treatment (asterisks). These SecA fragments cannot, however, be related to translocation since their appearance was not dependent on the presence of preprotein or SecYEG (lanes 5 and 6 and lane 8, respectively) or physiological temperature (not shown). Because their appearance did, however, require that either IMVs or liposomes be present in addition to nucleotide, these fragments may correspond to proteolytic digestion products of SecA conformationally altered by its associations with lipid and ATP. As previously reported for the 30-kDa domain of [125I]SecA (15), the 65-kDa and 30-kDa [35S]SecA domains were also protected from proteolysis in the presence of IMVs and adenylyl-imidophosphate, a nonhydrolyzable ATP analog (not shown). Furthermore, the translocation-related 65-kDa and 30-kDa membrane-inserted SecA domains, as well as many lipid-associated SecA domains, could be protected from trypsin concentrations of up to 10 mg/ml (Fig. 2B). Similar results were obtained when proteolysis was performed using chymotrypsin or proteinase K instead of trypsin (not shown).
Figure 2.
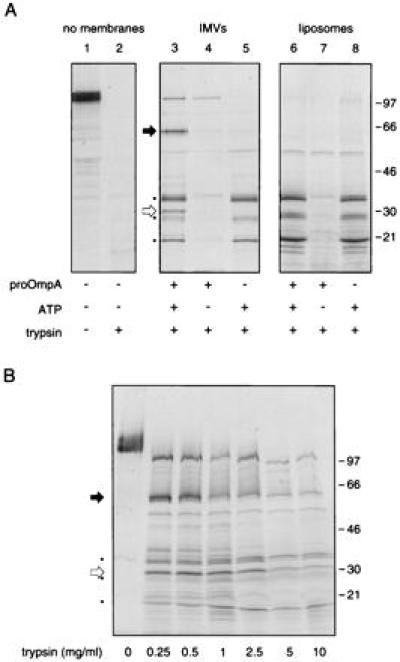
A 65-kDa domain of SecA is protected from proteolysis under translocation conditions. (A) The effect of trypsin treatment (1 mg/ml, 15 min, 0°C) on [35S]SecA was examined in the absence of membranes (lane 2) or in translocation reactions with either IMVs prepared from cells genetically enriched in SecYEGDFyajC [the integral membrane domain of preprotein translocase holoenzyme (10); lanes 3–5] or liposomes (lanes 6–8), as described. Reaction mixtures contained proOmpA and ATP (lanes 3 and 6) or either proOmpA or ATP alone (lanes 4 and 7 or 5 and 8, respectively). (B) The profile of protease-resistant [35S]SecA domains generated under translocation conditions was examined with 0–10 mg/ml trypsin. For each lane, the black arrow identifies the protease-protected SecA 65-kDa domain and the white arrow identifies the protease-protected SecA 30-kDa domain; molecular mass markers are shown on the right.
Identification of the Protease-Protected 65-kDa SecA Domain.
Because the protease-protected 65-kDa SecA domain was not observed in translocation reactions using C-terminally radiolabeled [125I]SecA (15), it seems unlikely that the protease-protected C-terminal 30-kDa domain is contained within the larger 65-kDa domain, but, rather, that the two represent unique portions of the 102-kDa SecA polypeptide. If this is indeed the case, then the 65-kDa protease-protected SecA domain would originate near the N terminus of the polypeptide. Amino acid sequencing of the 65-kDa fragment generated upon limited tryptic digestion of SecA showed this fragment to begin at the fifth amino acid residue of SecA (27). To determine whether the 65-kDa protease-protected fragment obtained from translocation reactions that contained [35S]SecA (Fig. 3, lane 2) corresponds to the 65-kDa fragment obtained from a limited trypsinolysis of [35S]SecA in solution (Fig. 3, lane 1), peptide mapping of each was performed using S. aureus V8 (Fig. 3). The almost identical digestion patterns confirm that the translocation-related protease-protected SecA 65-kDa domain originates near the N terminus.
Figure 3.
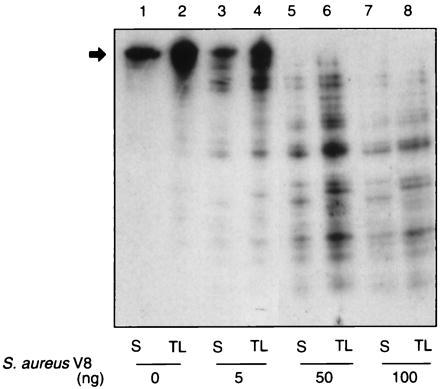
The protease-protected SecA 65-kDa domain lies near the N terminus of the protein. The [35S]SecA 65-kDa fragments resulting from limited trypsin treatment of SecA in solution (S, lanes 1, 3, 5, and 7) or protected from tryptic digestion under translocation conditions (TL, lanes 2, 4, 6, and 8) were analyzed by peptide mapping using S. aureus V8, as described. The black arrow identifies the position of the SecA 65-kDa domain.
As an independent means of mapping the protease-protected SecA 65-kDa domain, it was determined that affinity-purified anti-peptide antibodies to the N-terminal amino acid residues 8–20 (but not those to the C-terminal amino acid residues 877–890) recognized, albeit weakly, the 65-kDa band from complete translocation reactions containing IMVs, proOmpA, and ATP (not shown). The assignment of the band recognized by the SecA N-terminal antibodies as the 65-kDa protease-protected fragment was established by its absence when the translocation reactions were performed without either ATP or proOmpA.
Protease-Protection of the SecA 65-kDa Domain Occurs at SecYEG.
SecYEG is essential for the generation of both the SecA 65-kDa and 30-kDa protease-protected domains (see Fig. 2A; also ref. 15 and 24). To confirm that protection occurs at these sites, translocation experiments were performed with IMVs from cells that overexpress SecY, SecE, SecG, SecD, SecF, and YajC, integral membrane polypeptides of preprotein translocase holoenzyme (10), or IMVs from BL21 wild-type cells. [35S]SecA was mixed with increasing amounts of unlabeled SecA before being added to translocation reactions as a measure of the abundance of SecA insertion sites. In both cases, nonradioactive SecA competed with [35S]SecA for insertion sites that allow the formation of 65-kDa and 30-kDa protease-protected domains (Fig. 4 A and B). Quantitation of these data (Fig. 4C) shows that overexpression of SecYEGDFyajC provides additional sites for SecA membrane insertion, which leads to the formation of 65-kDa and 30-kDa fragments. Moreover, nonradioactive SecA competes in an identical fashion for the formation of the 65-kDa and 30-kDa protease-protected fragments in either membrane preparation. Thus, these data show that insertion of SecA at SecYEGDFyajC sites leads to protease-protection of both the 65-kDa and 30-kDa domains. The results also confirm that the binding and insertion of SecA at lipid sites (Fig. 4A, asterisks) lead, upon proteolysis, to the formation of other protease-protected fragments (see Fig. 2A). Lipid association of SecA is nonsaturable (8), as shown by the failure of nonradioactive SecA to compete for sites of formation of these fragments (Fig. 4 A and B). Furthermore, the SecA 65-kDa and 30-kDa domains formed using IMVs from either the BL21 wild-type strain or the SecYEGDFyajC-overexpressing strain are protected from proteolytic degradation, confirming that the protection is not the result of the overexpression of the membrane-embedded Sec proteins. Indeed, the 65-kDa and 30-kDa protease-protected fragments could also be generated using IMVs from E. coli strain KM9 (see below).
Figure 4.
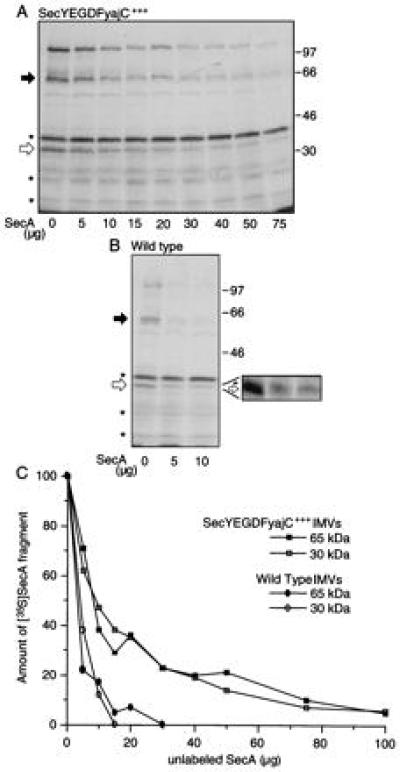
Protease protection of the SecA 65-kDa domain occurs at SecYEGDFyajC sites. [35S]SecA (5 × 104 cpm) was mixed with increasing amounts of unlabeled SecA (0–75 μg) and used in translocation reactions with IMVs (10 μg) from either SecYEGDFyajC-overexpressing cells (A) or wild-type cells (B), as described. For each lane, the black arrow identifies the protease-protected SecA 65-kDa domain and the white arrow identifies the protease-protected SecA 30-kDa domain; molecular mass markers are shown on the right. The expanded area of the gel serves to distinguish between the protease-protected 30-kDa fragment and a slightly larger band that is not related to translocation. (C) The radioactivity in protease-protected SecA 65-kDa and 30-kDa domains were determined densitometrically and expressed as a percentage of the amount of these fragments obtained in translocation reactions using [35S]SecA alone.
The 65-kDa Protease-Protected SecA Domain Inserts into the Membrane.
To determine whether the protease resistance of the SecA 65-kDa fragment was due to a protease-resistant conformation of SecA (assumed upon interaction with SecYEGDFyajC, ATP, and preprotein) or rather due to insertion into the membrane as reported for the protease-protected SecA 30-kDa domain (15), 1% Triton X-100 was added to translocation reactions containing [35S]SecA just prior to the addition of trypsin. Detergent disruption of membrane integrity allowed digestion of both the SecA 65-kDa and the 30-kDa domains (Fig. 5, lane 2). Protease-resistant SecA fragments of 38, 28, and 21 kDa (asterisks) were unaffected by the disruption of the membrane. Lipid association was required for the SecA conformational changes leading to the generation of these fragments, since complete digestion of SecA occurs in the absence of membranes (Fig. 2A, lane 2). To test whether the requirement for intact membranes for formation of protease-resistant SecA 65-kDa and 30-kDa domains simply reflected the binding of SecA to SecYEG at the membrane surface or rather indicated a more intimate relationship between SecA and the membrane, proteolytic protection of the SecA 65-kDa domain was compared in translocation reactions that were subjected to freeze/thaw either before trypsin treatment or, alternatively, in the presence of trypsin. In samples that were frozen and thawed before the addition of trypsin, protection of the SecA 65-kDa and 30-kDa domains was unaffected, as compared with samples not subjected to a cycle of freeze/thaw (compare Fig. 5, lanes 1 and 3). When, however, trypsin was added prior to membrane disruption by a freeze/thaw cycle, protease protection of the SecA-65 kDa and 30-kDa domains was greatly diminished (lane 4). Freeze/thaw in the presence of protease allows access of the protease into the IMV lumen during the reversible disruption of membrane integrity. Any membrane-traversing SecA domain would thus be available for digestion by the lumenally entrapped protease. The addition of protease subsequent to freeze/thaw would not allow access of the protease to the interior of the resealed IMVs, thereby retaining the protease inaccessibility of the SecA 65-kDa and 30-kDa fragments, as observed. These observations are consistent with a model in which the SecA 65-kDa and 30-kDa protease-protected fragments insert into and across the membrane. Other SecA fragments (asterisks) survived proteolysis even when membrane integrity was lost (lanes 2 and 4) and thus presumably originate from protease-resistant conformations of lipid-associated SecA.
Figure 5.

The protease-protected SecA 65-kDa domain inserts into the membrane. [35S]SecA (5 × 104 cpm) was used in translocation reactions with SecYEGDFyajC-enriched IMVs as described. After a 30-min incubation at 37°C, the samples were transferred to ice and either digested with trypsin (1 mg/ml, 15 min; lane 1); incubated with 1% Triton X-100 and then digested with trypsin (lane 2); frozen in liquid N2, thawed on ice (50 min), and then digested with trypsin (50 min; lane 3); or mixed with trypsin, immediately frozen, and thawed and then incubated for 50 min (lane 4). The black arrow identifies the protease-protected SecA 65-kDa domain, the white arrow identifies the protease-protected SecA 30-kDa domain, and molecular mass markers are shown on the right.
SecDFyajC Slows Membrane Deinsertion of the SecA 65-kDa Domain.
It has been reported that the protease-protected 30-kDa domain of [125I]SecA undergoes cycles of membrane insertion and deinsertion during preprotein translocation (15). To determine whether cycling was also seen for the protease-protected 65-kDa SecA domain, translocation reactions were initiated using [35S]SecA and IMVs prepared from E. coli strain KM9. Fifteen minutes later, an excess of unlabeled SecA was added and translocation was allowed to continue. Aliquots were removed at successive intervals and the level of protease-protected [35S]SecA 65-kDa domain was determined (Fig. 6A). Upon addition of unlabeled SecA, the level of the radiolabeled protease-protected SecA 65-kDa domain rapidly decreased, due to deinsertion of membrane-inserted [35S]SecA and subsequent exchange by the unlabeled SecA species. Similar behavior was observed for the protease-protected SecA 30-kDa domain, obtained from either [35S]SecA or [125I]SecA. Indeed, the kinetics of deinsertion of the protease-protected SecA 65-kDa and 30-kDa domains were identical and resembled those reported for the [125I]SecA 30-kDa fragment in earlier studies (15). In contrast, the levels of nonsaturable, lipid-associated [35S]SecA fragments, such as the 38-kDa fragment, were unaffected by an excess of unlabeled SecA.
Figure 6.
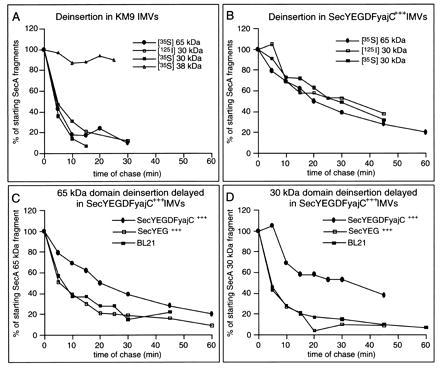
The protease-protected SecA 65-kDa domain undergoes membrane insertion and deinsertion. Nine combined translocation reactions containing [35S]SecA or [125I]SecA were performed in 0.9 ml. After a 15-min incubation at 37°C, 210 μg of unlabeled SecA was added. At 5- to 10-min intervals, 100-μl aliquots were removed, transferred to ice, and treated with trypsin. The samples were TCA precipitated, acetone “washed,” heated in sample buffer (3 min, 95°C), and examined by SDS/PAGE as above. SecA protease-protected fragments were quantitated densitometrically and expressed as a percentage of the band intensities at the time of introduction of unlabeled SecA. In terms of absolute levels, SecYEG-enriched IMVs contained higher amounts of protease-protected [35S]SecA 65-kDa and [125I]SecA 30-kDa domains as compared with wild-type IMVs, whereas even higher levels of these membrane-inserted SecA fragments were observed in IMVs obtained from SecYEGDFyajC-overproducing cells. (A) Translocation reactions using KM9 IMVs. (B) Translocation reactions using SecYEGDFyajC-enriched IMVs. (C) Levels of the protease-protected [35S]SecA 65-kDa fragment in IMVs prepared from wild-type BL21 cells or cells that overproduce either SecYEG or SecYEGDFyajC. (D) Levels of the protease-protected [125I]SecA 30-kDa fragment in IMVs prepared from wild-type BL21 cells or cells that overproduce either SecYEG or SecYEGDFyajC.
When similar experiments were performed using IMVs prepared from the SecYEGDFyajC-overproducing strain, both the [35S]SecA 65-kDa and 30-kDa protease-protected domains were chased at the same rate upon the addition of excess unlabeled SecA (Fig. 6B). Furthermore, similar deinsertion kinetics were obtained for the protease-protected [125I]SecA 30-kDa domain. Comparison of the rates of deinsertion in the two IMV preparations, however, revealed that the SecYEGDFyajC-enriched IMVs required six times longer to achieve the degree of deinsertion observed with KM9 IMVs. To determine whether this difference in the SecA insertion/deinsertion cycle was due to strain differences or to the overexpression of the membrane-embedded Sec components, preprotein translocation was initiated with [35S]SecA or [125I]SecA and then continued in the presence of excess unlabeled SecA, using IMVs prepared from wild-type BL21 cells or BL21 cells overexpressing either SecYEG or SecYEGDFyajC. The kinetics of SecA 65-kDa and 30-kDa domain deinsertion were indistinguishable in wild-type and SecYEG-enriched IMVs, and were similar to those obtained using IMVs prepared from KM9 cells. In contrast, significantly longer incubation periods were required for equivalent SecA 65-kDa and 30-kDa domain deinsertion in IMVs containing enhanced levels of SecYEGDFyajC (Fig. 6 C and D) consistent with earlier reports that SecDFyajC stabilized the inserted SecA 30-kDa domain (10, 30). As shown using IMVs from E. coli KM9 (Fig. 6A), lipid-associated SecA, assayed by the 38-kDa fragment (see Figs. 2, 4, and 5), did not deinsert in any of the three BL21 IMV preparations (not shown).
DISCUSSION
SecA exists in several distinct conformational states. Differences in SecA proteolytic digestion patterns have been observed depending on whether the protein is in solution, membrane-bound, or in the presence of ATP or preprotein (31, 32). During translocation reactions in the presence of SecYEG, nucleotide, and preprotein, a 30-kDa domain of [125I]SecA is protected from proteolytic digestion through insertion into the membrane (15). A closer examination of [125I]SecA reveals, however, that only surface-accessible tyrosine residues in the C-terminal third of the protein are radioiodinated. Metabolic [35S] labeling allows for a more uniform labeling (Fig. 1) and, hence, a broader view of SecA conformational states. With this more powerful probe, we now confirm and extend our prior findings of the dynamics of the 30-kDa C-terminal domain of SecA, report that a 65-kDa N-terminal domain of SecA has similar properties, and show that the low-affinity association of SecA with lipid allows for ATP-dependent conformational change(s) in which several protease-resistant regions are generated.
Our studies of [35S]SecA are in full agreement with our earlier studies employing [125I]SecA. When included in translocation reactions containing SecYEG, ATP, and preprotein, both forms of labled SecA are able to translocate preprotein and to protect a 30-kDa domain from externally added protease. The even distribution of radiolabel in [35S]SecA, however, allows for the detection of an additional protease-protected SecA 65-kDa domain. Protease protection of both SecA 65-kDa and 30-kDa domains have the same requirements (i.e., SecYEG, nucleotide, preprotein, and physiological temperature). In addition, the uniform distribution of radiolabel in [35S]SecA allows for differentiation between translocation-related, SecYEG-dependent SecA domains that are protected from proteolysis by the membrane permeability barrier and protease-resistant fragments that arise from nonspecific, low-affinity interactions between SecA and membrane lipids. Whereas SecA fragments of 65 kDa and 30 kDa were generated only in the presence of SecYEG, ATP, and preprotein, additional SecA fragments of 60, 38, 28, and 21 kDa were detected whenever reaction mixtures contained ATP and either liposomes or membranes. Because the generation of this latter set of SecA fragments is nonsaturable and did not require either SecYEG or preprotein, it cannot be related to translocation. Instead, it most likely results from proteolysis of lipid-associated SecA molecules in ATP-dependent conformations. Indeed, nucleotide-dependent differences in vesicle-bound SecA conformation have been reported (31, 32). Moreover, we have recently observed that two of these fragments, namely the 60-kDa and the 38-kDa species, are readily labeled by a lipid-restricted radioiodinated crosslinker (16). Although lipid-associated SecA is abundant in vitro, the in vivo abundance and physiological roles of this form of SecA are unknown.
Like the [125I]SecA 30-kDa domain (15), the protease-protected [35S]SecA 65-kDa and 30-kDa domains insert into the plane of the membrane and can be digested by protease from the periplasmic side of the inner membrane. Other researchers have also reported that SecA is accessible from the periplasm, although these studies did not directly relate the membrane-traversing SecA to translocation cycles (32, 33). Our observations suggest that the translocation-related SecA 65-kDa and 30-kDa domains cross the membrane and lie largely within the periplasmic space. Alternatively, the SecA 65-kDa and 30-kDa domains may insert far enough into the membrane (possibly within the membrane-spanning domains of SecYEG) to allow access of periplasmically localized protease to these SecA domains. The inserted 65-kDa and 30-kDa domains may have different degrees of periplasmic exposure. It is noteworthy that the membrane-inserted SecA 65-kDa and 30-kDa domains are largely shielded from the lipid phase of the membrane (16).
As for SecA, transmembrane orientation has also been proposed for the ATP-binding subunits of several other bacterial membrane transport systems. As members of the ATP-binding cassette (ABC) transporter superfamily, bacterial permeases utilize ATP energy to mediate the trafficking of a large variety of substrates. Eukaryotic members of the ABC transporter superfamily include the cystic fibrosis transmembrane conductance regulator and the P-glycoprotein involved in multidrug resistance (for review see refs. 34 and 35). Bacterial permeases can be composed of both an integral membrane protein complex and a hydrophilic ATP-binding protein. Like SecA, these ATP-binding subunits were thought to be peripheral membrane proteins. However, proteolytic digestion experiments using inverted and right-side-out membrane vesicles have revealed transmembrane topologies for the ATP-binding elements of the l-histidine transporter (HisP), the maltose transporter (MalK), and the polysialic acid transporter (KpsT) (36–38). Notably, increased proteolysis was observed with the E150G mutant of KpsT, which binds ATP and substrate but cannot hydrolyse the bound ATP and is thereby “locked” in a transmembrane state (38). This is similar to preprotein translocase studies using the analagous D209N SecA mutant, in which a blockage of ATP hydrolysis prevented the deinsertion of membrane-inserted SecA (30).
The inserted SecA 65-kDa and 30-kDa domains can deinsert, as revealed by a replacement of [35S]SecA by unlabeled SecA. Such replacement must entail both deinsertion of inserted [35S]SecA to the bound state at SecYEG sites on the cytoplasmic surface of the membrane and dissociation from these sites into solution. Because SecA is bound with high affinity to the membranes at these SecYEG sites under translocation conditions (8, 24), much of the deinserted and bound [35S]SecA may reinsert rather than disassociate from the membrane. Thus, our replacement assay provides only a minimum estimate of the true rate of deinsertion. Earlier studies using [125I]SecA reported that SecA membrane insertion/deinsertion cycling at SecYEG involves distinct ATP binding and hydrolysis events and is regulated by SecDF (15, 30). In agreement with these findings, we find that the membrane-inserted SecA 65-kDa and 30-kDa domains show similar, rapid kinetics of deinsertion using IMVs from E. coli KM9 or BL21 wild-type strains. Overproduction of SecYEG provides more sites for SecA binding and insertion (24) but did not affect the rate of chase of inserted SecA by unlabled SecA. Overproduction of SecDFyajC, however, significantly slowed the deinsertion of inserted SecA in the SecYEG-overproducing strain. The SecDFyajC-mediated effect on SecA deinsertion could be a result of stabilization of the inserted form of SecA or slowing of an active deinsertion process.
Using [35S]SecA, Tai and coworkers (39) have also recently examined the translocation-related SecA membrane insertion/deinsertion cycle. In most of their experiments, without proteolysis to generate distinct lipid-bound and SecYEG-inserted SecA fragments, Chen et al. could not observe chase of membrane-inserted SecA. In our current study, membrane-inserted [35S]SecA 65-kDa and 30-kDa domains could be clearly chased upon addition of an excess of unlabeled SecA, due to deinsertion and exchange of radiolabeled SecA by its unlabeled counterpart, by using four different IMV preparations. The levels of nonspecifically bound, lipid-associated [35S]SecA fragments remained unchanged during translocation reactions performed in the presence of excess unlabeled SecA, reflecting the inability of these non-translocation-related SecA fragments to dissociate from the membrane. Without proteolysis to differentiate between the two species of membrane-associated SecA, the abundant lipid-associated, nonsaturatably bound SecA might have masked the chase of SecA inserted at high-affinity SecYEG sites in the studies of Chen et al. (39). These authors do not address the amount of specifically bound, SecYEG-associated SecA in their system. Indeed, when Chen et al. performed proteolysis, they observed SecA 66-kDa and 28-kDa fragments that were maximally protected from proteinase K digestion in the presence of ATP and preprotein and that could be chased by excess unlabeled SecA in an energy-dependent manner. These SecA 66-kDa and 28-kDa proteinase K-generated fragments most likely correspond to the 65-kDa and 30-kDa fragments produced by trypsin digestion in the present study. Chen et al. (39) report that a significant amount of 66-kDa fragment is found in the supernatant. This may represent protease-protected membrane-inserted SecA fragments that dissociate because of extensive digestion of SecYEG, the site of SecA insertion. We have not examined this phenomenum in our system. In contrast to the present study, Chen et al. (39) observed only a moderate increase (less than 3-fold) in these protease-protected fragments upon addition of ATP and preprotein. The reasons for this difference is unclear, though we find reproducible differences in the levels of protease-protected, membrane-inserted fragments formed using IMVs generated from different strains (not shown). We have shown that insertion of the translocation-related SecA 65-kDa and 30-kDa fragments occurs at SecYEG (Fig. 4). In the present study, IMVs were prepared from E. coli with overexpressed SecYEGDFyajC. These IMVs display a 10- to 20-fold higher number of SecA binding sites as compared with the BL21 wild-type strain (10). Use of these IMVs highlights the presence of the protease-protected, translocation-related SecA 65-kDa and 30-kDa fragments. These bands were, however, also readily detected using IMVs prepared from either the wild-type BL21 parent strain (Fig. 4B) or from E. coli strain KM9 (Fig. 6A).
In accord with its ability to interact with almost all of the components required for preprotein translocation, SecA contains distinct regions dedicated to its different functions. SecA contains a high-affinity ATP binding domain encompassing amino acid residues 102–210 and a low-affinity ATP binding domain between amino acid residues 503 and 653 (18, 19). Only the high-affinity ATP binding site is essential for SecA insertion and deinsertion, whereas the low-affinity domain is required for preprotein translocation (30). One site for preprotein binding is located between amino acid residues 267 and 340 (12), whereas suppressors of signal-sequence mutations have been mapped to SecA residues 111, 373, and 488 (40). The protease-protected SecA 65-kDa domain, originating near the N terminus and extending to near amino acid residue 570, should contain both the high-affinity ATP binding site as well as the proposed sites for preprotein binding. Indeed, the SecA 65-kDa domain was labeled in cross-linking studies using [32P]ATP (not shown). It is possible, therefore, that membrane insertion of the SecA 65-kDa domain plays a direct role in preprotein translocation. Forward movement of preprotein may involve the SecA membrane insertion/deinsertion cycle, since ATP-driven translocation of preprotein occurs in a stepwise manner (14, 41). In solution, SecA exists as a dimer and may function as a dimer during active preprotein translocation (42, 43). Taken together, the protease-protected N-terminal 65-kDa domain and the C-terminal 30-kDa domain account for almost the full length of the 102-kDa SecA monomer. It is, however, unclear whether the membrane-inserted 65-kDa and 30-kDa domains are derived from the same SecA molecule or whether they originate from different halves of a single SecA dimer. Indeed, it is not yet known whether both monomers of a SecA dimer insert into the membrane. Until such questions are answered, it is difficult to assess the relative contributions of the SecA 65-kDa and 30-kDa domains to translocation. Future experiments must address these points for a clearer picture of the role of the SecA membrane insertion/deinsertion cycle in preprotein translocation to emerge.
Acknowledgments
This work was supported by the National Institute of General Medical Sciences. J.E. was supported by a long-term fellowship from the Human Frontiers Science Program Organization.
References
- 1.Schatz G, Dobberstein B. Science. 1996;271:1519–1526. doi: 10.1126/science.271.5255.1519. [DOI] [PubMed] [Google Scholar]
- 2.Wickner W, Driessen A J M, Hartl F-U. Annu Rev Biochem. 1991;60:101–124. doi: 10.1146/annurev.bi.60.070191.000533. [DOI] [PubMed] [Google Scholar]
- 3.Pugsley A P. Microbiol Rev. 1993;57:50–108. doi: 10.1128/mr.57.1.50-108.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Arkowitz R A, Bassilana M. Biochim Biophys Acta. 1994;1197:311–343. doi: 10.1016/0304-4157(94)90012-4. [DOI] [PubMed] [Google Scholar]
- 5.Ito K. Genes Cells. 1996;1:337–346. doi: 10.1046/j.1365-2443.1996.34034.x. [DOI] [PubMed] [Google Scholar]
- 6.Wiess J B, Ray P H, Bassford P J., Jr Proc Natl Acad Sci USA. 1988;85:8978–8982. doi: 10.1073/pnas.85.23.8978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brundage L, Hendrick J P, Schiebel E, Driessen A J M, Wickner W. Cell. 1990;62:649–657. doi: 10.1016/0092-8674(90)90111-q. [DOI] [PubMed] [Google Scholar]
- 8.Hartl F-U, Lecker S, Schiebel E, Hendrick J P, Wickner W. Cell. 1990;63:269–279. doi: 10.1016/0092-8674(90)90160-g. [DOI] [PubMed] [Google Scholar]
- 9.Lill R, Dowhan W, Wickner W. Cell. 1990;60:271–280. doi: 10.1016/0092-8674(90)90742-w. [DOI] [PubMed] [Google Scholar]
- 10.Duong F, Wickner W. EMBO J. 1997;16:2756–2768. doi: 10.1093/emboj/16.10.2756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Akita M, Sasaki S, Matsuyama S, Mizushima S. J Biol Chem. 1990;265:8164–8169. [PubMed] [Google Scholar]
- 12.Kimura E, Akita M, Matsuyama S-i, Mizushima S. J Biol Chem. 1991;266:6600–6606. [PubMed] [Google Scholar]
- 13.Tani K, Shiozuka K, Tokuda H, Mizushima S. J Biol Chem. 1989;264:18582–18588. [PubMed] [Google Scholar]
- 14.Schiebel E, Driessen A J M, Hartl F-U, Wickner W. Cell. 1991;64:927–939. doi: 10.1016/0092-8674(91)90317-r. [DOI] [PubMed] [Google Scholar]
- 15.Economou A, Wickner W. Cell. 1994;78:835–843. doi: 10.1016/s0092-8674(94)90582-7. [DOI] [PubMed] [Google Scholar]
- 16.Eichler J, Brunner J, Wickner W. EMBO J. 1997;16:2188–2196. doi: 10.1093/emboj/16.9.2188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Joly J C, Wickner W. EMBO J. 1993;12:255–263. doi: 10.1002/j.1460-2075.1993.tb05651.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Matsuyama S, Kimura E, Mizushima S. J Biol Chem. 1990;265:8760–8765. [PubMed] [Google Scholar]
- 19.Mitchell C, Oliver D B. Mol Microbiol. 1993;10:483–497. doi: 10.1111/j.1365-2958.1993.tb00921.x. [DOI] [PubMed] [Google Scholar]
- 20.Cunningham K, Lill R, Crooke E, Rice M, Moore K, Wickner W, Oliver D. EMBO J. 1989;8:955–959. doi: 10.1002/j.1460-2075.1989.tb03457.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Crooke E, Brundage L, Rice M, Wickner W. EMBO J. 1988;7:1831–1835. doi: 10.1002/j.1460-2075.1988.tb03015.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cabelli R J, Chen L, Tai P C, Oliver D B. Cell. 1988;55:683–692. doi: 10.1016/0092-8674(88)90227-9. [DOI] [PubMed] [Google Scholar]
- 23.Sambrook J, Fritsch D B, Maniatis T. Molecular Cloning: A Laboratory Manual. Cold Spring Harbor, NY: Cold Spring Harbor Lab. Press; 1989. [Google Scholar]
- 24.Douville K, Price A, Eichler J, Economou A, Wickner W. J Biol Chem. 1995;270:20106–20111. doi: 10.1074/jbc.270.34.20106. [DOI] [PubMed] [Google Scholar]
- 25.Klionsky D J, Brusilow W A, Simoni R D. J Bacteriol. 1984;160:1055–1066. doi: 10.1128/jb.160.3.1055-1060.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brundage L, Fimmel C J, Mizushima S, Wickner W. J Biol Chem. 1992;267:4166–4170. [PubMed] [Google Scholar]
- 27.Price A, Economou A, Duong F, Wickner W. J Biol Chem. 1996;271:31580–31584. doi: 10.1074/jbc.271.49.31580. [DOI] [PubMed] [Google Scholar]
- 28.Cleveland D W, Fischer S G, Kirschner M W, Laemmli U K. J Biol Chem. 1977;252:1102–1106. [PubMed] [Google Scholar]
- 29.Schmidt M G, Rollo E, Grodberg E, Oliver D B. J Bacteriol. 1988;170:3404–3414. doi: 10.1128/jb.170.8.3404-3414.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Economou A, Pogliano J A, Beckwith J, Oliver D B, Wickner W. Cell. 1995;83:1171–1181. doi: 10.1016/0092-8674(95)90143-4. [DOI] [PubMed] [Google Scholar]
- 31.Shinkai A, Mei L H, Tokuda H, Mizushima S. J Biol Chem. 1991;266:5827–5833. [PubMed] [Google Scholar]
- 32.den Blaauwen T, Fekkes P, de Wit J G, Kuiper W, Driessen A J M. Biochemistry. 1996;35:11994–12004. doi: 10.1021/bi9605088. [DOI] [PubMed] [Google Scholar]
- 33.Kim Y J, Rajapandi T, Oliver D. Cell. 1994;78:845–853. doi: 10.1016/s0092-8674(94)90602-5. [DOI] [PubMed] [Google Scholar]
- 34.Ames G F-L, Mimura C S, Shyamala V. FEMS Microbiol Rev. 1990;75:429–446. doi: 10.1111/j.1574-6968.1990.tb04110.x. [DOI] [PubMed] [Google Scholar]
- 35.Higgins C F. Annu Rev Cell Biol. 1992;8:67–113. doi: 10.1146/annurev.cb.08.110192.000435. [DOI] [PubMed] [Google Scholar]
- 36.Baichwal V, Liu D, Ames G F-L. Proc Natl Acad Sci USA. 1993;90:620–624. doi: 10.1073/pnas.90.2.620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schneider E, Hunke S, Tebbe S. J Bacteriol. 1995;177:5364–5367. doi: 10.1128/jb.177.18.5364-5367.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bliss J M, Silver R P. J Bacteriol. 1997;179:1400–1403. doi: 10.1128/jb.179.4.1400-1403.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chen X, Xu H, Tai P C. J Biol Chem. 1996;271:29698–29706. doi: 10.1074/jbc.271.47.29698. [DOI] [PubMed] [Google Scholar]
- 40.Fikes J D, Bassford P J., Jr J Bacteriol. 1989;171:402–409. doi: 10.1128/jb.171.1.402-409.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Uchida K, Mori H, Mizushima S. J Biol Chem. 1995;270:30862–30868. doi: 10.1074/jbc.270.52.30862. [DOI] [PubMed] [Google Scholar]
- 42.Akita M, Shinkai A, Matsuyama S, Mizushima S. Biochem Biophys Res Commun. 1991;174:211–216. doi: 10.1016/0006-291x(91)90507-4. [DOI] [PubMed] [Google Scholar]
- 43.Driessen A J M. Biochemistry. 1993;32:13190–13197. doi: 10.1021/bi00211a030. [DOI] [PubMed] [Google Scholar]